
Abstract
Patients at clinical high-risk (CHR) for psychosis show elevations in [18F]DOPA uptake, an estimate of dopamine (DA) synthesis capacity, in the striatum predictive of conversion to schizophrenia. Intrasynaptic DA levels can be inferred from imaging the change in radiotracer binding at D2 receptors due to a pharmacological challenge. Here, we used methylphenidate, a DA reuptake inhibitor, and [11C]-(+)-PHNO, to measure synaptic DA availability in CHR both in striatal and extra-striatal brain regions. Fourteen unmedicated, nonsubstance using CHR individuals and 14 matched control subjects participated in the study. Subjects underwent two [11C]-(+)-PHNO scans, one at baseline and one following administration of a single oral dose (60 mg) of methylphenidate. [11C]-(+)-PHNO BPND, the binding potential relative to the nondisplaceable compartment, was derived using the simplified reference tissue model with cerebellum as reference tissue. The percent change in BPND between scans, ΔBPND, was computed as an index of synaptic DA availability, and group comparisons were performed with a linear mixed model. An overall trend was found for greater synaptic DA availability (∆BPND) in CHR than controls (p = 0.06). This was driven entirely by ∆BPND in ventral striatum (−34 ± 14% in CHR, −20 ± 12% in HC; p = 0.023). There were no significant group differences in any other brain region. There were no significant differences in DA transmission in any striatal region between converters and nonconverters, although this finding is limited by the small sample size (N = 2). There was a strong and negative correlation between ΔBPND in VST and severity of negative symptoms at baseline in the CHR group (r = −0.66, p < 0.01). We show abnormally increased DA availability in the VST in CHR and an inverse relationship with negative symptoms. Our results suggest a potential early role for mesolimbic dopamine overactivity in CHR. Longitudinal studies are needed to ascertain the significance of the differential topography observed here with the [18F]DOPA literature.
Similar content being viewed by others
Introduction
The majority of individuals who are at clinical high risk for psychosis (CHR) experience attenuated positive symptoms—positive symptoms that do not meet criteria for syndromal psychosis due to being experienced with lesser conviction levels or with minimal or no functional impairment [1]. Approximately 30% of these individuals develop a syndromal psychosis over 2 years. Many of those who do not develop syndromal psychosis have other psychiatric conditions (e.g., anxiety, depression) or have persistent attenuated positive or negative symptoms [2]. Potential benefits of examining the neurobiology of psychosis at this stage is that pathophysiological processes can be separated from effects of illness, chronicity, treatment, and hospitalizations.
[18F]DOPA PET studies in CHR individuals have shown that dopamine (DA) synthesis capacity is elevated in the striatum in general and more specifically in the associative striatum (AST) in CHR subjects [3,4,5], in line with the evidence in schizophrenia [6]. Furthermore, the increase predicted conversion to psychosis [7]. A SPECT study using the alpha-methyl-para-tyrosine DA depletion paradigm and the D2/3 radioligand [123I]–IBZM in 14 high-risk patients and 15 control subjects demonstrated no differences in change in striatal- binding potential between the two groups, indicating similar D2/3 occupancy by DA [8]. The authors did, however, observe that those patients with higher baseline levels of positive symptoms demonstrated greater reduction in symptoms after DA depletion [8]. One study examined extra-striatal regions in the CHR period with [18F]DOPA and found increases in the midbrain area [9]. The [18F]DOPA uptake rate, Ki or \(K_{_i}^{cer}\), is a measure of a combination of aromatic-L-amino acid decarboxylase (AADC) activity and vesicular loading in the presynaptic terminal as well as DA turnover due to metabolism, while the depletion paradigm measures availability of intrasynaptic DA. Another imaging technique that examines intrasynaptic DA is the methylphenidate paradigm, used extensively in the PET literature [10]. By combining D2 radiotracer imaging with a methylphenidate challenge, which blocks the DA transporter and thus prevents reuptake of DA into the presynaptic terminal, one can infer intrasynaptic DA availability. Here we used methylphenidate in combination with [11C]-(+)-PHNO imaging.
[11C]-(+)-PHNO. [11C]-(+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin -9-ol ([11C]-(+)-PHNO) is a dopamine-3 receptor (D3R) preferring DA D2R/D3R agonist. Unlike D2R antagonist radioligands such as [11C]raclopride, [18F]fallypride, [11C]FLB457 or the SPECT ligand [123I] IBZM, the specific binding of [11C]-(+)-PHNO is highest in structures such as the globus pallidus (GP) and the ventral striatum (VST), where D3R density is high [11]. It was first radiolabeled with carbon-11 in 2005 [12], has robust binding in both striatal and extra-striatal regions including midbrain and thalamus [13, 14], and is sensitive to changes in DA release [15, 16]. An advantage of [11C]-(+)-PHNO over other D2/3 radiotracers such as [11C]FLB457, [11C]raclopride, and [18F]fallypride is that it allows imaging both striatal and extra-striatal areas, and the signal in all these regions can be quantified within a 2 h scan duration [17,18,19,20,21,22].
[11C]-(+)-PHNO has been previously used in CHR patients. Mizrahi et al. examined stress-induced release of DA in 12 CHR patients and matched controls and observed increased release in AST and sensorimotor, but not limbic, striatum in patients [23]. They did not report on extra-striatal regions. However, in a follow-up paper in which Mizrahi et al. examined stress-induced DA release in CHR patients with and without cannabis use [24], they reported that CHR subjects who were not using cannabis exhibited greater DA release in limbic, associative, and sensorimotor striatum, as well as substantia nigra, than did CHR subjects who were using cannabis, and that greater release of DA was related to higher baseline total negative symptom score on the Structured Interview for Psychosis-Risk Syndromes [SIPS; 25].
Here, we used [11C]-(+)-PHNO imaging combined with a methylphenidate challenge to examine striatal and extrastrial intrasynaptic DA availability in CHR patients compared to healthy controls. We aimed to clarify whether DA availability in the midbrain is enhanced as suggested by [18F]DOPA studies in CHR previously, and to further examine the topography of striatal DA abnormalities.
Methods
Subjects
Subjects were recruited at the Center of Prevention and Evaluation (COPE), an outpatient research program for the evaluation and treatment of attenuated positive symptoms at the New York State Psychiatric Institute (NYSPI)/Columbia University Irving Medical Center (CUIMC) in New York City. The NYSPI Institutional Review Board approved the research. All subjects provided written informed consent.
We recruited individuals between the ages of 18 and 30 who were required to have symptoms that met criteria for the SIPS Attenuated Positive Symptoms Psychosis-Risk Syndrome (APSS) delineated in the SIPS [26,27,28,29,30]. The APSS criteria require at least one positive symptom occurring at least once weekly in the past month and scored between 3 and 5. Symptoms must not be better explained by another psychiatric or medical condition. The syndrome is ruled out by past or present syndromal psychosis (i.e., at least one positive symptom scored 6, occurring for at least 1 h daily for 4 days weekly throughout 1 month, or causing severe disorganization, or endangering self or others). All patients in this cohort met criteria for the Progression subtype of the SIPS APSS syndrome, which additionally requires that at least one positive symptom is new or has worsened by one or more points in the past year. Consensus of individuals’ SIPS scores and diagnostic categories were established by certified administrators.
All participants were also evaluated with either the Diagnostic Interview for Genetic Studies [31] or Structured Clinical Interview for DSM-IV-I Disorders, Patient Edition (SCID-I/P; [32]).
Exclusion criteria included lack of proficiency in English; a current or lifetime DSM psychotic disorder, including affective psychoses; a DSM disorder better accounting for the clinical presentation; I.Q. < 70; medical conditions affecting the central nervous system; marked risk of harm to self or others; unwillingness to participate in research; geographic distance; or a DSM diagnosis of current (but not previous) substance or alcohol abuse or dependence. Use of any psychotropic medication was exclusionary, as was any use of any substance of abuse (confirmed via urine toxicology). We also recruited healthy control subjects who met similar criteria to CHR individuals except that they did not meet criteria for any psychosis-risk syndrome on the SIPS, did not meet criteria for any Axis I disorder on the SCID or Diagnostic Interview for Genetic Studies, were nonsubstance using, and had never taken psychiatric medications. The study sample is described in Table 1.
Subjects were screened with clinical laboratory tests, vital sign assessments, electrocardiography, physical and psychiatric examinations, and the Clinical Global Impression-Severity scale [33] before participation. Subjects were evaluated every 3 months for up to 2 years for potential conversion to syndromal psychosis.
PET and radiochemistry methods
[11C]-(+)-PHNO was prepared by N-acylation of the despropyl precursor with [11C]-propionyl chloride followed by reduction of the resulting amide with lithium aluminum hydride and purification by reverse-phase high-performance liquid chromatography [12]. All scans were 120 min, following a single, 2 min, bolus injection of [11C]-(+)-PHNO. PET studies were performed on a Siemens mCT hybrid PET/computed tomography (CT) scanner (Siemens, Knoxville, TN). An intravenous catheter was inserted in the antecubital vein for tracer injection. After being properly positioned on the scanner bed, each subject was fitted with a polyurethane head immobilizer system molded around the subject’s head (Soule Medical, Tampa, FL) to reduce head motion during the PET scan. Fifteen minutes before ligand injection, a 7 s CT scan was performed for attenuation correction. Following injection, three-dimensional emission data were acquired in a list mode, rebinned into a sequence of 21 frames of increasing duration (3 × 20 s, 3 × 1 min, 3 × 2 min, 2 × 5 min and 10 × 10 min), and, following correction for scatter, random coincidences, dead-time and attenuation, were reconstructed by filtered backprojection using manufacturer-supplied software (Siemens).
Each subject received two [11C]-(+)-PHNO PET scans separated by at least 5 h in order to minimize potential mass-carryover effects [Table S1; 14]. A 60 mg oral dose of methylphenidate was administered to all subjects 1 h before the second scan. This dose of methylphenidate has been shown to produce a robust displacement in radiotracer binding [10, 34]. All subjects received continuous EKG and vital sign monitoring for 3 h or until vital signs returned to baseline, whichever came later, after methylphenidate administration. A licensed and ACLS-certified physician was present at all times during methylphenidate administration and monitoring. Patients were assessed with the full SIPS before methylphenidate administration and with the positive symptoms of the SIPS 3 h after methylphenidate administration. As an additional precaution, all patients were admitted to an inpatient psychiatric research unit after receiving their dose of methylphenidate for close monitoring of symptoms and discharged the next morning.
A T1-weighted anatomical magnetic resonance imaging (MRI) scan was acquired for each subject and used to draw regions of interest (ROIs) for each individual. ROIs included AST (pre- and post-commissural caudate and pre-commissural putamen), sensorimotor striatum (SMST, post-commissural putamen), VST, GP, thalamus, a midbrain region including substantia nigra and ventral tegmental area (SN/VTA), and cerebellum as a reference tissue [11]. PET data were coregistered to subjects’ MRIs using a maximization of mutual information algorithm implemented in SPM12 [35]. ROIs were applied to the coregistered PET images. Left and right sides were averaged and time activity curves were generated as the average ROI activity in each frame.
PET data analysis
Reference tissue-based kinetic modeling was performed on all data using a basis function version of the simplified reference tissue method. The primary outcome measure was the binding potential relative to the nondisplaceable compartment (BPND) [36] at baseline and post-methylphenidate challenge in each ROI. The fractional change in BPND across conditions (∆BPND) was computed as the relative change between baseline BPND and BPND following methylphenidate administration:
ΔBPND was taken as an index of synaptic DA availability.
Statistical analyses
Group level differences in demographics, clinical variables, and scan parameters were examined using independent groups t-tests for continuous variables and Pearson Chi-square tests for categorical variables. BPND and ∆BPND were tested with a linear model in a mixed model framework with group as a fixed effect and ROI as repeated measure, using the gls function from the nlme package in R [37]. Relationships between clinical parameters and BPND were analyzed using Pearson correlation coefficients.
Results
Fourteen CHR individuals and 14 matched control subjects completed all study procedures (Table 1). One additional CHR participant did not undergo the post-methylphenidate PET scan due to a radiochemistry failure. Two additional control subjects who had completed screening were unable to participate due to one radiochemistry failure and one PET scanner technical failure. There were no significant differences in group demographics or scan parameters between the two groups. There were no significant differences in plasma levels of methylphenidate between the two groups. All subjects received both PET scans on the same day except for one control subject who received his methylphenidate administration and second PET scan 14 days after his baseline scan.
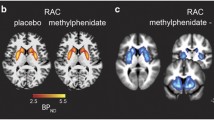
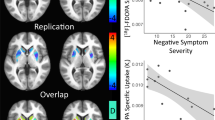
Table 2 and Figs. 1 and 2 show the PET results. Main effects of group and group by ROI interaction were not significant for BPND, nor were there significant differences between groups in any individual ROI. The main effect of methylphenidate (model intercept for ∆BPND) was significantly different than 0 (F(1, 156) = 78, p < 0.001) and main effect of group reached trend level (F(1, 156) = 3.63, p = 0.059). This was driven by ΔBPND in VST which was significantly greater in CHR (−34 ± 14%) than in healthy control subjects (−20 ± 12%; t = 2.29, p = 0.023). There were no significant differences in ∆BPND between groups in any other ROI. There was a strong correlation between ΔBPND in VST and baseline total negative symptoms as measured by the SIPS in the CHR group (r = 0.66, p < 0.01; Fig. 3) in which greater displacement was related to less negative symptoms. We also observed significant relationships between greater ΔBPND in midbrain and greater positive symptoms (r = −0.57, p = 0.03; Fig. 3) and a trend for greater ΔBPND in thalamus with greater disorganization symptoms as measured by the SIPS (r = −0.52, p < 0.06; Fig. 3), although none of these relationships would survive correction for multiple comparisons. No other clinical correlations achieved significance (Table S2).

Circular markers are CHR. Triangular markers are HC. Blue are baseline scans. Red are post-MP scans.

Blue circles are CHR; Red triangles are HC.

SN/VTA (R = −0.57, p = 0.03) and thalamus (R = −0.52, p < 0.06). Black markers are the data. The red line is the fit. The blue dashed lines are 95% confidence intervals.
We followed patients for up to 2 years. Two of the subjects developed syndromal schizophrenia within the 2 year follow-up time period, one at 3 months and one at 21 months. There were no significant differences in DA transmission in any striatal region between converters and nonconverters, although this finding is limited by the small sample size.
Given the importance of the mesolimbic system to depression [38], we examined whether there were differences in DA transmission among CHR individuals with and without a diagnosis of major depressive disorder (n = 7 with and n = 7 without) and found no differences (p = 0.45). We observed similar results when we included a patient with a diagnosis of dysthymic disorder in the group with depression (p = 0.61). As expected, when examining differences in DA transmission between CHR individuals with and without a diagnosis of depression and control subjects, we observed that CHR individuals still demonstrated greater DA transmission in VST (p = 0.02 for both). One of the subjects with MDD and the subject with dysthymia had active symptoms. All others were in remission.
Further, given that half of our sample met criteria for a depressive disorder at some point in their lifetime, and depressive symptoms could confound our observed relationship between negative symptoms and ΔBPND in VST, we examined relationships between PET data (ΔBPND in all regions) and total General symptoms (Table S2), which captures depressive symptoms in item G2 (“Dysphoric Mood”). We observed no relationships between ΔBPND in any region and total General symptoms or item G2.
Similarly, given other important findings of cannabis use in CHR individuals affecting DA release in striatal regions [24], we examined whether there were differences in DA transmission among CHR individuals with a previous diagnosis of any cannabis or other substance use disorder (N = 4; all four had cannabis use disorders; one had a comorbid past LSD use disorder and another had a comorbid, past alcohol use disorder) and control subjects. There were no significant differences in any striatal region.
Further, nine subjects had comorbid anxiety disorders, one had hypochondriasis, and one had an eating disorder.
The study procedures were very well tolerated. We observed minimal change in total positive symptoms related to the methylphenidate challenge (baseline: 15.36 (3.46), post methylphenidate: 15.43 (3.32)) in patients. Two subjects experienced nausea and two subjects vomited after receiving their first [11C]-(+)-PHNO injections. All were able to complete their scans. One patient developed mild, transient anxiety after receiving methylphenidate and another developed insomnia.
Discussion
This is the first study combining methylphenidate challenge with an agonist ligand, [11C]-(+)-PHNO, for the D2/3 receptors. Using this novel paradigm we observe no differences in baseline measures of receptor availability, which is consistent with prior studies [39], and a larger methylphenidate-induced [11C]-(+)-PHNO displacement in the VST in CHR individuals compared to healthy controls, indicative of excess DA availability in this striatal subregion. No abnormalities in DA transmission were observed in the other striatal subdivisions or in extra-striatal regions. The number of converters to syndromal psychosis (N = 2) was too small to derive useful information regarding group differences or relationships between DA parameters and symptoms in these subjects. We also observed an inverse relationship between VST DA transmission and severity of negative symptoms, but no relationship to positive symptoms, and no change in positive symptoms induced by methylphenidate. These results have potential implications for our current views on the development of DA dysfunction in schizophrenia [40].
In healthy controls, our results show a similar gradient of displacement across the striatal subdivisions as seen with the amphetamine challenge where VST and SMST show more displacement than AST [41]. Despite this similarity in healthy controls across these two paradigms, the main group difference we observe here in CHR patients is in the VST, rather than the AST, as we would have predicted from the [18F]DOPA literature in CHR and its concordance with the findings in schizophrenia, also showing excess amphetamine-induced DA release predominantly in the AST [3,4,5, 23]. Furthermore, our results differ as we observe no difference in synaptic DA at the level of the whole striatum [8]. There may be several reasons for these topographical differences.
First, we will consider clinical factors that may inform these differences. Subjects in the current study met criteria for the APSS, whereas patients in other studies could have also met criteria [3,4,5, 23] for the Brief Intermittent Psychotic Syndrome which is, by definition, a CHR diagnosis that is closer to syndromal psychosis than APSS. Therefore, it is possible that the apparent discrepancies between this and prior studies could reflect the differential effect of pathogenic mechanisms along the temporal development of psychotic disorders, where our subjects are earlier in the disease process, and less symptomatic. This is supported by the finding in the study of Mizrahi et al. that a stress task led to a significant increase in positive symptoms in CHR individuals [23], similar to psychostimulant challenge in individuals with syndromal psychosis [42], while in our study 60 mg of methylphenidate resulted in nearly no change in positive symptoms. If this interpretation is correct, it also suggests that DA dysregulation may first be observed in VST prior to other adjacent structures of the striatum and may propagate later to other more caudal and dorsal regions as the illness progresses toward frank psychosis. This progression was observed in a prior study with [18F]DOPA increasing from AST to SMST with a 2 year follow-up in patients who transitioned to schizophrenia [7]. It would also suggest that with progression the magnitude of VST DA excess may diminish. Such adaptations in the DA system may occur over time. For example, it has been suggested from preclinical studies that excess striatal D2 stimulation affects NMDA expression on VTA DA cells and reduces the activity of these cells projecting to the limbic regions [43]. This interpretation of the data merits further replication in larger cohorts as it suggests that the earliest dysregulation of DA may emerge in the VST, as proposed initially by the mesolimbic hypothesis of schizophrenia [40, 44]. It is also congruent with recent data from clinical and preclinical studies emphasizing the importance of excess glutamate in the hippocampus (and in particular the CA1 subfield) [45,46,47,48,49,50] leading to increased DA cell firing in VTA and increased DA release in the VST [51] by reducing tonic inhibition onto DA cells.
Understanding the potential contribution of DA abnormalities in the VST versus AST [52] has important implications for the understanding of psychosis and schizophrenia, since these striatal subregions differ in their topographical connections within the cortico-basal ganglia loops and their neurochemical markers [6]. While both structures may be involved in the prodromal phase of the disease emergence, our data, based on the lower symptomatic burden, suggest that VST may precede AST pathology.
Other cohort related differences may be due to medication effects. With the exception of the study of Mizrahi et al. [23], substance use and nonantipsychotic psychoactive medications were allowed in other studies [3,4,5], while only patients without any current psychoactive medication use and who were nonsubstance using were enrolled in the current study. In these studies, substance use may have compromised DA transmission in the VST but not in other subregions, since VST DA is particularly affected by drugs of abuse [53]. In the current study, there were no differences between CHR individuals with previous substance use disorders (primarily cannabis) and those without, but the small number of subjects involved in this observation and the fact that none of the patients were actively using any substances warrants caution in comparing this finding with other studies [24].
It is also possible that our finding of increased DA transmission in VST is related to depression, given the relevance of the mesolimbic DA system to reward dysfunction in depression [38]. Previous studies of amphetamine challenge in depression have reported either no differences in DA transmission in striatum [54, 55] or a trend level increase [56] when compared with control subjects. Therefore, we examined whether there were differences in DA transmission among CHR individuals with and without a diagnosis of major depressive disorder and found no differences. As expected, when examining differences in DA transmission between CHR individuals with and without a diagnosis of depression and control subjects, we observed that CHR individuals still demonstrated greater DA transmission in VST. In addition, there were no relationships between DA transmission and depressive symptoms as measured by the SIPS. Therefore, it is unlikely that the findings of this study are related to comorbid depressive disorders.
Second, technical differences in data acquisition could have led to the observed differences in results between studies. The Mizrahi et al. study [23] used 90 min of scanning with [11C]-(+)-PHNO which is adequate for reliable BPND estimation in most regions. We scanned for 120 min to allow for accurate estimation in regions like the VST and GP where washout is slow, but a possible limitation of our study is that long scans combined with low activity (5–6 mCi on average) may have led to less reliable BPND estimation in dorsal striatum, where washout is faster, than a shorter scan. To test this possibility, we repeated the analysis in SMST and AST with data truncated to 90 min duration. We find that 90 min BPND estimates are within 2 ± 3% of the 120 min measurements in both groups and both conditions, and that ∆BPND and group level statistical tests of ∆BPND are similarly unchanged. Thus, it is not the case that failure to detect group differences in dorsal striatum was due to long duration or low counts. Another difference between our study and that of Mizrahi et al. is the use of scanner with lower spatial resolution (FWHM ~ 4.5 mm at the center of the field of view on the mCT used in our study, compared to ~2.7 mm on the HRRT in Mizrahi et al.). The lower resolution will likely lead to lower BPND due to increased partial volume effect. We have shown previously though, that this effect is much less pronounced in ∆BPND, essentially, because the lost resolution cancels when the difference across conditions is normalized to the baseline value [57].
It has been shown that the mass doses of [11C]-(+)- PHNO used in this study (~2 μg unlabeled (+)-PHNO) may exceed the tracer dose assumption of peak receptor occupancy by the radiotracer of 5% or less at D3 receptors (not at D2) [58]. We have shown previously that this is likely to have a modest effect on measurement of occupancy by a competitive antagonist [59]. The present setting is different, as the radiotracer and competitor (DA) are both agonists and there may be effects at D3 receptors other than pure competition. Importantly, though, both groups were exposed to the same levels of PHNO mass, and so mass effects are unlikely to account either for the observed group differences in VST, or lack of differences in GP and SN/VTA, regions with large contributions of D3 binding to [11C]PHNO BPND.
Another difference between our study and prior literature is the psychostimulant challenge paradigm used here, which measures DA reuptake inhibition, as opposed to DA synthesis, storage and release capacity [3,4,5] or stress-induced DA release [23]. Differences in results could reflect differences in the cellular mechanisms of DA dysregulation captured by these different paradigms. Berry et al. examined DA synthesis capacity and methylphenidate-induced DA reuptake inhibition in the same subjects and observed no relationships [60, 61]. Alterations in DAT activity across the striatal subregions could explain our findings, however, no studies to date have examined the DAT across striatal subregions. This interpretation will need further testing with specific DAT tracers.
We observed no group differences in extra-striatal regions. In schizophrenia, data have been inconsistent and have suggested increased [18F]DOPA uptake [62], while we have previously observed a generalized deficit in DA release, especially in the DLPFC and the midbrain, with [11C]FLB457 imaging and the amphetamine-challenge paradigm [63]. A study by Schifani et al. used [11C]FLB457 and stress-induced release of DA and reported no differences in extra-striatal DA release between CHR patients and control subjects [64], similar to the finding in the current study. Tseng et al. used [11C]-(+)-PHNO and stress-induced release of DA and also observed no differences in extra-striatal DA release between CHR patients and control subjects [65]. Interestingly, Schifani et al. also reported no changes in positive symptoms in CHR subjects in response to their stress challenge, which is also consistent with the current study. More work is needed to gain a better understanding of extra-striatal DA function in CHR individuals, but these studies do not suggest widespread alterations.
We observed a strong correlation between ΔBPND in VST and baseline total negative symptoms in the CHR group (r = 0.66, p = 0.01) in which greater ligand displacement (i.e., DA transmission) was related to less negative symptoms. This is inconsistent with Mizrahi et al. who reported that greater stress-induced release of DA in limbic striatum, GP, and midbrain, but not AST, was related to higher baseline total negative symptom scores on the SIPS in CHR individuals [25]. However, the findings of the current study are consistent with a similar finding in a study of DA depletion with [11C]raclopride in syndromal schizophrenia [52]. The relationships between greater positive symptoms at baseline and ΔBPND in midbrain are interesting and consistent with findings from a study of nigral neuromelanin in both CHR and individuals with schizophrenia in which greater positive symptoms was related to greater levels of neuromelanin [66].
There are some limitations to this study. Very few subjects transitioned to psychosis upon follow-up and we have a small sample size. Thus, we need to be cautious in interpretation our data. Further, while the patients who were included in this study are characteristic of those in our clinic in terms of symptomatology [67], it is possible that the subjects who were actually able to participate in this rigorous study were not characteristic of CHR subjects in other ways. The evolution of patients in this risk category is mixed, and it may be that the excess DA in VST is unrelated to the emergence of schizophrenia. Second, we may not have had sufficient power to detect either a relationship to psychosis or even alterations in other regions. In VST, where the effect size for ∆BPND was large (d = 1.06), 15 subjects per group would be required to obtain power of 0.8 for a test significance level of 0.05, and therefore a replication study should be feasible with typical PET study sample sizes. The next largest effect size was in SMST (d = 0.33) which corresponds to a sample size of 146 subjects per group to obtain the same power and significance levels. Thus, it is clearly not practical to measure this difference with PET, nor is it clear that such small differences in displacement (29% vs 26%) are clinically meaningful. In addition, it is possible that the methylphenidate blood levels might have differentially changed over the course of the 120-min PET scans, which could have confounded the results. Finally, while the use of a reference tissue approach to analysis eliminates the need for invasive arterial catheterization and is commonly used in [11C]-(+)-PHNO studies [23, 24], we cannot rule out the possibility that there were effects of methylphenidate on cerebellum that differed by group. While this possibility cannot be tested rigorously without the use of an arterial input function, analysis of the cerebellum time activity curves showed no evidence of this (See Supplement). Nevertheless, this study demonstrates the safety of studying DA availability in CHR with methylphenidate as a challenge paradigm and reveals intriguing observations about the possible role of VST in the early stages of risk and in the genesis of negative symptoms.
In summary, we demonstrate here that methylphenidate challenge is associated with increased DA transmission in the VST in CHR individuals, though not in any other brain regions. We also demonstrated an inverse relationship between VST DA transmission and negative symptoms, but not with psychosis-like symptoms. This study, taken together with evidence from cross sectional studies in first-episode psychosis and other work in CHR individuals discussed herein, indicates that striatal DA alterations may progress in a temporo-spatial pattern, in a manner analogous and potentially pathophysiologically linked to the spread of hippocampal dysfunction in early psychosis [45, 51, 68, 69]. Striatal DA alterations may also precede extra-striatal alterations in the pathogenesis of psychotic disorders, although longitudinal follow-up is needed to ascertain what proportion of this sample will actually show other pathology. Future studies would benefit from longitudinal imaging with multiple ligands capable of fully sampling cortical and striatal regions (e.g., [11C]-(+)-PHNO and [11C]FLB457), combined with clinical evaluations to track disease trajectories. This will allow for the development of focused, stage dependent, therapeutic or preventative interventions.
References
Fusar-Poli P, Borgwardt S, Bechdolf A, Addington J, Riecher-Rossler A, Schultze-Lutter F, et al. The psychosis high-risk state: a comprehensive state-of-the-art review. JAMA Psychiatry. 2013;70:107–20.
Addington J, Cornblatt BA, Cadenhead KS, Cannon TD, McGlashan TH, Perkins DO, et al. At clinical high risk for psychosis: outcome for nonconverters. Am J Psychiatry. 2011;168:800–5.
Howes OD, Bose SK, Turkheimer F, Valli I, Egerton A, Valmaggia LR, et al. Dopamine synthesis capacity before onset of psychosis: a prospective [18F]-DOPA PET imaging study. Am J Psychiatry. 2011;168:1311–7.
Egerton A, Chaddock CA, Winton-Brown TT, Bloomfield MA, Bhattacharyya S, Allen P, et al. Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: findings in a second cohort. Biol Psychiatry. 2013;74:106–12.
Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P, et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry. 2009;66:13–20.
Weinstein JJ, Chohan MO, Slifstein M, Kegeles LS, Moore H, Abi-Dargham A. Pathway-specific dopamine abnormalities in schizophrenia. Biol Psychiatry. 2017;81:31–42.
Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry. 2011;16:885–6.
Bloemen OJ, de Koning MB, Gleich T, Meijer J, de Haan L, Linszen DH, et al. Striatal dopamine D2/3 receptor binding following dopamine depletion in subjects at ultra high risk for psychosis. Eur Neuropsychopharmacol. 2013;23:126–32.
Allen P, Luigjes J, Howes OD, Egerton A, Hirao K, Valli I, et al. Transition to psychosis associated with prefrontal and subcortical dysfunction in ultra high-risk individuals. Schizophr Bull. 2012;38:1268–76.
Volkow ND, Wang G, Fowler JS, Logan J, Gerasimov M, Maynard L, et al. Therapeutic doses of oral methylphenidate significantly increase extracellular dopamine in the human brain. J Neurosci. 2001;21:RC121.
Ginovart N, Willeit M, Rusjan P, Graff A, Bloomfield PM, Houle S, et al. Positron emission tomography quantification of [11C]-(+)-PHNO binding in the human brain. J Cereb Blood Flow Metab. 2007;27:857–71.
Wilson AA, McCormick P, Kapur S, Willeit M, Garcia A, Hussey D, et al. Radiosynthesis and evaluation of [11C]-(+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin-9 -ol as a potential radiotracer for in vivo imaging of the dopamine D2 high-affinity state with positron emission tomography. J Med Chem. 2005;48:4153–60.
Searle G, Beaver JD, Comley RA, Bani M, Tziortzi A, Slifstein M, et al. Imaging dopamine D3 receptors in the human brain with positron emission tomography, [11C]PHNO, and a selective D3 receptor antagonist. Biol Psychiatry. 2010;68:392–9.
Girgis RR, Xu X, Miyake N, Easwaramoorthy B, Gunn RN, Rabiner EA, et al. In vivo binding of antipsychotics to D(3) and D(2) receptors: a PET study in baboons with [(11)C]-(+)-PHNO. Neuropsychopharmacology. 2011;36:887–95.
Boileau I, Payer D, Chugani B, Lobo DS, Houle S, Wilson AA, et al. In vivo evidence for greater amphetamine-induced dopamine release in pathological gambling: a positron emission tomography study with [(11)C]-(+)-PHNO. Mol Psychiatry. 2013;19:1305–13.
Gallezot JD, Kloczynski T, Weinzimmer D, Labaree D, Zheng MQ, Lim K, et al. Imaging nicotine- and amphetamine-induced dopamine release in rhesus monkeys with [(11)C]PHNO vs [(11)C]raclopride PET. Neuropsychopharmacology. 2014;39:866–74.
Halldin C, Farde L, Hogberg T, Mohell N, Hall H, Suhara T, et al. Carbon-11-FLB 457: a radioligand for extrastriatal D2 dopamine receptors. J Nucl Med. 1995;36:1275–81.
Narendran R, Frankle WG, Mason NS, Rabiner EA, Gunn RN, Searle GE, et al. Positron emission tomography imaging of amphetamine-induced dopamine release in the human cortex: a comparative evaluation of the high affinity dopamine D2/3 radiotracers [11C]FLB 457 and [11C]fallypride. Synapse. 2009;63:447–61.
Chou YH, Halldin C, Farde L. Effect of amphetamine on extrastriatal D2 dopamine receptor binding in the primate brain: a PET study. Synapse. 2000;38:138–43.
Olsson H, Halldin C, Swahn CG, Farde L. Quantification of [11C]FLB 457 binding to extrastriatal dopamine receptors in the human brain. J Cereb Blood Flow Metab. 1999;19:1164–73.
Narendran R, Mason NS, May MA, Chen CM, Kendro S, Ridler K, et al. Positron emission tomography imaging of dopamine D2/3 receptors in the human cortex with [(1)(1)C]FLB 457: reproducibility studies. Synapse. 2011;65:35–40.
Sudo Y, Suhara T, Inoue M, Ito H, Suzuki K, Saijo T, et al. Reproducibility of [11 C]FLB 457 binding in extrastriatal regions. Nucl Med Commun. 2001;22:1215–21.
Mizrahi R, Addington J, Rusjan PM, Suridjan I, Ng A, Boileau I, et al. Increased stress-induced dopamine release in psychosis. Biol Psychiatry. 2012;71:561–7.
Mizrahi R, Kenk M, Suridjan I, Boileau I, George TP, McKenzie K, et al. Stress-induced dopamine response in subjects at clinical high risk for schizophrenia with and without concurrent cannabis use. Neuropsychopharmacology. 2014;39:1479–89.
Miller TJ, McGlashan TH, Rosen JL, Cadenhead K, Cannon T, Ventura J, et al. Prodromal assessment with the structured interview for prodromal syndromes and the scale of prodromal symptoms: predictive validity, interrater reliability, and training to reliability. Schizophr Bull. 2003;29:703–15.
McGlashan TH, Miller TJ, Woods SW, Hoffman RE, Davidson LA. Scale for the assessment of prodromal symptoms and states. In: Miller T, Mednick, SA, McGlashan, TH, Liberger, J, Johannessen, JO, editor Early intervention in psychotic disorders. Dordrecht: Kluwer Academic Publishers; 2001. p. 135–49.
Miller TJ, McGlashan TH, Rosen JL, Somjee L, Markovich PJ, Stein K, et al. Prospective diagnosis of the prodrome for schizophrenia: preliminary evidence of interrater reliability and predictive validity using operational criteria and a structured interview. Am J Psychiatry. 2002;159:863–65.
Miller TJ, McGlashan TH, Rosen JL, Cadenhead K, Cannon T, Ventura J, et al. Prodromal assessment with the Sstructured Interview for Prodromal Syndromes: Predictive validity, interrater reliability, and training to reliability. Schizophrenia Bull. 2003;29:703–15.
Rosen JL, Woods SW, Miller TJ, McGlashan TH. Prospective observations of emerging psychosis. J Nerv Ment Dis. 2002;190:133–41.
McGlashan TH, Walsh BC, Woods SW. Structured Interview for Psychosis Risk Syndromes. New Haven, CT: PRIME Research Clinic, Yale School of Medicine; 2014.
Nurnberger JI Jr, Blejar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J, et al. Diagnostic interview for genetic studies. Rationale, unique features, and training. Arch Gen Psychiatry. 1994;51:849–59.
First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition. (SCID-I/P). New York: Biometrics Research NYSPI; 2002.
Guy W. Clinical global impressions. In: Guy W, editor. ECDEU assessment manual for psychopharmacology: revised, ADM 76-338. Washington, DC: Department of Health, Education, and Welfare; 1976. p. 217–22.
Martinez D, Carpenter KM, Liu F, Slifstein M, Broft A, Friedman AC, et al. Imaging dopamine transmission in cocaine dependence: link between neurochemistry and response to treatment. Am J Psychiatry. 2011;168:634–41.
SPM Online Bibliography. http://www.fil.ion.ucl.ac.uk/spm/doc/biblio/. Accessed 2020.
Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–9.
Pinheiro J, Bates D, DebRoy S, Sarkar D, Team RC. nlme: linear and nonlinear fixed effects models. 2020. https://CRAN.R-project.org/package=nlme.
Nestler EJ, Carlezon WA Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–9.
Suridjan I, Rusjan P, Addington J, Wilson AA, Houle S, Mizrahi R. Dopamine D2 and D3 binding in people at clinical high risk for schizophrenia, antipsychotic-naive patients and healthy controls while performing a cognitive task. J Psychiatry Neurosci. 2013;38:98–106.
McCutcheon RA, Abi-Dargham A, Howes OD. Schizophrenia, dopamine and the striatum: from biology to symptoms. Trends Neurosci. 2019;42:205–20.
Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, et al. Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: amphetamine-induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab. 2003;23:285–300.
Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology. 1987;91:415–33.
Simpson EH, Kellendonk C. Insights about striatal circuit function and schizophrenia from a mouse model of dopamine D2 receptor upregulation. Biol Psychiatry. 2017;81:21–30.
Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–86.
Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78:81–93.
Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66:938–46.
Ho NF, Holt DJ, Cheung M, Iglesias JE, Goh A, Wang M, et al. Progressive decline in hippocampal CA1 volume in individuals at ultra-high-risk for psychosis who do not remit: findings from the longitudinal youth at risk study. Neuropsychopharmacology. 2017;42:1361–70.
Bossong MG, Antoniades M, Azis M, Samson C, Quinn B, Bonoldi I, et al. Association of hippocampal glutamate levels with adverse outcomes in individuals at clinical high risk for psychosis. JAMA Psychiatry. 2019;76:199–207.
Kraguljac NV, White DM, Reid MA, Lahti AC. Increased hippocampal glutamate and volumetric deficits in unmedicated patients with schizophrenia. JAMA Psychiatry. 2013;70:1294–302.
Provenzano FA, Guo J, Wall MM, Feng X, Sigmon HC, Brucato G, et al. Hippocampal pathology in clinical high-risk patients and the onset of schizophrenia. Biol Psychiatry. 2020;87:234–42.
Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci. 2011;12:585–601.
Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–9.
Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–38.
Parsey RV, Oquendo MA, Zea-Ponce Y, Rodenhiser J, Kegeles LS, Pratap M, et al. Dopamine D(2) receptor availability and amphetamine-induced dopamine release in unipolar depression. Biol Psychiatry. 2001;50:313–22.
Busto UE, Redden L, Mayberg H, Kapur S, Houle S, Zawertailo LA. Dopaminergic activity in depressed smokers: a positron emission tomography study. Synapse. 2009;63:681–9.
Schneier FR, Slifstein M, Whitton AE, Pizzagalli DA, Reinen J, McGrath PJ, et al. Dopamine release in antidepressant-naive major depressive disorder: a multimodal [(11)C]-(+)-PHNO positron emission tomography and functional magnetic resonance imaging study. Biol Psychiatry. 2018;84:563–73.
Slifstein M, Narendran R, Hwang DR, Sudo Y, Talbot PS, Huang YY. et al. Effect of amphetamine on [F-18]fallypride in vivo binding to D-2 receptors in striatal and extrastriatal regions of the primate brain: Single bolus and bolus plus constant infusion studies. Synapse. 2004;54:46–63.
Gallezot JD, Beaver JD, Gunn RN, Nabulsi N, Weinzimmer D, Singhal T, et al. Affinity and selectivity of [(1)(1)C]-(+)-PHNO for the D3 and D2 receptors in the rhesus monkey brain in vivo. Synapse. 2012;66:489–500.
Slifstein M, Abi-Dargham A, Girgis RR, Suckow RF, Cooper TB, Divgi CR, et al. Binding of the D3-preferring antipsychotic candidate F17464 to dopamine D3 and D2 receptors: a PET study in healthy subjects with [(11)C]-(+)-PHNO. Psychopharmacology. 2020;237:519–27.
Berry AS, Shah VD, Furman DJ, White RL 3rd, Baker SL, O’Neil JP, et al. Dopamine synthesis capacity is associated with D2/3 receptor binding but not dopamine release. Neuropsychopharmacology. 2018;43:1201–11.
Nour MM, McCutcheon R, Howes OD. The relationship between dopamine synthesis capacity and release: implications for psychosis. Neuropsychopharmacology. 2018;43:1195–96.
Kumakura Y, Cumming P, Vernaleken I, Buchholz HG, Siessmeier T, Heinz A, et al. Elevated [18F]fluorodopamine turnover in brain of patients with schizophrenia: an [18F]fluorodopa/positron emission tomography study. J Neurosci. 2007;27:8080–7.
Slifstein M, van de Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry. 2015;72:316–24.
Schifani C, Tseng HH, Kenk M, Tagore A, Kiang M, Wilson AA, et al. Cortical stress regulation is disrupted in schizophrenia but not in clinical high risk for psychosis. Brain. 2018;141:2213–24.
Tseng HH, Watts JJ, Kiang M, Suridjan I, Wilson AA, Houle S, et al. Nigral stress-induced dopamine release in clinical high risk and antipsychotic-naive schizophrenia. Schizophr Bull. 2018;44:542–51.
Cassidy CM, Zucca FA, Girgis RR, Baker SC, Weinstein JJ, Sharp ME, et al. Neuromelanin-sensitive MRI as a noninvasive proxy measure of dopamine function in the human brain. Proc Natl Acad Sci USA. 2019;116:5108–17.
Brucato G, Masucci MD, Arndt LY, Ben-David S, Colibazzi T, Corcoran CM, et al. Baseline demographics, clinical features and predictors of conversion among 200 individuals in a longitudinal prospective psychosis-risk cohort. Psychol Med. 2017;47:1923–35.
Small SA. Isolating pathogenic mechanisms embedded within the hippocampal circuit through regional vulnerability. Neuron. 2014;84:32–9.
Lieberman JA, Small SA, Girgis RR. Early detection and preventive intervention in schizophrenia: from fantasy to reality. Am J Psychiat. 2019;176:794–810.
Acknowledgements
We would like to acknowledge the patients who participated in this study. We thank Xiaoyan Xu, Rawad Ayoub and Jiayan Meng for excellent technical support. The project described was supported by the Doris Duke Charitable Foundation as well as the Brain and Behavior Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
RRG has recently received research support from Otsuka, Allergan, BioAdvantex and Genentech and has received advances/royalties from books published by Wipf and Stock and Routledge/Taylor and Francis. MS has consulted for Curasen Therapeutics. GB discloses that he receives royalties and/or advances from Routledge/Taylor and Francis and Prometheus Books. LSK has received research support from Amgen. JAL has received support administered through his institution in the form of funding or medication supplies for investigator-initiated research from Lilly, Denovo, Biomarin, Novartis, Taisho, Teva, Alkermes, and Boehringer Ingelheim, and is a member of the advisory board of Intracellular Therapies and Pierre Fabre. He neither accepts nor receives any personal financial remuneration for consulting, advisory board or research activities. He holds a patent from Repligen and receives royalty payments from SHRINKS: The Untold Story of Psychiatry. AAD reports serving on an advisory board for Sunovion, receiving an honorarium from Otsuka, receiving stock options from Terran Biosciences and System1 Biosciences, and receiving research support from Neurocrine and LB Pharmaceuticals in the previous year. No other authors report relevant conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Girgis, R.R., Slifstein, M., Brucato, G. et al. Imaging synaptic dopamine availability in individuals at clinical high-risk for psychosis: a [11C]-(+)-PHNO PET with methylphenidate challenge study. Mol Psychiatry 26, 2504–2513 (2021). https://doi.org/10.1038/s41380-020-00934-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-020-00934-w
- Springer Nature Limited
This article is cited by
-
How changes in dopamine D2 receptor levels alter striatal circuit function and motivation
Molecular Psychiatry (2022)