
Abstract
Although circadian and sleep disorders are frequently associated with autism spectrum disorders (ASD), it remains elusive whether clock gene disruption can lead to autistic-like phenotypes in animals. The essential clock gene Bmal1 has been associated with human sociability and its missense mutations are identified in ASD. Here we report that global Bmal1 deletion led to significant social impairments, excessive stereotyped and repetitive behaviors, as well as motor learning disabilities in mice, all of which resemble core behavioral deficits in ASD. Furthermore, aberrant cell density and immature morphology of dendritic spines were identified in the cerebellar Purkinje cells (PCs) of Bmal1 knockout (KO) mice. Electrophysiological recordings uncovered enhanced excitatory and inhibitory synaptic transmission and reduced firing rates in the PCs of Bmal1 KO mice. Differential expression of ASD- and ataxia-associated genes (Ntng2, Mfrp, Nr4a2, Thbs1, Atxn1, and Atxn3) and dysregulated pathways of translational control, including hyperactivated mammalian target of rapamycin complex 1 (mTORC1) signaling, were identified in the cerebellum of Bmal1 KO mice. Interestingly, the antidiabetic drug metformin reversed mTORC1 hyperactivation and alleviated major behavioral and PC deficits in Bmal1 KO mice. Importantly, conditional Bmal1 deletion only in cerebellar PCs was sufficient to recapitulate autistic-like behavioral and cellular changes akin to those identified in Bmal1 KO mice. Together, these results unveil a previously unidentified role for Bmal1 disruption in cerebellar dysfunction and autistic-like behaviors. Our findings provide experimental evidence supporting a putative role for dysregulation of circadian clock gene expression in the pathogenesis of ASD.
Similar content being viewed by others


Introduction
The circadian clock orchestrates rhythmic gene expression, neuronal activities, and animal behaviors based on the time of day [1, 2]. In cells circadian gene expression is driven by transcriptional-translational genetic feedback loops [3]. In mammals, the transcription factors CLOCK (Circadian Locomotor Output Cycles Kaput) or NPAS2 (Neuronal PAS domain protein 2) and BMAL1(Brain and Muscle ARNT-Like 1) form heterodimers and activate rhythmic transcription of Period (Per) and Cryptochrome (Cry) genes. The clock gene Bmal1 (Arntl or Mop3) is essential for proper functioning of the molecular clock. Bmal1 deletion leads to complete loss of circadian rhythms in cells and mice [4].
Autism spectrum disorders (ASDs) are a group of complex neurodevelopmental disorders that are diagnosed based on behavioral manifestations including impaired social interaction and communication, as well as stereotyped and repetitive body movements [5] (DSM-5, American Psychiatric Association, 2013). Although a causal role for circadian clock dysfunction in ASD pathogenesis remains elusive, circadian and sleep problems are frequent comorbidities with ASD [6]. De novo loss-of-function variants of the clock genes Per1, Per2, Timeless, Bmal1 and Npas2 have been identified in ASD [7,8,9] and multiple clock genes are included in the database of autism-associated genes [10]. Together, the clinical evidence suggests that circadian dysfunction may be associated with the pathogenesis of ASD in some patients. To date, however, no experimental paradigm has established a direct link between clock gene disruption and autistic-like phenotypes in animals.
When deleted, Bmal1 is the only core clock gene that leads to abolition of the molecular clock function and circadian arrhythmicity in mammals [4]. Interestingly, Bmal1 has recently been associated with human sociability [11], impairment of which is a hallmark of ASD. In addition, missense mutations of Bmal1 have been identified in ASD [9]. Besides its cardinal role in driving circadian gene transcription, BMAL1 also functions as a key regulator of protein synthesis (mRNA translation) [12]. Importantly, aberrant protein synthesis is thought as a key pathway that may lead to autistic phenotypes [13]. Dysregulation of mRNA translation is implicated in the pathogenesis of ASD-like behavioral phenotypes in animals [14, 15]. Together, these lines of evidence suggest a potential role for Bmal1 in the pathogenesis of ASD. Here using a multidisciplinary approach, we investigated whether genetic deletion of Bmal1 would lead to autistic-like phenotypes in mice. Our results identify a previously unidentified role for Bmal1 in regulating cerebellar gene expression and autistic-like animal behavior.
Materials and methods
Detailed Materials and Methods can be found in Supplementary Materials and Methods.
Animals
All mice were on a C57BL/6J background. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Minnesota.
Mouse behavioral tests
Unless otherwise indicated, all behavioral tests were performed under dim red light (~20 lux at cage level) between Zeitgeber time (ZT) 20~22 (8~10 h after light-off). All tests were video recorded and analyzed after the tests by researchers that were blinded to the group information.
Chronic metformin administration
Mice were injected i.p. with metformin (200 mg/kg, MP Biomedicals, Solon, OH) or equal volume of saline (10 µl x body weight in grams) at ZT 20 (4 h before light-ON) once daily for 10 d before being used for tests.
Brain tissue processing, immunostaining, and microscopic imaging analysis
Mice were sacrificed by cervical dislocation and decapitation and brains were quickly harvested. Brain tissue was separately processed for immunostaining. Images were analyzed using the Adobe Photoshop software (Adobe Systems Incorporated, San Jose, CA) and ImageJ software (version 1.52e).
Analysis of dendritic morphology of cerebellar Purkinje cells by Golgi-Cox staining
Rapid GolgiKit (FD NeuroTechnologies, MD) was used to perform Golgi-Cox staining. Dendrites of cerebellar Purkinje cells were analyzed based on published methods [16].
Acute cerebellar slice electrophysiology
Acute sagittal cerebellar slices (250 μm thick) were obtained from 3- to 4-month-old mice. Recordings were obtained from lobules V and VI Purkinje cells (PCs).
Protein extraction and western blotting analysis was performed as described [17]
RNA sequencing, Ribosome profiling, and bioinformatic analysis were performed as described [18].
Statistical analysis
An estimate of variation within each group was performed and variances between the groups were statistically compared before the mean values were statistically compared between groups. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications [14, 19]. All samples were included in the statistical analysis. Animals were allocated into experimental groups based on genotypes. In the same genotype group animals were allocated into subgroups using a Random Number Table. Statistical analysis was performed, and graphs were plotted using GraphPad Prism 7 (GraphPad Software, La Jolla, CA). Data are presented as individual values as well as mean ± standard error of mean (SEM) or mean ± SEM only. P < 0.05 was considered as statistically significant. Statistical details of experiments can be found in the Figure legends and/or in the Results. Descriptive statistical results are included in Supplementary Table 3.
Results
Bmal1 KO mice exhibit deficits in social interaction and recognition, excessive repetitive behaviors, and stereotypy
We first used the three-chamber test to measure sociability and preference for social novelty in Bmal1 KO mice and wild-type (WT) littermates (Fig. 1a). The WT mice spent significantly longer time in the chamber of and interacting with a caged stranger mouse (S), as compared to an empty wire cup (E), demonstrating robust sociability (P < 0.0001, Fig. 1a). In stark contrast, the KO littermates displayed impaired social approach behavior, as evidenced by the similar time spent in the two chambers and in sniffing the two-wire cages (P = 0.250, Fig. 1a). Next, the WT mice spent significantly more time in the chamber with the novel stranger (S2) than in the chamber with the familiar stranger (S1) and more time sniffing the novel stranger cup (P = 0.002, Supplementary Fig. 1a), indicating a preference for social novelty. Similar to the WT mice, the KO mice exhibited intact preference for social novelty (P = 0.007, Supplementary Fig. 1a). For both phases of the three-chamber test, the number of entries to the two side-chambers was not different in WT or KO mice (Supplementary Fig. 1b). However, the KO mice exhibited a greater number of overall entries compared with the WT mice (social approach: P < 0.0001, social novelty: P = 0.002, Supplementary Fig. 1b), which suggests hyperactivity in KO mice.

a Three-chamber test for mouse sociability. Top: a schematic diagram and representative heat maps of a Bmal1 WT mouse and a Bmal1 KO mouse in the three-chamber test. S: stranger 1, C: center, E: empty. The heat map indicates time spent on the location. Bottom: bar graphs indicating time spent in individual chambers (left, F (2, 57) chamber × genotype = 8.735, P = 0.001, two-way ANOVA) and time spent sniffing wire cages (right, F (1, 38) chamber × genotype = 5.451, P = 0.025, two-way ANOVA) during the three-chamber test. Note that the KO mice spent similar time in the S chamber as in the E chamber (Bmal1 KO: S vs. E, P = 0.421, post hoc Bonferroni’s comparison). The KO mice also spent similar time sniffing S cage compared to the E cage (Bmal1 KO: S vs. E, P = 0.250, post hoc Bonferroni’s comparison). n = 10–11 mice/group. b Reciprocal social interaction test. Left: schematic diagram indicates the setup of the test. Middle: bar graphs indicating time spent on nose - anogenital sniffing (t (20) = 4.265, P = 0.0004, Student’s t-test) and nose - nose sniffing (t (20) = 1.685, P = 0.108, Student’s t-test). Right: time spent in interactions including push-crawling and following activities (t (20) = 3.277, P = 0.004, Student’s t-test). n = 11 mice/group. c Olfactory habituation test. A schematic diagram is shown on the left. Sniffing time is shown on the right. W: water, C: cinnamon, B: butter, S: social odor (dirty cage swabs). Notably, time spent sniffing the social odor was markedly reduced, whereas time sniffing non-social odors was not changed in the KO mice compared with the WT mice (F (11,55) odor × genotype = 7.168, P < 0.0001, two-way ANOVA). n = 6 mice/group. Data are presented as mean ± standard error of the mean (SEM). ****P < 0.0001. d Analysis of mouse grooming. Top: spontaneous grooming. A diagram is on the left and bar graphs are on the right indicating grooming bouts (t (17) = 3.514, P = 0.003, Student’s t-test) and time (t (17) = 0.341, P = 0.372, Student’s t-test). n = 9–10 mice/group. Bottom: water-puff induced grooming. A diagram is shown on the left and bar graphs on the right indicate grooming bouts (t (13) = 3.839, P = 0.002, Student’s t-test) and time (t (13) = 2.365, P = 0.034, Student’s t-test). n = 7–8 mice/group. e Analysis of route tracing behavior. Route tracing is defined as a mouse repeatedly following the same route for at least three times on the cage floor. Top: A diagram is shown on the left and trace maps of route tracing episodes (20 s) are on the right. Bottom: bar graphs indicate numbers of bouts (t (15) = 3.010, P = 0.009, Student’s t-test), duration (t (15) = 3.786, P = 0.002, Student’s t-test), and percentage of time engaging in route tracing (t (15) = 3.833, P = 0.002, Student’s t-test). n = 8–9 mice/group. All data in (a), (b), (d), and (e) are presented as individual values and mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant (Supplementary Fig. 1; see Supplementary Table 3 for detailed statistics).
In a reciprocal social interaction test, the KO mice spent less time engaging in social interactions including anogenital sniffing, push-crawl, and following, compared with the WT mice (P = 0.0004 and P = 0.004 respectively, Fig. 1b). However, the time spent nose-nose sniffing was not different between the WT and the KO mice (P = 0.108, Fig. 1b). In an olfactory habituation-dishabituation test, mice were exposed to a series of non-social (water, cinnamon, or butter) and social odors (mouse cage swabs) (Fig. 1c). Neither the WT nor the KO mice exhibited a preference for the cinnamon odor compared to the water control. All mice spent a significantly longer time sniffing the butter odor compared to the water control, and the sniffing time was not different between the WT and the KO mice (~ 15 s) (Fig. 1c). Repeated exposure to butter odor resulted in diminished sniffing time in mice of both genotypes, indicating rapid habituation to the odor (Fig. 1c), indicating that the olfactory function is intact in KO mice. Importantly, however, the WT mice spent a longer time sniffing the cage swab (~32 s) compared with the butter odor. In contrast, the KO mice exhibited markedly reduced preference towards the cage swab (~15 s) compared with the WT mice (KO vs. WT, P < 0.0001, Fig. 1c). These results demonstrate olfactory social recognition is impaired in Bmal1 KO mice.
We next assessed mouse repetitive and stereotypic behaviors. The KO mice exhibited a significantly increased number of spontaneous grooming bouts (P = 0.003), but there was no difference in overall time spent grooming (P = 0.372, Fig. 1d). Likewise, water puff-induced grooming was also increased significantly in KO mice compared with the WT mice (bouts: P = 0.002, time: P = 0.034, Fig. 1d). Mouse route tracing is defined as repeated back-and-forth moving following the same routes. The number of bouts and duration of route tracing, as well as the percentage of time spent route tracing were increased in the KO mice compared with the WT mice (bouts: P = 0.009, duration: P = 0.002, percentage of time: P = 0.002, Fig. 1e), indicating increased behavioral stereotypy in KO mice. The KO mice displayed markedly reduced activities in marble-burying and nestlet shredding tests compared with the WT mice (marble burying: P = 0.0003, nest building: P = 0.016, Supplementary Fig. 1c and d), which may indicate a lack of interest in novel environments or objects in KO mice. No significant difference was found in the body weight between WT and KO mice (Supplementary Fig. 1e). Compared with the WT mice, the KO mice spent less time in the center and more time in the corner zones (center zone, P = 0.004; corner zone, P < 0.0001, Supplementary Fig. 1f) in the open field test, indicating anxiety-like behaviors in KO mice. Notably, the KO mice traveled a longer distance compared with the WT mice (P = 0.003, Supplementary Fig. 1f), which is consistent with hyperactivity demonstrated in the three-chamber test (Supplementary Fig. 1b). The KO mice exhibited shorter immobility time in the forced swim test (P = 0.016, Supplementary Fig. 1g) and decreased latency to feed in the novelty suppressed feeding test (P = 0.009, Supplementary Fig. 1h), counter indicative of depressive-like behaviors in these mice. Taken together, these results indicate that Bmal1 deletion leads to significant deficits in social interaction, olfactory social recognition, excessive repetitive behaviors, and stereotypy, all of which resemble core behavioral hallmarks in ASD.
Motor learning deficits and cerebellar pathological changes in Bmal1 KO mice
Impaired performance of basic motor skills is consistently reported in ASD [20]. In the rotarod test the KO mice needed significantly more pre-training trials than the WT mice to reach criterion (8 to 1 respectively, P = 0.001, Fig. 2a). During the training trials, the KO mice displayed markedly shorter latencies to fall and fell at slower rotating speeds compared with the WT mice (P < 0.0001, Fig. 2a). In contrast to the WT mice, the KO performance was not improved by training (Fig. 2a). Next, by mouse gait analysis we found that the stride length was decreased (P = 0.008) but the bases of support were not changed in the KO mice as compared to the WT mice (fore paw: P = 0.608, hind paw: P = 0.579, Supplementary Fig. 2a). Together, these results demonstrate significant deficits in motor learning and coordination in KO mice suggesting potential functional changes in the cerebellum.

a Mouse rotarod test. Left: A diagram of the rotarod test equipment. Middle: A bar graph indicates numbers of pre-trials (t (14) = 3.981, P = 0.001, Student’s t-test). Note that the Bmal1 KO mice required more pre-training trials before the rotarod test. Right: Line graphs indicate latencies to fall and speeds at fall. Note that the KO mice exhibited markedly shorter latency to fall and lower speed at falling. The performance in KO mice was not improved over the eight trials compared with the WT littermates (F (1,112) genotype = 169.7, P < 0.0001, two-way ANOVA). n = 8 mice/group. Data are shown as mean ± SEM. **P < 0.01, ****P < 0.0001. b Expression of Calbindin-D (28k), a Purkinje cell marker. Left: representative microscopic images of cerebellar lobule V immunolabeled for Calbindin-D (red) and counterstained by DAPI (blue). Scale bar = 50 μm. Right: quantitation of Purkinje cell density and soma length is shown in box plots. The whiskers indicate the range from minimum to maximum and the boxes indicate the range from the 25th to 75th percentiles. The lines in the box indicate the medians. Note that Purkinje cell density was increased in the KO cerebellum compared with WT cerebellum (t (103) = 5.493, P < 0.0001, Student’s t-test). n = 50–55 cells, 3-4 mice/group. c Golgi-Cox staining of Purkinje cells. Left: representative microscopic images of Golgi-Cox staining indicating dendritic morphology of Purkinje cells. Scale bar = 10 μm for low and 3 μm for high magnification images. Right: Quantitative analysis of density and morphology of dendric spines in box plots. The whiskers indicate the range from minimum to maximum and the boxes indicate the range from the 25th to 75th percentiles. The lines in the box indicate the medians. Note that the number of dendritic spines was increased (t (58) = 8.187, P < 0.0001, Student’s t-test), and more spines were of immature morphology in KO mice compared with WT mice (F (1,116) fraction × genotype = 155.5, P < 0.0001, immature fraction Bmal1 WT vs. Bmal1 KO: P < 0.0001, two-way ANOVA). n = 30 dendrite branches, 3 mice/group. ***P < 0.001, ****P < 0.0001 (Supplementary Fig. 2; see Supplementary Table 3 for detailed statistics).
To identify potential pathological changes in the cerebellum of Bmal1 KO mice, we first performed hematoxylin and eosin staining and found no significant difference in the thickness of the molecular layer but a moderate decrease in the thickness of the granular layer in KO mice compared with the WT mice (P = 0.005, Supplementary Fig. 2c). Next, immunostaining for Bmal1 indicated enrichment in the cerebellum of the WT mice, whereases no Bmal1 was detected in the brain of KO mice (Supplementary Fig. 2b). Interestingly, immunostaining for calbindin-D (28k), a specific marker for cerebellar Purkinje cells (PCs) [21], revealed a significant increase of PC density and soma length in the cerebellum of KO mice compared with the WT mice (density: P < 0.0001, soma length: P = 0.0004, Fig. 2b). Immunostaining for Fox2 identified a significant decrease of cerebellar interneurons in the molecular layer of the KO mice (P < 0.0001, Supplementary Fig. 2d). Immunostaining for NeuN did not detect any difference in the number of cerebellar granule cells between WT and KO mice (P = 0.479, Supplementary Fig. 2e). By Golgi–Cox staining we found the density of dendritic spines in the PC was increased in the KO mice compared with the WT mice (P < 0.0001, Fig. 2c). The fraction of immature dendritic spines (filopodial or branched) was increased, whereas the fraction of mature dendritic spines (stubby or mushroom shaped) was decreased in the PC of KO mice (P < 0.0001, Fig. 2c). Together, these results demonstrate abnormal PC density and dendritic spine morphology, suggesting developmental deficits in the cerebellum of Bmal1 KO mice.
Electrophysiological changes in cerebellar Purkinje cells of Bmal1 KO mice
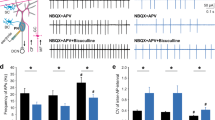
PCs receive an excitatory synaptic input from a single climbing fiber and several weaker inputs from parallel fibers (Fig. 3a and b). PCs also receive inhibitory inputs from the interneurons, stellate and basket cells (Fig. 3b). Excitatory postsynaptic currents (EPSCs) elicited by parallel fiber stimulation gradually increased in response to increasing stimulus intensity in WT and KO mice. However, an ~ 2-fold increase in EPSC amplitudes was found in the KO mice compared with the WT mice (P < 0.001, Fig. 3c). When two consecutive stimuli to parallel fibers were applied, the second response was larger than the first (pair pulse facilitation) and the pair pulse ratios (PPRs) were not different between WT and KO mice (P = 0.647, Supplementary Fig. 3a). Responses elicited by climbing fiber stimulation were characterized by their large amplitude (1-2 nA) and their all-or-none responses in WT and KO mice (Fig. 3d). Interestingly, a marked increase (~ 3-fold) in EPSC amplitudes was found in the KO mice compared with the WT mice (P < 0.001, Fig. 3d). When climbing fibers were stimulated the synaptic current was reduced in the second pulse (pair pulse depression) and PPR were not different between WT and KO mice (P = 1.000, Supplementary Fig. 3b). These results indicate that excitatory synaptic transmission onto PCs is altered in the Bmal1 KO mice due to postsynaptic mechanisms.

a Differential interference contrast (DIC) image of a sagittal cerebellar section of a WT mouse with lobules labeled with roman numbers. PCs were recorded from lobules V and VI on both sides of the primary fissure (pf, framed region). b Schematic representation of lobules V and VI (left) and synaptic inputs onto PCs (right) (ML: Molecular layer, PCL: Purkinje cell layer, GCL: granule cell layer, GC: granule cells, PC: Purkinje cell, BC: basket cells, SC: stellate cells). c Input-Output relationships of excitatory postsynaptic currents (EPSCs) evoked by parallel fiber stimulation (WT, n = 10 cells; KO, n = 14 cells; two-way ANOVA with post hoc Bonferroni’s comparisons). Representative traces are shown in the top inset. d Input-Output relationships of EPSCs evoked by climbing fiber stimulation (WT, n = 9 cells; KO, n = 6 cells; two-way ANOVA with post hoc Bonferroni’s comparisons). Representative traces are shown in the top inset. e Input-Output relationships of IPSCs obtained at Vh = +35 mV (WT, n = 16; KO, n = 16; two-way ANOVA with Bonferroni post hoc analysis). Representative traces are shown in the top inset. f Synaptic excitation: inhibition ratio (E:I ratio) evoked by electrical stimulation of parallel fibers (WT, n = 14 cells; KO, n = 17 cells; Mann-Whitney test; P = 0.677). Representative traces are shown in the top inset. Note that EPSCs are going inwards and IPSCs are going outwards. g Cell-attached recordings of spontaneous action potential firing in PCs. No blockers were added to the artificial cerebrospinal fluid. Left: representative traces of a WT (top panel) and a KO (bottom panel) Purkinje cell. Right: Quantitative analysis of PC spike rates (WT, n = 14 cells; KO, n = 14 cells; t-test, one-tailed P = 0.036). h Cell-attached recordings of spontaneous action potential firing in PCs. Picrotoxin 50 µM, CNQX 20 µM and AP5 50 µM were added to the ACSF to block GABAA, AMPA and NMDA receptors, respectively. Left: representative traces of a WT (top panel) and a KO (bottom panel) Purkinje cell. Right: Quantitative analysis of PC spike rates (WT, n = 16 cells; KO, n = 16 cells; t-test, two-tailed P = 0.026). Data are presented as mean ± standard error of mean (SEM) in (c), (d), and (e). Data are presented as individual values and mean ± SEM in (f), (g), and (h). *P < 0.05, **P < 0.01 (Supplementary Fig. 3; see Supplementary Table 3 for detailed statistics).
Next, disynaptic inhibitory postsynaptic currents (IPSCs) were produced by stimulating parallel fibers. The amplitude of IPSCs was about 2-fold higher in the Bmal1 KO mice compared with the WT mice (P < 0.001, Fig. 3e). We found no difference in the excitatory: inhibitory ratio (E:I ratio, EPSC amplitude/IPSC amplitude) between the WT and KO mice (P = 0.677, Fig. 3f). Firing activities of PCs form the sole output from the cerebellar cortex [22]. We found remarkable reduction (~40%) of spontaneous firing rates in the PCs of the KO mice compared with the WT mice (P = 0.036, Fig. 3g). Next, when both excitatory and inhibitory transmission were blocked, the spontaneous firing rates were not significantly affected by the blockers and the rate was also markedly reduced (~30%) in the KO mice compared with the WT mice (P = 0.026, Fig. 3h), indicating the reduction in spike rates was not due to altered synaptic inputs. We also analyzed the coefficients of variation (CV and CV2) of the interspike intervals (Supplementary Figs. 3c, 3d). Small values close to 0 indicate regular firing, whereas large values close to or >1 indicate irregular firing. Although there was a trend for an increase in CV and CV2 without blockers in the KO mice compared with the WT mice, the differences did not reach statistical significance (CV: P = 0.730, CV2: P = 0.421, Supplementary Fig. 3d). We further analyzed the event distribution of interspike intervals, which has been shown to be developmentally regulated [23]. Histograms of interspike interval reveal a more spread distribution in KO mice compared with that in WT mice (Supplementary Fig. 3e). As such, the cumulative probability of ISI exhibited a shift to the right in the KO mice (P < 0.001, Kolmogorov–Smirnov test, Supplementary Fig. 3f). We also performed the auto-correlograms of action potential firing. We observed the proportions of cells firing (1) at 0–40 Hz was similar in WT and KO; (2) at 40–80 Hz was higher in KO than in WT, (3) at 80–100 Hz was similar in WT and KO; (4) at 100–140 Hz was lower in KO than in WT. However, these differences were not statistically significant (χ2 test, n = 13 and 12 cells in WT and KO mice, respectively; P = 0.835 for 0–40 Hz, P = 0.160 for 40–80 Hz, P = 1.000 for 80–100 Hz, P = 0.322 for 100–140 Hz) (Supplementary Fig. 3g). Finally, evoked action potentials were not significantly altered across a range of injected currents in KO mice, indicating similar membrane excitability of PCs in the KO and WT mice (P = 0.932, Supplementary Fig. 3h). Together, the electrophysiological changes, including increased excitatory and inhibitory transmission onto PCs, increased strengths of oscillatory activity, are consistent with the observed immature morphology of PC spines. Taken together, these results indicate developmental deficits in the cerebellum.
Differential expression of Autism-associated genes in the cerebellum of Bmal1 KO mice
To investigate molecular mechanisms underlying the pathophysiological changes in Bmal1 KO cerebellum, genome-wide transcriptome profiling was performed using RNA-Seq. A total of 444 differentially expressed genes (DEGs) were identified in the KO cerebellum, comprising 264 upregulated and 180 downregulated genes compared with the WT cerebellum (Supplementary Fig. 4a, b, Supplementary Table 1). Among the DEGs, 7 upregulated and 12 downregulated genes were included in the SFARI autism gene database (Supplementary Table 1). To analyze interconnections of identified SFARI genes with other DEGs, protein-protein interaction networks were constructed using the STRING database and the relevant biological processes were defined (Supplementary Fig. 4c, d). Two upregulated gene networks were identified involving SFARI genes Ntng2 and Mfrp, which are related to neurocircuit organization and cell differentiation (Supplementary Fig. 4d). Two downregulated gene networks were identified involving SFARI genes Ntng1, Nr4a2, and Thbs1, which are related to nervous system development and cell-cell interactions (Supplementary Fig. 4d). To validate the RNA-Seq data, quantitative RT-PCR was performed using biological replicates and indeed significant upregulation of Ntng2 and Mfrp and significant downregulation of Ntng1, Nr4a2, and Thbs1 expression were measured in the cerebellum of KO mice as compared with the WT mice (Supplementary Fig. 4e), in accordance with the RNA-Seq results.
As expected, differential expression of multiple clock genes, including Per1, Per2, Per3, Clock, Npas2, and Nr1d1 was also detected by RNA-Seq, indicating that Bmal1 deletion disrupts clock gene network in the cerebellum (Supplementary Fig. 4f). Notably, multiple SFARI genes were involved in the disrupted network including Per1, Per2, Npas2, Nr1d1, and Aldh1a3 (Supplementary Fig. 4f). Expression levels of these genes were also verified by quantitative RT-PCR (Supplementary Fig. 4g) and by western blotting for the protein products (Supplementary Fig. 4h). Interestingly, spinocerebellar ataxia-related genes Atxn1, 3, and 7 were closely linked to the disrupted clock gene network, and their mRNA and/or proteins levels were found to be markedly reduced in the cerebellum of KO mice (Supplementary Fig. 4g, h), which may underlie motor learning and coordination deficits in KO mice (Fig. 2a). Together, these results demonstrate that the expression of multiple autism or ataxia-associated genes is dysregulated in the cerebellum of Bmal1 KO mice.
Altered translational landscape and dysregulated translational control pathways in the cerebellum of Bmal1 KO mice
As Bmal1 regulates mRNA translation [12], ribosome profiling was performed to investigate potential translational changes in the cerebellum of Bmal1 KO mice. Using RNA-Seq (Novaseq) of ribosome protected fragments (as a proxy for translation), we measured genome-wide translational efficiency by normalizing to mRNA RNA-Seq data (translation/transcription; Fig. 4a). Novaseq produced high quality reads for footprint and mRNA libraries, as evidenced first by the read distribution within the 3 frames (Supplementary Fig. 5a), secondly by the canonical distribution of footprint size (28–32nt) (Supplementary Fig. 5b), thirdly by the canonical periodicity of ribosomal footprints across mRNA coding and non-coding regions (Supplementary Fig. 5c) and fourthly by the R2 of Reads Per Kilobase of transcript per Million mapped reads (RPKM) between two biological replicates, which is >0.9 for both footprints and total mRNA (Supplementary Fig. 5d). The squared Pearson product-moment correlation coefficient R2 of RPKM between replicates was >0.97 for both footprints and total mRNA (Supplementary Fig. 5d), demonstrating reproducibility. Bmal1 deletion in the cerebellum led to an altered translational landscape with 304 differentially translated genes (DTGs) (R2 = 0.897; 202 upregulated and 102 downregulated genes) (Fig. 4b, Supplementary Table 1). Among the DTGs, 6 upregulated and 6 downregulated genes appear in the SFARI database (Supplementary Table 1). Interestingly, ingenuity canonical pathway analysis of the DTGs revealed changes in major translational control pathways including the eIF2 signaling, as well as eIF4E and p70 S6K signaling. As expected, circadian rhythm signaling was also identified amongst the top pathways (Fig. 4b, Supplementary Table 2).

a A diagram indicates experimental design to assess genome-wide translational efficiency of mRNAs using ribosome profiling in whole cerebellum tissue. RPKM: Reads Per Kilobase of transcript per Million mapped reads. b Top: log2 Translational Efficiency (TE) Plots (P < 0.05 and 1.5 ≥ ratio ≥ 0.667; n = 2 mice/group for footprints and mRNA). DTGs: Differentially Translated Genes, R2: squared Pearson product-moment correlation coefficient. Bottom: Ingenuity Pathway Analysis (IPA) of DTGs. c Left: Immunoblots indicating activities of the mTORC1/S6K1, eIF2α, and ERK MAPK/p-eIF4E pathways in the cerebellum using phosphorylated S6K1, S6, eIF2α, ERK and eIF4E as indicators. β-actin was used as a loading control. Right: Quantitation of protein levels of phospho-S6K1 (t (6) = 2.604, P = 0.041), phospho-S6 (t (6) = 4.072, P = 0.007), phospho-eIF2α (t (6) = 2.618, P = 0.040), phospho-ERK (t (6) = 0.629, P = 0.553) and phospho-eIF4E (t (6) = 2.479, P = 0.048). Student’s t-test, n = 4 mice/group. d Top: Representative bright field microscopic images of sagittal cerebellar sections immunolabeled for phospho-S6. Scale bar = 500 μm. Bottom: Quantitation of phospho-S6 levels in different cerebellar lobules (F (7,224) lobule × genotype = 3.476, P = 0.002, two-way ANOVA). n = 15 sections, 3 mice/group. In (c) and (d), data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001, n.s., not significant. e Left: Representative confocal microscopic images of cerebellar sections double immunolabeled for Calbindin-D(28k) (green) and phospho-S6 (p-S6, red). Cell nuclei were counterstained by DAPI (blue). Framed regions are magnified to the right. Scale bar = 20 μm. Right: cellular colocalization analysis of Calbindin-D(28k) and p-S6. n = 51–59 cells, 3 mice/group. Note that Calbindin-D(28k) and p-S6 were colocalized in the Purkinje cell soma and the p-S6 level was increased in the KO mice (Supplementary Figs. 4 and 5; see Supplementary Table 3 for detailed statistics).
To validate the ribosome profiling results, western blotting was performed to detect activities of the translational control pathways in the cerebellum (Fig. 4c). As expected, Bmal1 was eliminated in Bmal1 KO cerebellum. Hyperactivation of the mTORC1/S6K1 pathway was detected, as demonstrated by increased levels of phospho-S6K1(Thr389) and phospho-S6 (Ser240/244) (P = 0.041, P = 0.007 respectively, Fig. 4c). Moderate but significant increases of phosphorylation of eIF2α (P = 0.040) and eIF4E (P = 0.048) were also detected (Fig. 4c). By immunostaining. Pervasive upregulation of phospho-S6 was found in all lobules in the KO mice as compared with the WT mice (P < 0.0001, Fig. 4d), consistent with the western results. Interestingly, double labeling for phospho-S6 and Calbindin-D (28k) revealed cellular colocalization of the two proteins in the PC soma, indicating enriched mTORC1 activities in PCs (Fig. 4e). Together, these results demonstrate an altered translational landscape and dysregulated translational control pathways in the cerebellum of Bmal1 KO mice.
Metformin reverses mTORC1 hyperactivation and rescues core behavioral deficits in Bmal1 KO mice
Metformin treatment (Fig. 5a) reversed the impaired sociability phenotypes in the Bmal1 KO, as evidenced by the significantly increased time spent in the stranger mouse (S) chamber (S vs. E, Bmal1 KO Metformin, P < 0.0001), and in sniffing the wire cage containing S compared to the empty cage (E) (S vs. E, Bmal1 KO Metformin, P < 0.0001, Fig. 5b). Metformin treatment did not affect the numbers of entries to both chambers in either the WT or the KO mice (P = 0.951, Supplementary Fig. 6a). Additionally, metformin treatment did not affect social novelty or numbers of entries in the three-chamber test in either mice genotype (social novelty: P = 0.362, number of entries: P = 0.895, Supplementary Fig. 6a). In the reciprocal social interaction test, metformin significantly increased total time of social interactions (P = 0.0005, Fig. 5c) but did not change anogenital or nose-nose sniffing time in the KO mice (Bmal1 KO Saline vs. Bmal1 KO Metformin, P > 0.99, Supplementary Fig. 6b).

a A diagram indicating the paradigm used for metformin treatment and tests. Metformin (200 mg/kg, i.p.) or saline (10 µl x body weight in grams, i.p.) was administered once a day at ZT 20 (8 h after light-off) for 10 d to Bmal1 WT and KO mice. Behavioral tests were started at Day 11. Metformin treatment was continued throughout the tests. b Three-chamber tests for mouse sociability. Top: Representative heat maps indicating time spent on the location from the four groups. S1: stranger 1, E: empty. Bottom: bar graphs indicate time spent in chambers and in sniffing. Note that metformin treatment reversed the sociability deficits in the Bmal1 KO mice (Bmal1 KO Saline: S1 vs. Empty, P > 0.99; Bmal1 KO Metformin: S1 vs. Empty, P < 0.0001, two-way ANOVA and post hoc Bonferroni’s comparison). n = 6–9 mice/group. c Reciprocal social tests. Bar graphs indicate time spent in social interactions. Note that metformin treatment significantly increased the interaction time in KO mice (push-crawl and following) (F (3,35) = 7.672, P = 0.0005, Bmal1 WT Metformin vs. Bmal1 KO Metformin: P = 0.931, one-way ANOVA). n = 8–11 mice/group. d Spontaneous grooming analysis. Note that metformin treatment decreased the number of grooming bouts in the Bmal1 KO mice (F (3,22) = 14.660, P < 0.0001, Bmal1 KO Saline vs. Bmal1 KO Metformin: P = 0.004, one-way ANOVA). n = 5–8 mice/group. e Route tracing analysis. Left: Trace maps of route tracing episodes (20 s). Right: Three bar graphs indicate that metformin treatment significantly decreased the number of bouts, duration, and percentage of time engaging in route tracing in Bmal1 KO mice (Bmal1 WT Metformin vs. Bmal1 KO Metformin: P > 0.999). n = 5 mice/group. f Left: representative western blots indicate decreased levels of phospho-S6K1 and phospho-S6 after metformin treatment in the cerebellum of Bmal1 KO mice. Right: Quantitative analysis of the blots. n = 3 mice/group. g Cell-attached recordings of spontaneous action potential firing in PCs. Picrotoxin 50 µM, CNQX 20 µM and AP5 50 µM were added to the ACSF to block GABAA, AMPA, and NMDA receptors, respectively. Left: representative traces from a Purkinje cell in each group. Right: Quantitative analysis of PC spike rates (WT saline, n = 16; KO saline, n = 15; WT metformin, n = 12; KO metformin, n = 11; one-way ANOVA with post hoc Bonferroni’s comparisons). Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n.s., not significant (Supplementary Fig. 6; see Supplementary Table 3 for detailed statistics).
Next, metformin treatment reduced the number of grooming bouts in the KO mice (Bmal1 KO Saline vs. Bmal1 KO Metformin, P = 0.004, Fig. 5d) but did not alter total grooming time (P > 0.99, Supplementary Fig. 6c). In the route tracing analysis, metformin treatment significantly reduced the number of bouts, duration, and percentage of time in route tracing in the KO mice to levels that were not different compared with the WT mice (Bmal1 KO Saline vs. Bmal1 KO Metformin, bouts: P = 0.005, duration: P = 0.0006, percentage of time: P = 0.004, Fig. 5e). Next, metformin treatment significantly reversed the increased levels of phospho-S6K1 (P < 0.0001), phospho-S6 (P < 0.0001) and phospho-eIF4E (P = 0.016) in the KO cerebellum (Fig. 5f and Supplementary Fig. 6d). Metformin treatment, however, did not rectify the aberrant levels of phospho-eIF2α, Atxn1, Atxn3, Nr1d1, or Npas2 (Supplementary Fig. 6d). After metformin treatment, the spontaneous firing rates, EPSC and IPSP amplitudes in cerebellar PCs were no longer different between WT and KO mice (Fig. 5g and Supplementary Fig. 6e–i). Together, these results demonstrate that metformin reverses mTORC1 hyperactivation in the cerebellum and rescues major behavioral and cellular deficits in Bmal1 KO mice.
Conditional Bmal1 deletion in cerebellar PCs recapitulates autistic-like phenotypes in Bmal1 KO mice
Next, we crossed Bmal1flx/flx mice with L7-Cre mice to obtain Bmal1flx/flx: L7-Cre mice, in which Bmal1 was specifically deleted in cerebellar PCs. In the three-chamber test, the Bmal1flx/flx: L7-Cre mice spent significantly longer time in the chamber of and interacting with a caged stranger mouse (S), as compared to an empty wire cup (E), demonstrating intact sociability, similar as in the Bmal1flx/flx littermates (S1 vs. E, P = 0.029, Supplementary Fig. 7a). The Bmal1flx/flx mice spent significantly more time in the chamber with the novel stranger (S2) than in the chamber with the familiar stranger (S1) and more time sniffing the novel stranger cup (P < 0.05, Fig. 6a), indicating a preference for social novelty. On the contrary, the Bmal1flx/flx: L7-Cre mice spent similar time in the chamber of and interacting with S2 and S1 (P = 0.19), indicating an impaired preference for social novelty (Fig. 6a). The numbers of entries to the two side chambers were not different between the Bmal1flx/flx: L7-Cre and the Bmal1flx/flx mice (social approach: P = 0.438, social novelty: P = 0.882, Supplementary Fig. 7b). Circadian wheel-running behavior was not disrupted in the Bmal1flx/flx: L7-Cre mice (Supplementary Fig. 7c). In the open field test no difference in time in center and corner zones and total travel distance was found between Bmal1flx/flx: L7-Cre and the Bmal1flx/flx mice (Supplementary Fig. 7d). The Bmal1flx/flx: L7-Cre mice exhibited a significantly increased water puff-induced grooming time and number of bouts compared with the Bmal1flx/flx mice (time: P = 0.024, bouts: P = 0.004, Fig. 6b), consistent with the Bmal1 KO mice. The Bmal1flx/flx: L7-Cre mice also buried more marbles than the Bmal1flx/flx mice (P = 0.0005, Fig. 6c) in the marble burying test, indicating excessive repetitive behaviors in these mice. In the rotarod test, the Bmal1flx/flx: L7-Cre displayed markedly shorter latencies to fall and fell at slower rotating speeds compared with the Bmal1flx/flx mice (latency: P = 0.0001, speed: P = 0.0004, Fig. 6d), indicating impaired motor coordination.

a Three-chamber test for mouse social behavior. Left: Representative heat maps of a Bmal1flx/flx mouse and a Bmal1flx/flx:L7-Cre mouse in the three-chamber social novelty test. The heat maps indicate time spent on the location. S1: stranger 1, S2: stranger 2. Right: bar graphs indicating time spent in individual chambers (F (2, 57) chamber × genotype = 4.997, P = 0.01, two-way ANOVA) and time spent sniffing wire cages (F (1,38) chamber × genotype = 2.191, P = 0.147, two-way ANOVA) during the three-chamber test. S1: stranger 1, C: center, S2: stranger 2. Note that the Bmal1flx/flx mice spent a longer time in the S2 chamber compared with the S1 chamber and a longer time sniffing the S2 cage compared with the S1 cage. In contrast, the Bmal1flx/flx:L7-Cre mice spent similar time in the S1 chamber as in the S2 chamber (S1 vs. S2, P = 0.190) and similar time sniffing S1 cage compared to the S2 cage (S1 vs. S2, P > 0.999, post hoc Bonferroni’s comparison). n = 9–12 mice/group. b Analysis of mouse grooming. Bar graphs indicate that the total grooming time (t (18) = 2.469, P = 0.024, Student’s t-test) and the number of grooming bouts (t (13) = 3.356, P = 0.004, Student’s t-test) were increased in the Bmal1flx/flx:L7-Cre mice compared with the Bmal1flx/flx mice. n = 10 mice/group. c Marble burying test. The bar graph indicates that the total number of buried marbles was increased in the Bmal1flx/flx:L7-Cre mice compared with the Bmal1flx/flx mice (t (20) = 4.127, P = 0.0005, Student’s t-test). n = 10–12 mice/group. d Mouse rotarod test. The line graph on the left indicates the time latency to fall and the graph on the right indicates the rotating speed at fall. Note that the Bmal1flx/flx:L7-Cre mice exhibited shorter latencies to fall (F (1,66) genotype = 16.64, P = 0.0001, two-way ANOVA) and fell at lower speeds (F (1,25) genotype = 16.75, P = 0.0004, two-way ANOVA). n = 4–5 mice/group, 3 trials/day. e Representative confocal microscopic images of cerebellar lobule V immunolabeled for Calbindin-D(28k) (green) and Bmal1 (red). Cell nuclei were counterstained by DAPI (blue). Scale bar = 50 μm. Numbers of Bmal1 positive PCs are shown to the right. f Input-Output relationships of EPSCs evoked by parallel fiber stimulation (Bmal1flx/flx, n = 16; Bmal1flx/flx:L7-Cre, n = 11; two-way ANOVA with Bonferroni post hoc analysis). Representative traces are shown in the top inset. g Input-Output relationships of IPSCs obtained at a Vh = +35 mV (Bmal1flx/flx, n = 13; Bmal1flx/flx:L7-Cre, n = 13; two-way ANOVA with Bonferroni post hoc analysis). Representative traces are shown in the top inset. h Cell-attached recordings of PCs spontaneous action potential firing. Picrotoxin 50 µM, CNQX 20 µM, and AP5 50 µM is added to the ACSF in order to block GABAA, AMPA, and NMDA receptors, respectively. Left: representative traces of a WT (top panel) and a KO (bottom panel) Purkinje cell. Right: Quantitative analysis of Purkinje cell spontaneous action potential firing (Bmal1flx/flx, n = 16; Bmal1flx/flx:L7-Cre, n = 17; t-test, one-tailed p = 0.0300). i Left: Representative bright field microscopic images of sagittal cerebellar sections immunolabeled for phospho-S6. Scale bar = 500 µm. Right: Quantitation of phospho-S6 levels in different cerebellar lobules (F (1,128) genotype = 299.6, P < 0.0001, two-way ANOVA). n = 9 sections, 3 mice/group. j Golgi–Cox staining of Purkinje cell dendrites. Left: representative microscopic images indicating dendritic morphology of Purkinje cells. Scale bar = 10 μm. Right: Box plots indicate quantitative analysis of density and morphology of dendric spines. Note that the number of dendritic spines was increased (t (102) = 3.213, P = 0.002, Student’s t-test), and more spines exhibited immature morphology in the Bmal1flx/flx:L7-Cre mice compared with the Bmal1flx/flx mice (F (1,204) fraction × genotype = 17.93, P < 0.0001, immature fraction Bmal1flx/flx vs. Bmal1flx/flx: L7-Cre: P = 0.016, two-way ANOVA). n = 48–56 dendrite branches, 4 mice/group. Data are presented as mean ± SEM with or without individual values. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant. (Supplementary Fig. 7; see Supplementary Table 3 for detailed statistics).
Immunolabeling for Bmal1 and Calbindin indicated that Bmal1 expression was diminished in the PCL but not changed in the GL (P < 0.0001, Fig. 6e), confirming specific abolition of Bmal1 expression in cerebellar PCs in the Bmal1flx/flx: L7-Cre mice. In electrophysiological recordings, we measured a ~2-fold increase in the amplitudes of EPSCs elicited by parallel fiber stimulation in the Bmal1flx/flx: L7-Cre mice compared with the Bmal1flx/flx mice (P < 0.001, Fig. 6f). Responses elicited by climbing fiber stimulation were not significantly different between the Bmal1flx/flx: L7-Cre and the Bmal1flx/flx mice (P = 0.833, Supplementary Fig. 7e). The amplitude of IPSCs induced by stimulating parallel fibers was about 2-fold higher in the Bmal1flx/flx: L7-Cre mice compared with the Bmal1flx/flx mice (P < 0.001, Fig. 6g). The E:I ratio (EPSC amplitude/IPSC amplitude) was not different between the Bmal1flx/flx: L7-Cre mice and the Bmal1flx/flx mice (P = 0.571, Supplementary Fig. 7f). Lastly, we found a significant reduction (~30%) of firing rates in the PCs of the Bmal1flx/flx: L7-Cre mice compared with the Bmal1flx/flx mice with or without blocking synaptic inputs into PCs (P = 0.03, Fig. 6h and P = 0.048, supplementary Fig. 7g). Together, these results indicate enhanced excitatory and inhibitory synaptic transmission but reduced spontaneous firing activities of the PCs in Bmal1flx/flx: L7-Cre mice.
Next, significant upregulation of phospho-S6 was detected in all lobules in the Bmal1flx/flx: L7-Cre mice compared with the Bmal1flx/flx mice (P < 0.0001, Fig. 6i), indicating that Bmal1 deletion only in PCs leads to mTORC1 hyperactivation in PCs. Lastly, using Golgi-Cox staining we found that the density of dendritic spines in the PC was increased in the Bmal1flx/flx: L7-Cre KO mice compared with the Bmal1flx/flx mice (P = 0.002, Fig. 6j). The fraction of immature dendritic spines was increased but the fraction of mature dendritic spines was decreased in the PCs of these mice (P = 0.016, Fig. 6j), suggesting Bmal1 deletion leads to developmental deficits in the dendritic spines of cerebellar PCs. Together, these results demonstrate that Bmal1 deletion only in cerebellar PCs recapitulates behavioral and cellular changes seen in Bmal1 KO mice.
Discussion
In the current study, we investigated a role for the essential clock gene Bmal1 in cerebellar dysfunction and autistic-like behaviors in mouse models. These findings establish a novel role for the clock gene Bmal1 in cerebellar physiology and social behavior.
Interestingly, a number of neural deficits involving the forebrain have been reported in the Bmal1 KO mice, some of which are consistent with pathophysiological changes reported in ASD patients. Bmal1 KO mice exhibit impaired contextual fear and spatial memory, as well as decreased LTP in hippocampal slices. Impairments of hippocampus-dependent learning and memory are found in Bmal1 KO and forebrain conditional Bmal1 KO mice [24, 25]. Consistent with this, cognitive deficits are found in about half of ASD patients [5]. Bmal1 KO mice exhibit striking astrogliosis by 2.5 months of age [26]. Interestingly, increased astrogliosis has been observed in postmortem ASD forebrain and cerebellar tissues [27, 28]. The forebrain neural architecture appears to be normal without significant changes in the hippocampal layers, but presynaptic axon terminal degeneration and reduced functional connectivity in the forebrain are found in Bmal1 KO mice at 6 months of age [26]. Of note, local and long-range functional connectivity is reduced proportionally in ASD [29]. Although a role for neurodegeneration in ASD is still under debate, emerging evidence indicates that some genetic ASD conditions (e.g., Fragile X syndrome) are associated with both neurodevelopment and neurodegeneration [30]. Taken together, these reported pathophysiological changes in the forebrain of Bmal1 KO mice are consistent with ASD-like changes and may also contribute to the behaviral deficits of Bmal1 KO mice we observed in the current study.
Increasing evidence indicates the involvement of the cerebellum in ASD pathogenesis [31]. Motor deficits are associated with the social, communicative, and behavioral impairments that define ASD [20]. The cerebellum is one of the most consistent sites of abnormality in ASD. One of the most consistent findings from postmortem studies of ASD individuals concerns PCs. PC abnormalities in numbers and sizes are found in postmortem autistic cerebellum [32]. In the current study, we found the Bmal1 KO mice displayed substantial deficits in motor learning and coordination, which led us to focus on the cerebellar changes in these mice. Indeed, we found pervasive upregulation of phospho-S6 in all lobules in the cerebellum of Bmal1 KO mice compared with WT mice. As different electrophysiological properties in PCs are found in different lobules [33], we focused our electrophysiological studies on PCs on lobules V/VI to minimize possible differences among different lobules. As PCs provide the sole output from the cerebellar cortex, aberrant neuronal activities in PCs may underlie behavioral defects in Bmal1 KO mice. Of note the reduction in PC firing rates in Bmal1 KO mice is consistent with the changes observed in some well-established mouse models of autism [34].
Using the Bmal1flx/flx: L7-Cre mice, we demonstrate that Bmal1 deletion in cerebellar PCs can cause significant autistic-like behaviors, along with prominent phenotypes at the cellular and molecular levels, similar to those in Bmal1 KO mice, including: (1) hyperactivation of mTORC1 and immature dendritic spine morphology in cerebellar PCs; (2) aberrant electrophysiological properties, including reduced spontaneous firing rate, enhanced synaptic transmission in cerebellar PCs; (3) autistic-like behaviors, including impaired preference for social novelty, excessive repetitive behavior, and impaired motor learning. Together, these results strongly support that cerebellar dysfunction caused by Bmal1 deletion underlies the autistic-like phenotypes in Bmal1 mutant mice. As Bmal1flx/flx: L7-Cre mice exhibit intact circadian rhythms, these results also demonstrate that a complete loss of circadian rhythms is not required to cause autistic-like phenotype by Bmal1 disruption.
Several differences in behavioral deficits, however, were observed between Bmal1 KO and Bmal1flx/flx: L7-Cre mice. First, Bmal1 KO mice exhibit impaired sociability but intact preference for social novelty. In contrast, Bmal1flx/flx: L7-Cre mice exhibit intact sociability but impaired preference for social novelty. The discrepancy could be due to the contribution of other brain regions (e.g., the forebrain) to the regulation of mouse social behavior. Further studies are needed to investigate a role for brain-region specific Bmal1 expression in the regulation of social behaviors. Second, Bmal1 KO mice but not Bmal1flx/flx: L7-Cre mice exhibit hyperactivity in the open field and three chamber tests. These results collectively suggest hyperactivity and anxiety-like behavior in Bmal1 KO mice, but they are not observed in the Bmal1flx/flx: L7-Cre mice. Consistent with our results, another study reported increased exploratory activity in the open filed test in Bmal1 KO mice [35]. Third, Bmal1 KO mice exhibit a lack of interest in the marble-burying and nestlet shredding tests, but Bmal1flx/flx: L7-Cre mice exhibit exacerbated marble-burying activities. This could be due to increased anxiety and impaired motor coordination in the Bmal1 KO mice. Fourth, Bmal1 KO mice are arrhythmic, but the circadian rhythms are largely intact in Bmal1flx/flx: L7-Cre mice. Thus, besides the contributions of other brain regions, systemic changes in the levels of neuroendocrine factors that are normally regulated by the circadian clock may also contribute to the behavioral differences between Bmal1 KO and Bmal1flx/flx: L7-Cre mice [36].
Dysregulated translational control by mTORC1 hyperactivation may be the biochemical mechanism that links Bmal1 deletion to ASD pathogenesis. Bmal1 is a key regulator of protein synthesis (mRNA translation) [12]. By ribosome profiling we identified changes in multipe translational control pathways in Bmal1 KO cerebellum, including the mTORC1 signaling, which was further confirmed by western blotting and immunostaining. mTORC1 hyperactivation may in turn dysregulate gene expression, alter activities of neuronal circuits, and cause autism-like behavioral changes in mice. Recently, we and others have successfully used metformin to reverse core phenotypes in mouse and fly models of fragile X syndrome [19, 37]. Moreover, recent clinical reports suggest that metformin can constitute a targeted therapeutic in young and adult fragile X syndrome patients [38, 39]. We found that metformin treatment revised the hyperactivation of mTORC1 without significantly altering other translational control pathways (e.g., eIF2), suggesting hyperactivation of mTORC1 is responsible for dysregulated translation and autistic-like behaviors in Bmal1 KO mice. Of note, metformin treatment does not correct the disrupted functions of the circadian clock, as disrupted expression of clock genes (Per1, Per2, Nr1d1, etc) was not affected by metformin treatment, suggesting that restoring the entire clock functions is not essential for improving ASD-like phenotypes in Bmal1 KO mice.
We have found the mTORC1/eIF4E pathway regulates fundamental functions (resetting, period, amplitude, etc) of the central and peripheral circadian clocks in cells and animals [17, 40, 41]. At the same time, the activities of the mTORC1/eIF4E axis are orchestrated by the circadian clock and exhibit robust diurnal oscillations in the SCN and peripheral tissues [17, 42]. Concurrently, mTOR and its regulators such as TSC1, TSC2 and PTEN, are frequently dysregulated in neurodevelopmental disorders including autism [43]. In experimental studies, conditional Tsc1 deletion in PCs can lead to mTORC1 hyperactivation and autism-like behaviors in mice [44]. Mice lacking the eIF4E repressor 4E-BPs (eIF4E binding proteins, mTORC1 downstream effectors) or overexpressing eIF4E show autistic-like phenotypes [14, 15]. Conversely, rectifying exacerbated translation by genetic removal of the mTORC1 target S6K1, or by repressing eIF4E phosphorylation, corrects autistic-like phenotypes in fragile X syndrome mice [45, 46]. Thus, dysregulation of the mTORC1/eIF4E axis disrupts the circadian clock function and engenders ASD-like phenotypes in animal models, indicating a potential crosstalk mechanism between circadian timing and autism pathogenesis via the mTORC1/eIF4E axis.
In conclusion, the current study provides the first experimental evidence demonstrating that disruption of the essential clock gene Bmal1 globally or only in the cerebellar PCs leads to autistic-like phenotypes in animals. Our results establish the Bmal1 KO mouse as a new genetic model of autism and shed light on a putative role of circadian clock gene disruption in the pathogenesis of ASD.
References
Herzog ED, Hermanstyne T, Smyllie NJ, Hastings MH. Regulating the Suprachiasmatic Nucleus (SCN) Circadian Clockwork: Interplay between Cell-Autonomous and Circuit-Level Mechanisms. Cold Spring Harb Perspect Biol. 2017;9:a027706.
Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–75.
Rosbash M. The implications of multiple circadian clock origins. PLoS Biol. 2009;7:e62.
Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–17.
Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383:896–910.
Lorsung E, Karthikeyan R, Cao R. Biological timing and neurodevelopmental disorders: a role for circadian dysfunction in autism spectrum disorders. Front Neurosci. 2021;15:642745.
Nicholas B, Rudrasingham V, Nash S, Kirov G, Owen MJ, Wimpory DC. Association of Per1 and Npas2 with autistic disorder: support for the clock genes/social timing hypothesis. Mol Psychiatry. 2007;12:581–92.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
Yang Z, Matsumoto A, Nakayama K, Jimbo EF, Kojima K, Nagata K, et al. Circadian-relevant genes are highly polymorphic in autism spectrum disorder patients. Brain Dev. 2016;38:91–99.
Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, et al. SFARI Gene 2.0: a community-driven knowledge base for the autism spectrum disorders (ASDs). Mol Autism. 2013;4:36.
Bralten J, Mota NR, Klemann C, De Witte W, Laing E, Collier DA, et al. Genetic underpinnings of sociability in the general population. Neuropsychopharmacology. 2021;46:1627–34.
Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, Nathan A, et al. The Circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell. 2015;161:1138–51.
Kelleher RJ 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–6.
Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–7.
Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, et al. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013;493:411–5.
Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–8.
Cao R, Gkogkas CG, de Zavalia N, Blum ID, Yanagiya A, Tsukumo Y, et al. Light-regulated translational control of circadian behavior by eIF4E phosphorylation. Nat Neurosci. 2015;18:855–62.
Amorim IS, Kedia S, Kouloulia S, Simbriger K, Gantois I, Jafarnejad SM, et al. Loss of eIF4E phosphorylation engenders depression-like behaviors via selective mRNA translation. J Neurosci. 2018;38:2118–33.
Gantois I, Khoutorsky A, Popic J, Aguilar-Valles A, Freemantle E, Cao R, et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat Med. 2017;23:674–7.
Dziuk MA, Gidley Larson JC, Apostu A, Mahone EM, Denckla MB, Mostofsky SH. Dyspraxia in autism: association with motor, social, and communicative deficits. Dev Med Child Neurol. 2007;49:734–9.
Whitney ER, Kemper TL, Rosene DL, Bauman ML, Blatt GJ. Calbindin-D28k is a more reliable marker of human Purkinje cells than standard Nissl stains: a stereological experiment. J Neurosci Methods. 2008;168:42–47.
De Zeeuw CI, Hoebeek FE, Bosman LW, Schonewille M, Witter L, Koekkoek SK. Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci. 2011;12:327–44.
Kawamura Y, Nakayama H, Hashimoto K, Sakimura K, Kitamura K, Kano M. Spike timing-dependent selective strengthening of single climbing fibre inputs to Purkinje cells during cerebellar development. Nat Commun. 2013;4:2732.
Wardlaw SM, Phan TX, Saraf A, Chen X, Storm DR. Genetic disruption of the core circadian clock impairs hippocampus-dependent memory. Learn Mem. 2014;21:417–23.
Snider KH, Dziema H, Aten S, Loeser J, Norona FE, Hoyt K, et al. Modulation of learning and memory by the targeted deletion of the circadian clock gene Bmal1 in forebrain circuits. Behav Brain Res. 2016;308:222–35.
Musiek ES, Lim MM, Yang G, Bauer AQ, Qi L, Lee Y, et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest. 2013;123:5389–5400.
Crawford JD, Chandley MJ, Szebeni K, Szebeni A, Waters B, Ordway GA. Elevated GFAP protein in anterior cingulate cortical white matter in males with autism spectrum disorder. Autism Res. 2015;8:649–57.
Laurence JA, Fatemi SH. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum. 2005;4:206–10.
Khan S, Gramfort A, Shetty NR, Kitzbichler MG, Ganesan S, Moran JM, et al. Local and long-range functional connectivity is reduced in concert in autism spectrum disorders. Proc Natl Acad Sci USA. 2013;110:3107–12.
Hickman RA, O’Shea SA, Mehler MF, Chung WK. Neurogenetic disorders across the lifespan: from aberrant development to degeneration. Nat Rev Neurol. 2022;18:117–24.
Wang SS, Kloth AD, Badura A. The cerebellum, sensitive periods, and autism. Neuron. 2014;83:518–32.
Bauman ML, Kemper TL. The neuropathology of the autism spectrum disorders: what have we learned? Novartis Found Symp. 2003;251:112–22.
Zhou H, Lin Z, Voges K, Ju C, Gao Z, Bosman LW, et al. Cerebellar modules operate at different frequencies. Elife. 2014;3:e02536.
Yang YM, Arsenault J, Bah A, Krzeminski M, Fekete A, Chao OY, et al. Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Mol Psychiatry. 2020;25:2017–35.
Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20:1868–73.
Leliavski A, Shostak A, Husse J, Oster H. Impaired glucocorticoid production and response to stress in Arntl-deficient male mice. Endocrinology. 2014;155:133–42.
Monyak RE, Emerson D, Schoenfeld BP, Zheng X, Chambers DB, Rosenfelt C, et al. Insulin signaling misregulation underlies circadian and cognitive deficits in a Drosophila fragile X model. Mol Psychiatry. 2017;22:1140–8.
Dy ABC, Tassone F, Eldeeb M, Salcedo-Arellano MJ, Tartaglia N, Hagerman R. Metformin as targeted treatment in fragile X syndrome. Clin Genet. 2018;93:216–22.
Biag HMB, Potter LA, Wilkins V, Afzal S, Rosvall A, Salcedo-Arellano MJ, et al. Metformin treatment in young children with fragile X syndrome. Mol Genet Genom Med. 2019;7:e956.
Ramanathan C, Kathale ND, Liu D, Lee C, Freeman DA, Hogenesch JB, et al. mTOR signaling regulates central and peripheral circadian clock function. PLoS Genet. 2018;14:e1007369.
Cao R, Robinson B, Xu H, Gkogkas C, Khoutorsky A, Alain T, et al. Translational control of entrainment and synchrony of the suprachiasmatic circadian clock by mTOR/4E-BP1 signaling. Neuron. 2013;79:712–24.
Cao R, Anderson FE, Jung YJ, Dziema H, Obrietan K. Circadian regulation of mammalian target of rapamycin signaling in the mouse suprachiasmatic nucleus. Neuroscience. 2011;181:79–88.
Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84:275–91.
Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488:647–51.
Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76:325–37.
Gkogkas CG, Khoutorsky A, Cao R, Jafarnejad SM, Prager-Khoutorsky M, Giannakas N, et al. Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 2014;9:1742–55.
Acknowledgements
We thank S. Fatemi and I. Gantois for critical reading of the manuscript. The work was supported by grants from NIH (NS118026 and DK109714 to RC; MH119355 to AA and PK.; NS097312, DA048822 to AA; NS022920 and NS045667 to HTO; GM114142 and CA217297 to VXJ), from the Winston and Maxine Wallin Neuroscience Discovery Fund, from the University of Minnesota Medical School Foundation and from Whiteside Institute for Clinical Research to RC.
Author information
Authors and Affiliations
Contributions
DL, CN, PK, AA, HTO, CGG, and RC designed research; DL, CN, KS, EL, ISA, KC, SP, and RC performed research; DL, CN, JL, KF, CGG, and RC analyzed data; DL, CN, EL, JCG, VXJ, PK, AA, HTO, CGG and RC wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Liu, D., Nanclares, C., Simbriger, K. et al. Autistic-like behavior and cerebellar dysfunction in Bmal1 mutant mice ameliorated by mTORC1 inhibition. Mol Psychiatry 28, 3727–3738 (2023). https://doi.org/10.1038/s41380-022-01499-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01499-6
- Springer Nature Limited
This article is cited by
-
Neural function of Bmal1: an overview
Cell & Bioscience (2023)
-
The comparative effectiveness of metformin and risperidone in a rat model of valproic acid-induced autism, Potential role for enhanced autophagy
Psychopharmacology (2023)
-
Krüppel-like factor 7 deficiency causes autistic-like behavior in mice via regulating Clock gene
Cell & Bioscience (2022)
-
The trilateral interactions between mammalian target of rapamycin (mTOR) signaling, the circadian clock, and psychiatric disorders: an emerging model
Translational Psychiatry (2022)
-
Signalling pathways in autism spectrum disorder: mechanisms and therapeutic implications
Signal Transduction and Targeted Therapy (2022)