
Abstract
Social memory processing requires functional CA2 neurons, however the specific mechanisms that regulate their activity are poorly understood. Here, we document that SorCS2, a member of the family of the Vps10 family of sorting receptors, is highly expressed in pyramidal neurons of CA2, as well as ventral CA1, a circuit implicated in social memory. SorCS2 specifically localizes to the postsynaptic density and endosomes within dendritic spines of CA2 neurons. We have discovered that SorCS2 is a selective regulator of NMDA receptor surface trafficking in hippocampal neurons, without altering AMPA receptor trafficking. In addition, SorCS2 regulates dendritic spine density in CA2 neurons where SorCS2 expression is enriched, but not in dorsal CA1 neurons, which normally express very low levels of this protein. To specifically test the role of SorCS2 in behavior, we generated a novel SorCS2-deficient mouse, and identify a significant social memory deficit, with no change in sociability, olfaction, anxiety, or several hippocampal-dependent behaviors. Mutations in sorCS2 have been associated with bipolar disease, schizophrenia, and attention deficient-hyperactivity disorder, and abnormalities in social memory are core components of these neuropsychiatric conditions. Thus, our findings provide a new mechanism for social memory formation, through regulating synaptic receptor trafficking in pyramidal neurons by SorCS2.
Similar content being viewed by others


Introduction
While it is well established that the hippocampus critically regulates declarative and spatial memory, more recent studies, using targeted inactivation or activation of CA2 pyramidal neurons, have defined this region as essential for social memory [1, 2]. Indeed, longstanding genetic inactivation of CA2 neurons, or acute silencing of the CA2 to ventral CA1 circuit [3], leads to a pronounced deficit in social memory, without impairment in sociability or other hippocampal-dependent behaviors, such as contextual or spatial memory. While these neurons are critical and express receptors for “social” neuropeptides including oxytocin and vasopressin [4, 5], the molecular mechanisms regulating neuronal activity and circuitry that involves CA2, and that contribute to social memory, have not been well defined.
The Vps10 family receptors, including sortilin, SorLA, SorCS1, SorCS2, and SorCS3, are transmembrane proteins with related N-terminal Vps10 ectodomains. These receptors play important roles in protein trafficking and we and others have identified roles in signal transduction, as receptors for proneurotrophins [6,7,8]. However, the prominent and largely nonoverlapping neuronal expression pattern of the SorCS subgroup (SorCS1-3) suggests that they may have distinct, regional functions in the hippocampus. In contrast, Sortilin and SorLA are more uniformly expressed in all hippocampal areas [9]. Genome-wide association studies have implicated SorCS receptors (SNP or CNV) with autism, schizophrenia (SCZ), biopolar disease (BD), attention-deficit hyperactivity disorder (ADHD), and Alzheimer’s disease in humans [10,11,12,13], and aggression in other species [14]. These associations, together with the localization of SorCS proteins at synapses [15, 16], suggest that they may play potential roles in pathogenesis, perhaps by modulating synaptic activity.
SorCS proteins have been established to play critical roles in subcellular trafficking. SorCS1 interacts with APP to alter the processing of APP to amyloid-β peptide (Aβ) [13, 17], and synaptic receptor trafficking, specifically for the AMPAR subunit Gria 2 and neurexin. Upon SorCS1 deletion, synaptic levels of the AMPAR Gria 2 are reduced, impairing glutamatergic transmission at excitatory synapses [16]. SorCS3 is also detected at synapses, where it interacts biochemically with PICK1, an adaptor implicated in glutamate receptor trafficking, although no direct interactions of SorCS3 with glutamate receptors have been demonstrated [18]. A recent report suggests that like sortilin, SorCS2 can interact with TrkB receptors [15].
To better define the functions of SorCS2, we report the generation and functional characterization of a SorCS2-deficient mouse model to query the relevance of this receptor in hippocampal-dependent processes. Within the hippocampus, SorCS2 is most highly expressed in CA2 and regulates dendritic spine density of CA2 pyramidal neurons. We demonstrate that SorCS2 is localized to the postsynaptic density and interacts specifically with NMDAR subunits, but not the AMPAR. SorCS2 selectively regulates the trafficking of NMDAR to the plasma membrane of CA2 dendrites as evidenced by immunoelectron microscopy. The behavioral consequences of SorCS2 deletion include a significant social memory deficit, but no changes in sociability or other hippocampal-dependent tasks. These findings uncover a role for SorCS2 as a critical mediator of social memory formation through regulating the synaptic trafficking of the NMDAR in CA2 pyramidal neurons.
Materials and methods
Perfusion and preparation of sections for immunofluorescence staining
Mice were anesthetized and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde (Electron Microscopy Sciences, 30525-89-4). Brains were postfixed overnight at 4 °C, cryoprotected, and 30 µm sections were obtained. Sections were incubated in blocking buffer (5% BSA + 0.3% Triton-100) and avidin/biotin blocking kit (Vector Laboratories, SP-2001) at room temperature (RT), and then incubated with either anti-SorCS2 antibody (1:1000, R&D System, AF4237) or anti-PCP4 antibody (1:1000, Sigma-Aldrich, HPA005792) for 18 h at 4 °C. Secondary antibodies used were biotinylated donkey anti-sheep IgG (for SorCS2 staining, at 1:400, Jackson ImmunoResearch, 713-065-147), or donkey anti-rabbit IgG (for PCP4 staining, at 1:400, Jackson ImmunoResearch, 711-006-152). Cy3-conjugated streptavidin (1:1000, Jackson ImmunoResearch, 016-160-084) was used to visualize the images. For SorCS2 and PCP4 co-staining, the secondary antibodies used were donkey anti-sheep 594 (for SorCS2 staining, Jackson ImmunoResearch, 713-585-147) and donkey anti-rabbit 488 (for PCP4 staining, Jackson ImmunoResearch, 711-545-152).
Immunoprecipitation and western blot analysis
Dissected tissues were minced and lysed in lysis buffer (1x Tris-buffered saline, 1% NP-40, 1% Triton-100, 0.1% SDS, 1 mM PMSF, 10% glycerol, and protease inhibitor cocktail (Sigma-Aldrich, P8340) for 30 min on ice. Lysates were further triturated using a 30 g needle, and supernatants collected following centrifugation at 14,000 rpm for 5 min. Cleared lysate samples were then incubated with anti-SorCS2 antibody (4–5 ug/mg total protein) for 18 h at +4 °C. To collect the immunoprecipitates, protein G-Sepharose beads (Invitrogen, 101241) were added to the supernatant for 90 min at 4 °C, and immunoprecipitates collected by centrifugation for 5 min at 5000 rpm. Beads were then washed in ice-cold lysis buffer, and immunocomplexes were resolved by SDS-PAGE. Following transfer, western blots were developed using incubation with primary antibodies, then with HRP-conjugated secondary antibodies and developed with the ECL kit (Amersham, RPN2106).
Neuronal culture, biotinylation of cell surface, and imaging of SorCS2
Neuronal culture
Hippocampi were dissected from E17.5 sorCS2−/− and sorCS2+/+ embryos and placed in Hanks balanced salt solution (Gibco, 14175079) containing 0.37% sucrose. After 10 min treatment with trypsin (Gibco, 25300054), neurons were triturated with a Pasteur pipette in plating medium (10% fetal bovine serum (Gibco,10099-147), 1 mM pyruvate (Gibco, 11360070), 0.37% glucose (Sigma-Aldrich, G8769), Minimal Essential Medium (Gibco, 41500-034)). Dissociated neurons, collected by centrifugation for 5 min at 800 rpm, were plated on nitric acid treated and poly-d-lysine (Sigma-Aldrich, P7886) coated coverslips. After incubating in neurobasal media (Gibco, 21103049) with 1 mM sodium pyruvate, 6 mM Glutamax (Gibco, 35050061), 10% fetal bovine serum, 0.5% glucose, and 50 μg ml−1 primocin (InvivoGen, ant-pm-1), the media was exchanged for neurobasal supplemented with B27 (Gibco, 17504044), 1 mM pyruvate, 2 mM glutamine (Gibco, 25030081), 50 μg ml−1 primocin, and 4 μM cytosine-1-β-D-arabinofuranoside (Sigma-Aldrich, 855855).
Cell surface biotinylation
DIV15 (days in vitro 15) neurons were carefully washed with ice-cold PBS plus (PBS containing 1 mM CaCl2 and 0.5 mM MgCl2), and incubated with 0.5 mg/ml sulfo-NHS-LC-biotin (Thermo Fisher Scientific, 21338) dissolved in PBS for 30 min at 4 °C. Free biotin was then quenched with PBS containing 50 mM Tris pH 7.4 for 10 min at 4 °C, washed with PBS plus, and lysed in RIPA buffer (1% NP-40, 0.1% SDS, 0.1% deoxycholate, 150 mM NaCl, 1 mM EDTA, and 10 mM Tris pH 7.4) with protease inhibitors. Clarified lysates were then immunoprecipitated with streptavidin-conjugated beads. The immunocomplexes were then subjected to western blot to detect NR2A, NR2B, NR1, and GluR1 using specific antibodies (Thermo Fisher Scientific, catalog numbers PA5-35377, PA3-105, PA3-102, PA1-46451, respectively).
SorCS2 immunostaining
DIV18 neurons were fixed with 4% paraformaldehyde/4% sucrose and permeabilized and blocked with 5% fetal bovine serum with 0.1% Triton X-100 in PBS for 30 min at RT. Primary antibodies were added at 1:800 dilution for anti-SorCS2 (R&D systems, AF4237), and 1:1000 for anti-MAP2 (Abcam, ab32454), anti-PSD-95 (Sigma-Aldrich, MAB1596), and anti-Bassoon (Enzo Life Sciences,SAP7F407). The proteins were visualized using Alexa-conjugated secondary antibodies (Life Technologies) and 405 mm fluorescent DyLight (Jackson ImmunoResearch Laboratories). Images were acquired using Structured Illumination Microscope (N-SIM) Nikon Eclipse Ti (Nikon Instruments Inc. NY, USA) using 100× Apo TIRF lens with 1.4 N.A. (Nikon Instruments Inc. NY, USA) and Andor iXon3 DU-897E EMCCD camera (Andor Technology Ltd, CT,USA). Images were reconstructed using SIM mode in Nikon Elements software and Z-stacks were collapsed into maximum intensity projection images. MAP2 outline was drawn and represented as ROI. The puncta were quantified using object count feature in Nikon Elements software and plotted using Graphpad Prism 6.
Electron microscopy
Electron microscopy
Sections were processed for electron microscopy as previously described [19]. Briefly, 12-month-old sorCS2+/+ and sorCS2−/− mice were overdosed with sodium pentobarbital (150 mg/kg, i.p.) and perfused transcardially with 2% heparin–saline followed by 30 ml 3.75% acrolein and 2% PFA in 0.1 M phosphate buffer (PB; pH 7.4). The brains were removed from the skull, postfixed in 2% acrolein and 2% PFA in PB for 30 min, and then placed in PB. Coronal sections through the brains were cut (40 µm thick) using a Vibratome (Leica Microsystems, Illinois, USA) and stored in cryoprotectant at −20 °C until use. For NR2B immunolabeling, dorsal hippocampal sections (n = 3 animals per genotype, two sections per animal; between Bregma −2.00 and −2.20 mm [20]) were rinsed in PB to remove cryoprotectant and then incubated in 1% sodium borohydride in PB for 30 min to remove active aldehydes. Sections were washed in 8–10 changes of PB until all the gaseous bubbles disappeared and then placed in 0.1 M Tris-buffered saline (TS; pH 7.6). Sections were incubated sequentially in: (i) 0.5% BSA in TS (30 min); (ii) rabbit anti-NR2B (Thermo Fisher Scientific, PA3-105) in 0.1% BSA in TS for 1 day at RT (~23 °C) and then 4 days at 4 °C. For immunogold detection of NR2B, sections were rinsed in 0.02 M PBS (pH 7.4), incubated in blocking buffer (0.8% BSA, 0.2% gelatin, 0.02% BSAc in 0.02 M PBS) for 30 min, and placed overnight in a 1:50 dilution of donkey anti-rabbit IgG with bound 10-nm colloidal gold (Electron Microscopy Sciences, cat# 25704) diluted in blocking buffer. The gold particles were fixed to the tissue in 2% glutaraldehyde in 0.02 M PBS and rinsed in PBS followed by 0.2 M sodium citrate buffer (pH 7.4). The bound silver–gold particles were enhanced using a silver intensification kit (SEKL15 Batch 009952 BBI Solutions, Cardiff, England) for 7 min.
Sections were rinsed in 0.1 M PB and then postfixed in 2% osmium tetroxide in PB for 1 h, dehydrated, and embedded with Epon 812 (Electron Microscopy Sciences) between 2 sheets of Aclar plastic. Sections for hippocampus were selected, mounted on EMBed chucks (Electron Microscopy Sciences), and trimmed to 1–1.5 mm-wide trapezoids. The CA2 region was selected using the fornix and pyramidal cell layer morphology as guides. Ultrathin sections (~70 nm thick) within 0.1–0.2 µm to the tissue-plastic interface were cut on a Leica Ultracut ultratome (Leica Microsystems, Il, USA), collected into copper mesh grids (EMS), and counterstained with uranyl acetate and Reynold’s lead citrate. Sections were viewed and photographed using a FEI Tecnai Biotwin electron microscope (FEI company, Or, USA) equipped with a digital camera (Advanced Microscopy Techniques, software version 3.2).
Ultrastructural image analysis
The data analysis procedure is similar as previously described [21]. The images were collected at a magnification of 18,500. The dendritic profiles contained regular microtubular arrays and were usually postsynaptic to axon terminal profiles [22]. Dendritic spines were small (about 0.1–0.2 µm in diameter), abutted terminals, and sometimes emanated from dendritic shafts [22]. Silver-intensified immunogold (SIG) labeling for NR2B appeared as black electron-dense particles. The criteria for field selection and the measures to avoid false-negative labeling of smaller profiles, variability between animals in each experimental group, and differential reagent sensitivity comparing SIG and immunoperoxidase labeling were performed as described before [19, 22]. Fifty dendrites and 25 dendritic spines from each animal were sampled for electron microscopic analysis.
The subcellular distribution and density for NR2B-SIG particles were determined as previously described [23]. For this, the NR2B-SIG particle localization was categorized as (i) on the plasma membrane, (ii) near the plasma membrane (particles not touching but within 70 nm from the plasma membrane), or (iii) cytoplasmic. The investigator performing the quantification of SIG particles was blinded to the genotypes. MCID imaging analysis software was used to determine the cross-sectional diameter, perimeter, surface area, form factor, and major and minor axis lengths of each immunolabeled dendrite. Density of SIG particles is expressed as: (i) number of SIG particles on plasma membrane/perimeter, (ii) number of SIG particles near plasma membrane/perimeter, (iii) number of SIG particles in cytoplasm/area, and (iv) total SIG particles/area.
All spine profiles that were in contact with axon terminals were further counted and classified as NR2B-labeled (containing at least 1 SIG particle) or non-NR2B-labeled spines. SIG particles in NR2B-labeled spines were additionally classified as cytoplasmic, on/near the plasma membrane, or on/near the synapse.
Rapid Golgi impregnation and analysis of dendritic spines
Rapid Golgi impregnation
The Golgi–Cox method for Golgi impregnation is as described [24]. Dissected brains were immersed in Golgi–Cox solution I for 10 days at RT protected from light, with a solution change after the initial 12 h. Brains were then switched to 30% sucrose in H2O for 4 days at 4 °C in the dark, with a solution change after 12 h. Brains were then cut coronally at 150 µm, and mounted on 0.3% gelatin coated slides, brushed with sucrose, and allowed to air dry for at least 2 days. Slides were immersed in H2O for 3 × 10 min with gentle shaking, transferred into Golgi–Cox solution II [24] for 5–10 min at RT followed by 3 × 5 min washes in H2O. Sections were mounted, dehydrated, and cleared with Histoclear (3 × 5 min), and DPX (mixture of Distyrene, Plasticizer, and Xylene) mounting medium was used to coverslip sections.
Dendritic spine analysis
Neurons were labeled using the Golgi–Cox method as described previously [24]. Golgi-impregnated sections were numbered in a blinded manner prior to quantitative analysis. Dendritic spines of CA2 and CA1 pyramidal neurons were counted at 100× magnification using a Zeiss HAL100 microscope (Carl Zeiss Microscopy, LLC, USA). Isolated dendrites were selected, and the total number of dendritic spines along 10 μm length was counted.
Behavioral experiments
The animals involved in this study are sorCS2−/− and their wild-type littermates, which have been back crossed to C57BL/6 for 10 generations or more. Animal care and experiments were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medicine. All efforts were made to minimize animal suffering. All experimental animals are caged in defined (12 h) light–dark conditions and given ad libitum access to food and water. The experiments were performed during the light period. On the day of test, mice were transferred to the test room for 1 h prior to the actual test for habituation. For sociability and social memory tests, both sexually naïve male and female animals (3–3.5 months old) were used. Male animals (3–3.5 months old) were used for other behavioral tests. All animals were grouped in a random manner to reduce bias. During behavioral experimental run, animals of different genotypes are staggered so that no two animals from the same group are evaluated consecutively.
Initial sample size for three-chamber social memory test was determined using power test (power = 0.8) based on effect size and standard deviation estimated from a previously published protocol [25]. Then subsequent sample sizes were determined using power test (power = 0.8) based on the observed effect size and standard deviation of the initial test.
Three-chamber sociability and novelty test
This test was performed according to [26] with modification. In brief, the three equal-sized compartments were used. On day 1, a 5-min sociability trial was conducted. A littermate was placed in the left or right compartment (systematically alternated) and the test subject was placed into the center compartment. The time that the test subject spent investigating each compartment (snout within 2 cm of the mesh barrier) was measured. On day 2, a 5-min social novelty test was conducted in which a littermate was placed in either the left or right compartment, and a novel animal (C57BL/6, 3 months old) was placed in the other compartment. The test subject was placed in the center compartment, and the investigation time of both novel animal and familiar animal was measured.
Two-trial social discrimination test
This test was conducted as described [27]. On the day of testing, all animals were moved to the testing room 1 h prior to the start of actual testing. Subject animals were then allowed to habituate for 10 min inside the chamber. During trial 1, a stimulus animal was introduced into the chamber for 5 min, and videotaped. The stimulus animal was then designated as the “familiar” animal and removed for 1 h. After 1 h, during Trial 2, either the familiar animal or a novel animal was introduced into the chamber, and videotaped.
Five-trial social memory assay
The assay was conducted as previously described [28]. Briefly, on the day of testing, all animals were moved to the testing room 1 h prior to the actual testing starts. Subject animals are then allowed to habituate for 10 min inside the chamber. The subject animals were presented with a novel mouse during the course of four successive 5-min trials (habituation); then on the fifth trial, another novel stimulus mouse was introduced. The interaction time during the 5 trials was videotaped and analyzed.
Buried food test
This test was conducted blindly as described [1]. At ~18–19 h prior to actual testing, subject animals were food-restricted, but not water-restricted, overnight to improve sensitivity. On the day of testing, after 1 h habituation to their new environment, sweetened cereals (5 pieces, ~1 g) of Kellogg’s were randomly placed in the corner of a clean cage under 1.5 cm of regular woodchip bedding. The subject mouse was then placed into the center of the cage and allowed to investigate for the buried food. Finding the cereal was defined as having both front paws on the cereal and beginning to eat. The latency to find the buried cereal was recorded with a stopwatch.
Olfactory habituation/dishabituation test
This test was performed as described [29]. On the day of testing, subject animals were moved to the testing room and allowed to acclimate to their new environment for 1 h. The sequence of odor consisted of two nonsocial odors (almond extract and banana extract (McCormick, 1:100 dilution)), and two social odors (obtained from mice of the same sex as the subject). The social odors 1 and 2 were from two cages containing same number of mice that have not been changed for at least 3 days. Immediately before use for the nonsocial odors, a cotton-tipped applicator was soaked in the diluted odorant and presented to the subject by suspension from the cage lid, ~3 cm above the bedding; for the social odors, the cage bottom (containing mice) was swiped in a zigzag fashion several times using a cotton-tipped applicator, then any bedding was shaken off from the cotton tip, and suspended from the cage lid as described above. Each odorant was presented for 2 min with an intertrial interval of 1 min for three trials (habituation) before switching to the next novel odorant (dishabituation). The cumulative time that the subject mouse spent sniffing the tip (orienting towards the cotton tip with its nose ≤2 cm) during the 2-min trial was recorded and analyzed blindly.
Open-field test
The open-field test was carried out in an apparatus consisting of a (40 × 40 × 49 cm) clear Plexiglas arena with a white floor marked with time tape dividing it into 12 equal quadrants. The experiment was monitored with a digital camera connecting to a video recorder and a computer under the control of the EthoVision tracking system. For each subject mouse, its behavior was recorded over a 20-min session. Anxiety levels were measured by the relative amount of exploration devoted to the center quadrants relative to those located adjacent to the walls of the arena. The data were quantified by two indices: (i) percentage time spent in the center quadrants and (ii) percentage of entries into the center quadrants. An entry into a given quadrant was only registered if all four paws were placed inside the quadrant.
Fear conditioning
The fear conditioning test was carried out as described [30] with continuous video recording during the behavioral testing. On conditioning day (day 1), following a 2.5-min acclimation period to the conditioning chamber, mice received three conditioning trials consisting of a 30-s presentation of a (5 kHz, 70-dB) tone (CS) that co-terminated with a 0.7-mA foot shock (US) delivered through the grid floor during the last 1.0 s of the tone. Each conditioning trial was separated by a 40-s intertrial interval. Following conditioning, mice were returned to their home cages. Mice were videotaped during CS presentations for subsequent quantification of behavior. Time spent “freezing” prior to and during the presentation of the tone CS was measured during the CS presentation as well as during a 30-s baseline period prior to the first tone trial. This latter measure served as an assay for both unconditioned effects on general activity levels. Following tone testing, mice were returned to their home cages. Memory for the context and the tone were evaluated on day 2 and 3, respectively (~24 and 48 h following conditioning). For the context test, mice were placed in the conditioning chamber and allowed to explore for 1 min, after which freezing to the context was assessed for the remaining 4.5 min. For the tone test, mice were placed in a novel chamber (circular in shape, with red walls, and scented with lemon odor), allowed to acclimate to the chamber for 2.5 min, and then presented with 3 tones (30 s, 5 kHz, 70 dB, ITI = 40 s). Freezing was evaluated during the 2.5 min acclimation period, during each presentation of the tone CS, and during the 40-s intertrial interval. Following memory tests, animals were returned to their home cage.
Statistical analysis
To be statistically adequate, we used power = 0.80 to determine the sample size on the basis of the smallest effect we wished to measure, to ensure that sample sizes were large enough to detect the effects of interest as an essential part of study design. For animal experiments, the animals were grouped in a random manner to reduce bias, and during behavioral experimental running, animals of different genotypes are staggered so that no two animals from the same group are evaluated consecutively. For all behavioral tests, all data were auto-scored by Ethovision system in real time, so that no human bias was introduced. Thus, no blinding process was involved here. For dendritic spine, buried food, and olfactory habituation analysis, the genotype of each animal was blinded to the experimenter who conducted the quantification, and the two different genotypes listed as group A or B. For all the behavioral tests, the data met the assumptions of the tests. The variation estimated within each group was compared, and it was similar between the groups that were being statistically compared. When two means were compared, statistical analysis was performed using two-tailed Student’s t tests, while multiple comparisons were made using one-way ANOVA with Newman–Keuls post hoc test or two-way ANOVA with Bonferroni post hoc test. Data are reported as +/−SEM.
Results
SorCS2 is highly expressed in CA2 and localized to postsynaptic sites
Prior studies document high levels of the SorCS2 transcript in CA2 [9, 31], and we evaluated the expression of the protein at a light microscopic level. In the murine cortex and subcortical regions, SorCS2 is highly expressed in region CA2 of the adult hippocampus, confirmed using a CA2 region-specific marker, PCP4 [32] (Figs. 1a, b, S1, and S2). SorCS2 immunoreactivity was noted in the dentate gyrus, and in cortical layer V, although at lower levels (Fig. 1a). SorCS2 was detected primarily in CA2 pyramidal cell bodies, and in basal and apical dendrites (Fig. 1a, high power image). In cultured hippocampal neurons (DIV15), SorCS2 immunoreactivity was detectable in cell bodies and dendrites, but not in axons (Fig. 1c). To gain further insight into subcellular localization, we used structured illumination microscopy (SIM) to localize SorCS2 to mature spines, colocalizing with PSD-95 (arrow), but not with the presynaptic marker Bassoon (Fig. 1d). Quantification demonstrated about 94% colocalization of SorCS2 with PSD-95 (Fig. 1e). Using immunoelectromicroscopy we confirmed that SorCS2 was localized to the postsynaptic density and in vesicles within dendrites (Fig. 1f). This regional localization of SorCS2 suggests a potential role in the intracellular trafficking of postsynaptic proteins.

a Hippocampal immunostaining for SorCS2 indicates predominant expression in CA2 region and modest expression in layer V of cortex. Higher power inset localizes SorCS2 in cell bodies and dendrites. Nonimmune IgG was used as a negative control. Representative data from 8 independent experiments with similar results. Scale: 200 μm. Scale for insert: 50 μm. b SorCS2 colocalizes with PCP4, a CA2 pyramidal neuron marker (n = 3 animals). Scale: 50 μm. c DIV15 hippocampal neurons, costained for SorCS2 or SMI312 (axonal marker) or MAP2 (dendritic marker). Scale: 30 μm. d SorCS2 is enriched in dendrites and found at the PSD. 3D-SIM image of a representative dendrite showing SorCS2 (green) colocalized with PSD-95 (red) (upper panel), but not with Bassoon (red) (lower panel). Representative colocalized puncta are highlighted by white arrows. Scale bar: 1 μm. e Quantification of SorCS2 localization in dendritic spines and colocalization with PSD-95. Nearly 80% of dendritic spines contain SorCS2 and of these SorCS2 positive spines, 74% have SorCS2 colocalized with PSD-95 (n = 9 neurons, 271 spines). Data are reported as ±SEM. f ImmunoEM was used to confirm that SorCS2 immunoreactivity is associated with the postsynaptic density in the dendritic spine (arrow) contacted by an unlabeled terminal (uT) and in a multivesicular body (mvb) in the adjoining dendrite. uT unlabeled terminal, mVb labeled multivesicular body, DCV dense core vesicle. Scale: 500 nm.
SorCS2 is a glycosylated, proteolytically processed protein
SorCS2 is a type 1 transmembrane protein with a predicted molecular mass of 123 kDa. In the brain areas examined, SorCS2 is detected as two forms of ~123 kDa (SorCS2-F) and ~105 kDa (SorCS2-T), with similar abundance using an ectodomain specific antibody (αECD) (Fig. 2a, left panel). A prior study reports cleavage of the SorCS2 ectodomain by TACE (tumour necrosis factor alpha-converting enzyme) [33] yielding a soluble 105 kDa species. Another study reports cleavage of the ectodomain, which remains noncovalently associated with the C-terminus in glia [34]. To determine whether the two forms represent effects of differential glycosylation or ectodomain cleavage, blots were probed using a cytoplasmic domain specific antibody (αICD), and detect only the 123 kDa band (Fig. 2a, right panel) and a band of ~18 kDa (SorCS2-C) (Fig. S3a), suggesting that proteolytically processing generates a cleaved ectodomain (~105 kDa) and a C-terminal fragment (~18 kDa). The treatment of lysates with N-glycanase resulted in more rapidly migrating forms of ~110 kDa and ~92 kDa (Fig. S3b), supporting the conclusion that SorCS2 is a proteolytically processed and glycosylated protein in vivo.

a An ectodomain specific SorCS2 antibody (α-ECD) detects the cleaved extracellular (SorCS2-T) and the full length protein (SorCS2-F). Anticytoplasmic domain (α-ICD) antibody detects SorCS2-F, but not SorCS2-T. Representative data from three independent experiments with similar results. b, c Characterization of sorCS2−/− mouse. The deletion of SorCS2 was confirmed by western blotting (b) and immunofluorescence staining (c). Representative graphs from 4 (b) and 6 (c) independent experiments with similar results. Scale: 200 μm. d, e Stereologic analysis of CA2 pyramidal neurons in sorCS2−/− and sorCS2+/+ mice using CA2 specific marker, PCP4. Representative staining images (d) and quantification of PCP4 positive pyramidal neurons of CA2 of sorCS2−/− and sorCS2+/+ brains (e). sorCS2−/− = 4, sorCS2+/+ = 3; n.s. = not significant, t test. Data are reported as ±SEM. Scale: 25 μm (d).
Generation of sorCS2 −/− mice
To investigate the physiological functions of SorCS2, we generated a sorCS2−/− knockout mouse in which exon 2 of the gene was flanked by LoxP sites (Fig. S4a, b). The efficiency of deletion, after crossing with deleter strains was examined by western blot and immunofluoresence staining. Using lysates from sorCS2−/− hippocampi, cortices, and cerebella, expression of SorCS2 (123 and 108 kDa) was not detectable (Fig. 2b) and lysates from sorCS2+/− mice demonstrated ~50% reduction in protein level as compared with the lysates from sorCS2+/+ littermates (Fig. 2b). Using immunofluorescence microscopy, we were unable to detect SorCS2 expression in the brains of sorCS2−/− mice (Figs. 2c, S1, and S5).
As Vps10 family members have been implicated in mediating apoptosis [7, 34], we performed stereologic analysis in CA2 using the marker PCP4 to detect CA2 pyramidal neurons. We observed no difference in the CA2 area, and no change in the location or boundaries of PCP4 staining in the sorCS2−/− mice as compared with sorCS2+/+ (Fig. 2d, e). These findings suggest that SorCS2 does not alter CA2 cell survival, migration, or differentiation.
SorCS2 interacts with NMDA receptors, but not the AMPA receptors
Subcellular localization of SorCS2 to the postsynaptic region (Fig. 1) [16], together with the known role of SorCS family members in intracellular trafficking, suggest that it may interact with and modulate surface localization of synaptic proteins. We examined potential interactions of SorCS2 and NMDARs, given the importance of these receptors in mediating synaptic plasticity in CA2 [15]. Co-immunoprecipitation analysis of C57BL/6 hippocampal lysates using anti-SorCS2 and western blot with anti-NR2B showed that SorCS2 co-precipitates with NMDAR subunit NR2B, as compared with the IgG control (Fig. 3a, left panel). To confirm the specificity of this interaction, hippocampal lysates of sorCS2−/− and sorCS2+/+ brains were subjected to immunoprecipitation. In wildtype, but not sorCS2−/− lysates, the NMDAR subunits NR2A and NR2B are co-precipitated (Fig. 3a, middle and right panels), demonstrating that in physiological conditions, SorCS2 interacts with NMDAR subunits. However, this interaction appears specific for NMDAR as the SorCS2 antibody does not specifically coprecipitate AMPAR as determined using anti-GluR1 antibody (Fig. S6).

a SorCS2 interacts with NMDAR. Hippocampal lysates were subjected to co-immunoprecipitation using anti-SorCS2 antibody and NMDAR subunits detected using anti-NR2B antibody (left panel). To confirm that the interaction is specific, sorCS2−/− hippocampal lysates were included. Upon SorCS2 deletion, NMDAR subunits, NR2B, and NR2A, can not be detected (middle and right panels). ctl, IgG control. Representative data from three independent experiments with similar results. b–d Cell surface biotinylation assay. DIV15 sorCS2−/− and sorCS2+/+ neurons were biotinylated and the biotinylated proteins were pulled down for western blotting. The NMDAR was detected using antibodies to subunits NR2B (b), NR2A (c), or NR1 (d), respectively. Quantification (right panels) revealed ~50% reduction of NMDAR at the sorCS2−/− cell surface as compared with sorCS2+/+ (n = 3 independent experiments; * < 0.05, ** < 0.01, n.s. = not significant, t test). Data are reported as ±SEM. e Deletion of SorCS2 does not alter AMPAR cell surface levels. After biotinylation, the cell surface AMPAR was detected using anti-GluR1 antibody. No significant difference was observed between sorCS2+/+ and sorCS2−/− neurons (right panel) (n = 3; * < 0.05, ** < 0.01, n.s. = not significant, t test). Data are reported as ±SEM. f, g Representative electron micrographs of dendrites (f) and synapses (g) in CA2 showing that NR2B silver-intensified immunogold (NR2B-SIG) particles are less frequently found on plasma membrane (PM) and spines (sp) of CA2 dendrites (Den) in sorCS2−/− mice (right panel) than sorCS2+/+ mice (left panel) at 12 months age. f White arrows, NR2B-SIG particle on PM; white arrowhead, NR2B-SIG particle near PM; arrows, NR2B-SIG particle in the cytoplasm. g Unlabeled terminals (uT) are shown for comparison. White arrow, NR2B-SIG particle on or near the synapses; white arrowhead, NR2B-SIG particle on or near dendritic PM; arrows, NR2B-SIG particle in the cytoplasm. Scale bar: 500 nm. 50 dendrites of each animal were analyzed. h Ultrastructural analysis shows that density of NR2B-SIG particles on PM is reduced significantly in sorCS2−/− mice compared with sorCS2+/+. No difference is observed near PM (nr PM). 50 dendrites of each animal were analyzed. i sorCS2−/− mice show significantly greater density of NR2B-SIG particles in the cytoplasm of CA2 dendrites than sorCS2+/+. Total density of NR2B-SIG within CA2 dendrites is similar between genotypes. j Graph showing that the percentages of NR2B-SIG containing sp in CA2 are not statistically different between sorCS2+/+ and sorCS2−/− mice. 300 sp of each animal were analyzed. k Graph showing the relative distribution of NR2B-SIG particles in CA2 sp. sorCS2+/+ mice have more NR2B-SIG particles associated with synapses and perisynaptic membranes (membrane), but less retained within the cytoplasm compared with sorCS2−/− mice. Twenty-five labeled sp of each animal were analyzed. h–k n = 3 animals/genotype; * < 0.05, ** < 0.01, *** < 0.001, t test. Data are presented as mean ± SEM.
SorCS2 regulates the cell surface distribution of NMDA receptors, but not AMPA receptors
Recent studies indicate that SorCS1 selectively interacts with neurexin, neuroligin, and AMPAR to regulate their surface localization and synaptic function [16]. To determine if SorCS2 regulates cell surface distribution of NMDAR, we carried out the cell surface biotinylation experiments in cultured hippocampal neurons (DIV15). In sorCS2−/− neurons, the cell surface levels of NMDAR subunits NR2B, NR2A, and NR1 were decreased to ~50% of the levels of sorCS2+/+ neurons (Fig. 3b–d, respectively). However, the cell surface levels of the AMPAR were not affected in sorCS2−/− neurons (Fig. 3e). The loss of surface receptors is not due to an effect on total levels of NR2B, NR2A, or NR1 because no changes in the whole lysates were observed (Fig. 3, input). These results suggest that SorCS2 regulates the trafficking NMDAR to the cell surface, but does not impact the total cellular level of these receptor subunits. Further analysis using expression of either full length SorCS2, or a mutant construct lacking the SorCS2 intracellular domain, in sorCS2−/− neurons suggests that the intracellular domain of SorCS2 is required for NMDA receptor surface distribution (Fig. S7). The interaction of SorCS2 with NMDAR subunits (NR2A and NR2B) does not appear to require additional proteins that are selectively exressed at the postsynaptic region, as co-immunoprecipitation of these subunits with SorCS2 is detected following expression in heterologous cells (Fig. S8a, b).
To directly determine whether SorCS2 is involved in NR2B trafficking, we utilized immunoelectron microscopy to quantitate the distribution of NR2B immunoreactivity in the dendrites (Fig. 3f) and spines (Fig. 3g) of CA2 neurons in sorCS2+/+ and sorCS2−/− mice. Immunostaining of NR2B on the plasma membrane of sorCS2−/− CA2 dendrites is decreased significantly compared with sorCS2+/+ mice at 12 months of age (Fig. 3h). In contrast, cytoplasmic NR2B-SIG is significantly increased in sorCS2−/− mice, although the total density of dendritic NR2B-SIG in both genotypes is comparable (Fig. 3i). Moreover, SorCS2 deficiency did not alter the percentage of NR2B-SIG-labeled CA2 dendritic spines (Fig. 3j), but there were fewer NR2B-SIG labeled at the synapses and more were retained in the cytoplasm of spines in sorCS2−/− mice as compared with sorCS2+/+ mice (Fig. 3k). These results demonstrate that deficiency of SorCS2 leads to significantly fewer NR2B containing NMDARs trafficked to dendritic and synaptic surfaces of CA2 neurons. From these studies, we conclude that SorCS2 is a key regulator of NR2B dendritic trafficking in CA2 neurons.
SorCS2 regulates dendritic spine density
The postsynaptic expression of SorCS2, together with its role in promoting synaptic localization of NR2B, suggest that it may play a role in dendritic spine formation and morphology. We evaluated apical dendritic spine density of CA2 pyramidal neurons of sorCS2−/− mice and wild-type littermates at P50 (Fig. 4a, b) and P105 (Fig. 4a, c). We observed a significant reduction in dendritic spine density in both ages in sorCS2−/− compared with sorCS2+/+ mice (Fig. 4b, c). To study the specificity of the effect of SorCS2 on CA2 pyramidal neurons, we also evaluated the CA1 pyramidal neurons, which express lower levels of SorCS2 (Fig. 1a and S1). We found no significant differences in spine density between sorCS2−/− and sorCS2+/+ CA1 neurons (Fig. 4d, e).

a Representative images of Golgi-stained dendritic spines of CA2 neurons from sorCS2+/+ and sorCS2−/− mice. The extent of co-localizatoin of SorCS2 positive neurons with the CA2 marker PCP4 is 94.5% (Fig. S2). b, c Deletion of SorCS2 resulted in reduction in spine density in CA2 neurons in both ages in sorCS2−/− mice: P50 (b) and P105 (c). Dendritic spines of 13–15 neurons/mouse of three mice per genotype were counted at 100×. Total spine number along a 10-µm-long dendrite was measured. d, e Deletion of SorCS2 did not influence the dendritic spine density of CA1 neurons in both ages. n = 3 animals per genotype. * < 0.05; ** < 0.01; *** < 0.001; t test. Data are reported as ±SEM.
SorCS2 is essential for social recognition
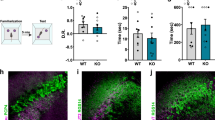
Prior studies have linked the expression of forebrain NMDAR to social memory [35, 36] and we next evaluated whether SorCS2 affects social memory. We performed three-chamber tests for sociability and social memory. Sociability was determined by the preference for a chamber containing an unfamiliar mouse versus an empty chamber. sorCS2−/− mice were comparable to sorCS2+/+ mice in reference for the compartment containing the unfamiliar mouse, indicating sorCS2−/− mice have normal sociability (Figs. 5a and S9a). Social recognition was then measured by the time that a subject mouse interacted with a novel unrelated mouse versus the time interacting with a familiar mouse. In contrast to sorCS2+/+ mice, sorCS2−/− mice showed no significant preference for the novel animal (Figs. 5b and S9b). To confirm this social recognition deficit, we conducted a more specific direct interaction test of social memory. In this two-trial test, a subject mouse was first exposed to a novel unrelated mouse in trial 1. After 1 h intertrial interval, in trial 2 the subject mouse either reexposed to the same mouse as in trial 1, or exposed to a novel unrelated mouse. The time spent exploring the previously encountered mouse versus the time spent exploring a novel unrelated mouse was recorded to determine the social memory. Unlike sorCS2+/+, sorCS2−/− mice spent similar exploration times during trials 1 and 2 (Fig. 5c, in trial 2, subject mice were reexposed to the same mouse as in trial 1; Fig. 5d, in trial 2, subject mice were exposed to a novel unrelated mouse). To exclude the possibility that this deficit is due to lack of interest in novelty, we performed the novel object recognition test and found that sorCS2−/− mice had a normal preference for a novel object as assayed by two different novel object recognition protocols (Fig. S10a–c), indicating that the social memory deficit was not due to a lack of interest in novelty.

a sorCS2−/− mice exhibit normal sociability. Sociability was determined by comparing sorCS2−/− mice (n = 15) with sorCS+/+ mice (n = 17) in preference for a chamber containing an unfamiliar mouse versus an empty chamber. b sorCS2−/− mice exhibit a deficit in social recognition. Social recognition was measured by the increased time a subject mouse spent interacting with a novel unrelated mouse compared with the time spent with a familiar mouse using the three-chamber test. c, d Two-trial direct interaction test. Subject mice were exposed to a novel unrelated mouse in trial 1 for 5 min, and after a 1-h interval, reexposed to the same mouse as in trial 1, or exposed to a second novel unrelated mouse. Social memory was measured by the time of exploring the previously encountered mouse (c) versus the time of exploring a novel unrelated mouse (d). sorCS2−/− mice spent similar and unchanging exploration times during trials 1 and 2 (+/+ = 17, −/− = 15). e Five-trial social memory assay. A novel unrelated mouse was presented to a subject mouse for four successive trials. On the fifth trial, another novel unrelated mouse was introduced. The social memory was measured by the habituation during the first four trials and dishabituation in the fifth trial. sorCS2−/− mice showed no significant habituation or dishabituation (+/+ = 17, −/− = 15). f, g Deletion of SorCS2 did not influence olfaction. Two tests were performed: the ability to find the buried food (f), and to detect or discriminate either nonsocial or social odors (g) (+/+ = 12, −/− = 10). h sorCS2−/− and sorCS2+/+ animals have no difference in acquisition (+/+ = 11, −/− = 12). i, j SorCS2 does not alter other hippocampal-dependent memory. Two tests were performed: context-fear conditioning (i) and cue-fear conditioning (j) (+/+ = 11, −/− = 12). k–m Anxiety-like behavioral test. In open-field test, sorCS2−/− displayed no differences in total running distance (k), the number of entries into the center (l), and the percentage of time spent in the center (m), as compared with sorCS2+/+ mice (+/+ = 17, −/− = 15). Statistical significance was assessed for all behavioral tests by one-way ANOVA, two-way RM ANOVA, or t test where appropriate. Data are reported as ±SEM. (*** < 0.001; n.s. = not significant).
To further assess social memory, we performed a more stringent five-trial social memory assay. Here the same novel mouse was presented to a subject mouse for four successive trials with 10-min intertrial intervals. On the fifth trial, a new unrelated mouse was introduced. Social memory was measured by the decreased exploration time (habituation) during the first four trials and increased exploration time (dishabituation) in the fifth trial. sorCS2−/− mice showed no significant habituation or dishabituation as compared with sorCS2+/+ mice (Fig. 5e). Thus these results confirm that SorCS2 is required for social memory.
Because of the importance of olfaction in social memory, we examined whether SorCS2 deficiency influences the detection of nonsocial or social odors [29]. sorCS2−/− and sorCS2+/+ mice demonstrated similar ability to detect the presence of food buried under cage bedding (nonsocial odor detection) (Fig. 5f). Furthermore, we conducted an olfactory habituation/dishabituation test and found that both sorCS2+/+ and sorCS2−/− mice showed similar abilities to detect or discriminate either nonsocial or social odors (Fig. 5g). Taken together, we conclude that the deficit in social memory in sorCS2−/− mice was not due to a defect in sensing social or nonsocial odors.
To test if the deletion of SorCS2 altered other forms of hippocampal-dependent memory, we assessed contextual fear and cue-dependent memory using a standard fear paradigm [30]. First we examined the acquisition of sorCS2−/− animals and found there are no differences as compared with sorCS2+/+ animals across the three-tone periods (Fig. 5h). sorCS2−/− mice showed no significant difference in context-dependent memory retrieval as compared with sorCS2+/+ mice (Fig. 5i). In addition, there was no significant difference in cue-dependent fear memory retrieval (Fig. 5j). In the open-field test to determine anxiety-like behaviors, sorCS2−/− and sorCS2+/+ mice displayed no differences in two standard measurements: the number of entries into the center compartment (Fig. 5l) and the percentage of time spent in the center compartment (Fig. 5m). Also, the motor ability between sorCS2−/− and sorCS2+/+ mice is comparable (Figs. 5k and S11). These data suggest that the deletion of SorCS2 leads to specific deficits in social memory, but not other hippocampal-related behaviors.
Discussion
Social memory is an important component of complex social behavior, which integrates social, spatial, and temporal information. This process depends upon discrimination of novel and familiar individuals. The hippocampal CA2, where sorCS2 is highly expressed, is a critical hub for social memory processing [1]. Here we report a novel function of SorCS2 in regulating social memory formation. We found that SorCS2 specifically interacts with NMDAR subunits, but not AMPAR. The impaired synaptic membrane distribution of NMDAR in sorCS2−/− neurons is likely to alter synaptic plasticity, and impact dendritic spine density of pyramidal neurons that express high levels of SorCS2, such as CA2 neurons, without affecting spine density of hippocampul neurons expressing low levels of SorCS2. Collectively, these results indicate that SorCS2 is a critical mediator of social memory formation by regulating the trafficking of synaptic receptors.
In the hippocampal trisynaptic pathway, information is processed from entorhinal cortical inputs to the dentate gyrus, and then to CA3 and CA1 [37], with distinct functions of these subregions in learning and memory [38, 39]. However, the role of the relatively small CA2 region has only recently been described to mediate social memory [1], and the importance of the projection to ventral CA1 has only recently been mapped [3]. Few molecular pathways have been implicated in the regulation of this memory processing, for instance, the vasopressin 1b receptor (Avpr1b). Genetic deletion of Avpr1b results in abnormal social memory [40], and delivery of Avpr1b agonists potentiates CA2 synaptic responses, which rely on NMDAR activation, and calcium and calcium/calmodulin-dependent protein kinase II activity [41]. These studies indirectly evaluated the role of NMDAR activation, through the use of chemical blockade. Our observations that sorCS2−/− mice have significant social memory deficit without affecting the sociability and other hippocampal-dependent behaviors, and have no alteration on Avpr1b cell surface expression (Fig. S12a, b), provide a new mechanism in which SorCS2 selectively regulates NMDAR trafficking to the postsynaptic site.
The dynamic trafficking of synaptic proteins regulates the function and plasticity of synapses [42]. For example, the transport, synaptic insertion, and removal of NMDAR and AMPAR are tightly regulated to control synaptic efficacy [43]. Using a proteomic approach, Savas et al. have identified that SorCS1 acts as a regulator of AMPAR trafficking. In sorCS1−/− neurons, the decrease of AMPAR in cell surface significantly impairs glutamatergic synaptic transmission [16]. Here we provide evidence that SorCS2 selectively interacts with NMDAR, but not AMPAR, to regulate the postsynaptic levels of NMDAR. NMDAR function is known to be a crucial component of oxytocin and vasopressin-mediated activation of CA2 [25, 40]. In hippocampal slices, the specific NMDAR inhibitor (AP5) blocks the activation of CA2 pyramidal neurons mediated by Avpr1b and Oxtr [41], indicating that postsynaptic NMDAR is required for social memory. It was previously not known how NMDAR trafficking to postsynaptic membrane was regulated to mediate the synaptic plasticity of CA2 neurons. Our data provide a critical mechanism, by which SorCS2 regulates NMDAR synaptic localization to facilitate synaptic function and social memory. It is important to note that other hippocampal regions, including ventral CA1 also express SorCS2 [44], although at reduced levels compared with CA2. While CA2 excitatory input to vCA1 modulates social memory [3], the current studies do not exclude additional effects of SorCS2 on these vCA1 pyramidal neurons.
Our current study demonstrates that SorCS2 regulates the postsynaptic membrane localization of NMDA receptors, therefore controlling synaptic activity and strength. CA2 pyramidal neurons differ from CA1 and CA3 neurons, as they are more resistant to damage, and importantly, they do not exhibit experimentally induced synaptic plasticity as readily as those in CA1 or CA3 [45]. These differences may be due to the increased spine calcium buffering [46]. NMDA receptors are essential for controlling Ca+2 influx and synaptic strength [47], and thus the insertion of NMDA receptors in dendritic spines may regulate normal glutamatergic neurotransmission and synaptic plasticity [48]. Several studies have also demonstrated that impaired Ca+2 homeostasis can result in late-onset synaptic degeneration associated with multiple neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington disease [49]. Taken together, our results are in line with these findings, and provide a potential mechanism by which SorCS2 regulates NMDA receptor localization and function to affect social memory formation.
Utilizing an independently generated SorCS2 knockout model which targets exon 15 of SorCS2, Glerup et al. recently demonstrated that SorCS2 interacts with and trafficks two neurotrophin receptors: p75 and TrkB [15]. This study postulates that SorCS2 interacts with p75 to form a complex to bind proneurotrophins, therefore facilitating long-term depression, whereas the interaction of SorCS2 with TrkB receptors may elicit long-term potentiation by acting to transport TrkB receptors to postsynaptic densities [15]. Our results provide a further mechanism by which SorCS2 regulates hippocampal function by regulating NMDAR trafficking.
Recently, several members of the Vps10 family proteins have been implicated as genetic risk factors for several neurodegenerative diseases, including AD and PD. Alterations in SorCS1 have been associated with AD, possibly due to the effects on processing of APP into Aβ peptide, which is crucial for the development of AD [17, 25]. Genome-wide association studies have implicated SorCS2 in the etiology of BD [50], SCZ [11], and ADHD [51]. Social memory is a component of these disorders [52, 53], and remarkably, the CA2 area is altered in SCZ, BD, as well as in neurodegenerative conditions [54]. Impaired intracellular trafficking is a common signature of both neuropsychiatric and neurodegenerative disease [55, 56], and our studies identify SorCS2 as an important novel regulator in NMDAR-dependent synaptic activities, and suggest its important function in related psychiatric disorders with a social memory-deficit endophenotypes.
References
Hitti FL, Siegelbaum SA. The hippocampal CA2 region is essential for social memory. Nature. 2014;508:88–92.
Smith AS, Williams Avram SK, Cymerblit-Sabba A, Song J, Young WS. Targeted activation of the hippocampal CA2 area strongly enhances social memory. Mol Psychiatry. 2016;21:1137–44.
Meira T, Leroy F, Buss EW, Oliva A, Park J, Siegelbaum SA. A hippocampal circuit linking dorsal CA2 to ventral CA1 critical for social memory dynamics. Nat Commun. 2018;9:4163.
Dudek SM, Alexander GM, Farris S. Rediscovering area CA2: unique properties and functions. Nat Rev Neurosci. 2016;17:89–102.
Young WS, Li J, Wersinger SR, Palkovits M. The vasopressin 1b receptor is prominent in the hippocampal area CA2 where it is unaffected by restraint stress or adrenalectomy. Neuroscience. 2006;143:1031–9.
Hermey G. The Vps10p-domain receptor family. Cell Mol Life Sci. 2009;66:2677–89.
Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427:843–8.
Deinhardt K, Kim T, Spellman DS, Mains RE, Eipper BA, Neubert TA, et al. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci Signal. 2011;4:ra82.
Hermey G, Plath N, Hübner CA, Kuhl D, Schaller HC, Hermans-Borgmeyer I. The three sorCS genes are differentially expressed and regulated by synaptic activity. J Neurochem. 2004;88:1470–6.
Grupe A, Li Y, Rowland C, Nowotny P, Hinrichs AL, Smemo S, et al. A scan of chromosome 10 identifies a novel locus showing strong association with late-onset Alzheimer disease. Am J Hum Genet. 2006;78:78–88.
Christoforou A, McGhee KA, Morris SW, Thomson PA, Anderson S, McLean A, et al. Convergence of linkage, association and GWAS findings for a candidate region for bipolar disorder and schizophrenia on chromosome 4p. Mol Psychiatry. 2011;16:240–2.
Lionel AC, Crosbie J, Barbosa N, Goodale T, Thiruvahindrapuram B, Rickaby J, et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med. 2011;3:95ra75.
Reitz C, Tokuhiro S, Clark LN, Conrad C, Vonsattel JP, Hazrati LN, et al. SORCS1 alters amyloid precursor protein processing and variants may increase Alzheimer’s disease risk. Ann Neurol. 2011;69:47–64.
Li Z, Zheng M, Abdalla BA, Zhang Z, Xu Z, Ye Q, et al. Genome-wide association study of aggressive behaviour in chicken. Sci Rep. 2016;6:30981. https://doi.org/10.1038/srep30981.
Glerup S, Bolcho U, Mølgaard S, Bøggild S, Vaegter CB, Smith AH, et al. SorCS2 is required for BDNF-dependent plasticity in the hippocampus. Mol Psychiatry. 2016;21:1740–51.
Savas JN, Ribeiro LF, Wierda KD, Wright R, DeNardo-Wilke LA, Rice HC, et al. The sorting receptor SorCS1 regulates trafficking of neurexin and AMPA receptors. Neuron. 2015;87:764–80.
Hermey G, Schmidt N, Bluhm B, Mensching D, Ostermann K, Rupp C, et al. SorCS1 variants and amyloid precursor protein (APP) are co-transported in neurons but only SorCS1c modulates anterograde APP transport. J Neurochem. 2015;135:60–75.
Breiderhoff T, Christiansen GB, Pallesen LT, Vaegter C, Nykjaer A, Holm MM, et al. Sortilin-related receptor SORCS3 is a postsynaptic modulator of synaptic depression and fear extinction. PLoS ONE. 2013;8:e75006.
Milner TA, Waters EM, Robinson DC, Pierce JP. Degenerating processes identified by electron microscopic immunocytochemical methods. Methods Mol Biol. 2011;793:23–59.
Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd ed. Hong Kong: Academic Press; 1996.
Marques-Lopes J, Lynch MK, Van Kempen TA, Waters EM, Wang G, Iadecola C, et al. Female protection from slow-pressor effects of angiotensin II involves prevention of ROS production independent of NMDA receptor trafficking in hypothalamic neurons expressing angiotensin 1A receptors. Synapse. 2015;69:148–65.
Peters A, Palay SL, Webster H deF. The fine structure of the nervous system: neurons and their supporting cells. 3rd ed. New York: Oxford University Press; 1991.
Van Kempen TA, Narayan A, Waters EM, Marques-Lopes J, Iadecola C, Glass MJ, et al. Alterations in the subcellular distribution of NADPH oxidase p47(phox) in hypothalamic paraventricular neurons following slow-pressor angiotensin II hypertension in female mice with accelerated ovarian failure. J Comp Neurol. 2016;524:2251–65.
Yang J, Harte-Hargrove LC, Siao CJ, Marinic T, Clarke R, Ma Q, et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014;7:796–806.
Kaidanovich-Beilin O, Lipina T, Vukobradovic I, Roder J, Woodgett JR. Assessment of social interaction behaviors. J Vis Exp. 2011;e2473. https://doi.org/10.3791/2473.
DeVito LM, Konigsberg R, Lykken C, Sauvage M, Young WS 3rd, Eichenbaum H. Vasopressin 1b receptor knock-out impairs memory for temporal order. J Neurosci. 2009;29:2676–83.
Kogan JH, Frankland PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000;10:47–56.
Ferguson JN, Young LJ, Hearn EF, Matzuk MM, Insel TR, Winslow JT. Social amnesia in mice lacking the oxytocin gene. Nat Genet. 2000;25:284–8.
Yang M, Crawley JN. (2009) Simple behavioral assessment of mouse olfaction. Curr Protoc Neurosci. https://doi.org/10.1002/0471142301.
Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–3.
Oetjen S, Mahlke C, Hermans-Borgmeyer I, Hermey G. Spatiotemporal expression analysis of the growth factor receptor SorCS3. J Comp Neurol. 2014;522:3386–402.
Lein ES, Callaway EM, Albright TD, Gage FH. Redefining the boundaries of the hippocampal CA2 subfield in the mouse using gene expression and 3-dimensional reconstruction. J Comp Neurol. 2005;485:1–10.
Hermey G, Sjøgaard SS, Petersen CM, Nykjaer A, Gliemann J. Tumour necrosis factor alpha-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem J. 2006;395:285–93.
Glerup S, Olsen D, Vaegter CB, Gustafsen C, Sjoegaard SS, Hermey G, et al. SorCS2 regulates dopaminergic wiring and is processed into an apoptotic two-chain receptor in peripheral glia. Neuron. 2014;82:1074–87.
Jacobs S, Wei W, Wang D, Tsien JZ. Importance of the GluN2B carboxy-terminal domain for enhancement of social memories. Learn Mem. 2015;22:401–10.
Jacobs S, Tsien JZ. Adult forebrain NMDA receptors gate social motivation and social memory. Neurobiol Learn Mem. 2017;138:164–72.
van Strien NM, Cappaert NL, Witter MP. The anatomy of memory: an interactive overview of the parahippocampal-hippocampal network. Nat Rev Neurosci. 2009;10:272–82.
Nakashiba T, Young JZ, McHugh TJ, Buhl DL, Tonegawa S. Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science. 2008;319:1260–4.
Suh J, Rivest AJ, Nakashiba T, Tominaga T, Tonegawa S. Entorhinal cortex layer III input to the hippocampus is crucial for temporal association memory. Science. 2011;334:1415–20.
Wersinger SR, Kelliher KR, Zufall F, Lolait SJ, O’Carroll AM, Young WS 3rd. Social motivation is reduced in vasopressin 1b receptor null mice despite normal performance in an olfactory discrimination task. Horm Behav. 2004;46:638–45.
Pagani JH, Zhao M, Cui Z, Avram SK, Caruana DA, Dudek SM, et al. Role of the vasopressin 1b receptor in rodent aggressive behavior and synaptic plasticity in hippocampal area CA2. Mol Psychiatry. 2015;20:490–9.
Choquet D, Triller A. The dynamic synapse. Neuron. 2013;80:691–703.
Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–43.
Giza JI, Kim J, Meyer HC, Anastasia A, Dincheva I, Zheng CI, et al. The BDNF Val66Met prodomain disassembles dendritic spines altering fear extinction circuitry and behavior. Neuron. 2018;99:1356.
Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–14.
Zhou M, Baudry M. Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J Neurosci. 2006;26:2956–63.
Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, et al. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci. 2011;31:9170–8.
Petralia RS, Al-Hallaq RA, Wenthold RJ. Trafficking and targeting of NMDA receptors. In: Van Dongen AM, editor. Biology of the NMDA receptor. Boca Raton, FL: CRC Press/Taylor & Francis; 2009.
Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100.
Lane RF, Steele JW, Cai D, Ehrlich ME, Attie AD, Gandy S. Protein sorting motifs in the cytoplasmic tail of SorCS1 control generation of Alzheimer’s amyloid-β peptide. J Neurosci. 2013;33:7099–107.
Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2008;13:197–207.
Alemany S, Ribasés M, Vilor-Tejedor N, Bustamante M, Sánchez-Mora C, Bosch R, et al. New suggestive genetic loci and biological pathways for attention function in adult attention deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2015;168:459–70.
Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;52:139–53.
Martinowich K, Schloesser RJ, Manji HK. Bipolar disorder: from genes to behavior pathways. J Clin Invest. 2009;119:726–36.
Nakazawa T, Hashimoto R, Sakoori K, Sugaya Y, Tanimura A, Hashimotodani Y, et al. Emerging roles of ARHGAP33 in intracellular trafficking of TrkB and pathophysiology of neuropsychiatric disorders. Nat Commun. 2016;7:10594.
Palmisano R, Heimann P, Shaw C, Troakes C, Schmitt-John T, Bartsch JW. Endosomal accumulation of APP in wobbler motor neurons reflects impaired vesicle trafficking: Implications for human motor neuron disease. BMC Neurosci. 2011;12:24.
Acknowledgements
We thank the members of the Hempstead and Lee Laboratories for helpful discussions and advice about the manuscript, and the Neuroanatomy EM Core facilities in the Feil Family Brain and Mind Research Institute for EM. We also thank Dr Jianhong Luo from Zhejiang University for providing the NMDAR 2A and 2B plasmids. This work was supported by NIH grants P01 HD023315 and NS064114 to BLH, NS052819 to FSL, and DA08259 to TM, Shaanxi Province Natural Science Basic Research Foundation of China (2019JM-051) and National Natural Science Foundation of China (81971275) to JY.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, J., Ma, Q., Dincheva, I. et al. SorCS2 is required for social memory and trafficking of the NMDA receptor. Mol Psychiatry 26, 927–940 (2021). https://doi.org/10.1038/s41380-020-0650-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-020-0650-7
- Springer Nature Limited
This article is cited by
-
Celsr2 regulates NMDA receptors and dendritic homeostasis in dorsal CA1 to enable social memory
Molecular Psychiatry (2024)
-
Loss of SORCS2 is Associated with Neuronal DNA Double-Strand Breaks
Cellular and Molecular Neurobiology (2023)
-
Finding memo: versatile interactions of the VPS10p-Domain receptors in Alzheimer’s disease
Molecular Neurodegeneration (2022)
-
Altered Calcium Permeability of AMPA Receptor Drives NMDA Receptor Inhibition in the Hippocampus of Murine Obesity Models
Molecular Neurobiology (2022)
-
Single neonatal dexamethasone administration has long-lasting outcome on depressive-like behaviour, Bdnf, Nt-3, p75ngfr and sorting receptors (SorCS1-3) stress reactive expression
Scientific Reports (2021)