
Abstract
The Fragile X Mental Retardation Protein (FMRP) is an RNA-binding protein essential to the regulation of local translation at synapses. In the mammalian brain, synapses are constantly formed and eliminated throughout development to achieve functional neuronal networks. At the molecular level, thousands of proteins cooperate to accomplish efficient neuronal communication. Therefore, synaptic protein levels and their functional interactions need to be tightly regulated. FMRP generally acts as a translational repressor of its mRNA targets. FMRP is the target of several post-translational modifications (PTMs) that dynamically regulate its function. Here we provide an overview of the PTMs controlling the FMRP function and discuss how their spatiotemporal interplay contributes to the physiological regulation of FMRP. Importantly, FMRP loss-of-function leads to Fragile X syndrome (FXS), a rare genetic developmental condition causing a range of neurological alterations including intellectual disability (ID), learning and memory impairments, autistic-like features and seizures. Here, we also explore the possibility that recently reported missense mutations in the FMR1 gene disrupt the PTM homoeostasis of FMRP, thus participating in the aetiology of FXS. This suggests that the pharmacological targeting of PTMs may be a promising strategy to develop innovative therapies for patients carrying such missense mutations.
Similar content being viewed by others


Introduction
The Fragile X Mental Retardation Protein (FMRP) is an RNA-binding protein highly expressed in the central nervous system (CNS) and evolutionarily conserved from drosophila to human [1]. Structurally, FMRP presents three canonical RNA-binding domains including two centrally-located hnRNP K homology (KH) domains (KH1, KH2) and one C-terminal arginine–glycine–glycine (RGG) box [2, 3] (Fig. 1). These domains respectively allow FMRP to specifically bind its target mRNAs through kissing-complex motifs [4] and G-quartet loops [5]. More recently, a non-canonical KH domain (KH0) at the N-terminus of FMRP has been reported. This domain is possibly involved in both protein–protein and protein–RNA interactions [6, 7]. FMRP also contains two N-terminal tandem Agenet modules (Agenet1, Agenet2) involved in chromatin and histone binding [6, 8] (Fig. 1).

Schematic representation of the functional domains of FMRP including two N-terminal Agenet domains involved in chromatin and histone binding, three N-terminal (KH0, KH1 and KH2) and one C-terminal (RGG box) RNA-binding domains, a nuclear localization signal (NLS, residues 111–152) and a nuclear export signal (NES, residues 425–441). FMRP protein–protein interactions mainly occur at the N-terminus. The amino acids that are post-translationally modified are differentially highlighted (blue: sumoylation; green: phosphorylation; purple: methylation). While FMRP is also ubiquitinated, the specific residues modified by this PTM are unknown. The FMR1 missense mutations reported in FXS patients are highlighted in red, including the R138Q mutation, reported in three unrelated individuals.
FMRP is mainly localized in the cytoplasm and is a component of large messenger ribonucleoprotein (mRNP) particles associated with mRNAs and polyribosomes [9]. As an RNA-binding protein, FMRP participates in the transport, stability, localization and the regulation of local translation of several mRNAs critical to neuronal development and function [10, 11]. In the mammalian brain, FMRP has been estimated to bind ~4% of the mRNAs including almost one third of these mRNAs coding for both pre- and postsynaptic proteins [12]. More recently, it has been reported that the repertoire of FMRP mRNA targets differs between brain regions [13]. In neurons, FMRP is a key component of neuronal RNA granules in which mRNAs are transported along axons and dendrites to the base of active synapses and released in an activity-dependent manner to allow for local translation [1, 14]. The targeting of mRNAs and subsequent regulation of their local translation are crucial processes to synapse elimination and maturation and thus, fundamental to establish a functional neuronal network in the developing brain. FMRP represses the translation of its target mRNAs through different mechanisms. For instance, the cytoplasmic FMRP-interacting protein 1 (CYFIP1) is recruited by FMRP to specific mRNAs where it interacts with eIF4E, thus interfering with the formation of the eIF4F complex required for translation initiation [15]. Given the association of FMRP with polyribosomes, a second model suggests that FMRP causes ribosome stalling during translation elongation [12, 16]. Other reports indicate that FMRP inhibits translation by interacting the long non-coding RNA BC1 [17] or recruiting the miRNA-RISC complex onto its mRNA targets [18,19,20,21,22,23]. More recently, it has been reported that FMRP also acts as a positive modulator of translation when it interacts with the RNA motif SoSLIP (Sod1 mRNA stem loops interacting with FMRP) [24, 25].
Although mainly cytoplasmic, FMRP contains a nuclear localization signal (NLS) and nuclear export signal (NES), allowing FMRP to shuttle between the nucleus and the cytoplasm [26,27,28] (Fig. 1). Besides its ability to bind mRNAs, FMRP forms homodimers [29] and interacts with several proteins, including other cytoplasmic RNA-binding proteins involved in mRNA metabolism, nuclear proteins, molecular motors, synaptic proteins and cytoskeletal remodelling proteins [30]. Many of these interactions, including those with the FMRP paralogs FXR1P and FXR2P [31, 32], NUFIP [33], 82-FIP [34], CYFIP1 and CYFIP2 [35], have been mapped onto the N-terminal domain of FMRP [36]. However, some of these protein–protein interactions, such as the binding of the kinesin-like protein KiF3C [37] and the pre-synaptic calcium channels Cav2.1 and Cav2.2 [38, 39], occur distally within the C-terminal domain of FMRP [37,38,39,40,41].
FMRP was first identified in the context of the Fragile X syndrome (FXS) [42]. FXS is the most frequent form of inherited intellectual disability (ID) and a leading monogenic cause of autism without effective therapies available [14]. This rare genetic disorder (1:4000 males; 1:7000 females) results from mutations in the FMR1 gene leading to the loss-of-function of the FMRP protein. Besides mild-to-severe ID degrees, FXS patients also display a wide spectrum of neurological alterations ranging from learning deficits to hyperactivity, anxiety, autistic-like behaviours and seizures [10, 11]. In neurons, FMRP participates in synapse maturation and elimination. Accordingly, the loss of the FMRP function in FXS patients leads to a pathological hyper-abundance of long thin protrusions called filopodia [43,44,45]. Such defects result from abnormal postsynaptic maturation and/or failure in the synapse elimination process [46]. A similar excess of thin immature dendritic spines is found in Fmr1 knockout (Fmr1−/y) mouse models for FXS [47] and correlates with alterations in synaptic transmission and plasticity as well as social and cognitive behaviours [11, 48, 49]. In particular, the absence of the FMRP-dependent translational repression at synapses in Fmr1−/y neurons leads to an exaggerated form of synaptic plasticity called long-term depression (LTD) that depends on the activity of the metabotropic glutamate receptor 5 (mGlu5R) and requires protein synthesis [50, 51].
Given the role of FMRP in the synaptic network formation, a tight and dynamic regulation of the FMRP function is essential. Such a spatiotemporal control is mainly governed by post-translational modifications (PTMs). PTMs generally refer to the enzymatic covalent but reversible modification of proteins concomitant with or following protein synthesis. PTMs participate in the regulation of protein folding, subcellular localization, enzymatic activity, stability/turnover, aggregation and interaction with other molecules (for a recent review, see ref. [52]). Growing evidence also indicates that several PTMs simultaneously occur and cooperate to dynamically regulate complex biological processes [52]. Interestingly, the dysregulation of PTM homoeostasis contributes to the development of several pathological conditions, including cancer and neurological disorders [53,54,55,56]. In mammalian cells, phosphorylation and ubiquitination are the most extensively characterized PTMs. However, in the last decades, other PTMs such as sumoylation, acetylation and palmitoylation emerged as key regulators of protein function and became the object of intense investigation. Several studies show that FMRP is a target of phosphorylation [16, 57], ubiquitination [58] and methylation [59], clearly demonstrating that PTMs play an essential role in tuning the function of FMRP in neurons. More recently, our team demonstrated that FMRP is also modified by the sumoylation system in the mammalian brain, adding an additional level of complexity in the spatiotemporal regulation of the FMRP function [60].
Here, we provide an overview of the current knowledge on the PTMs regulating the FMRP function and discuss how the interplay between these PTMs contributes to the physiological regulation of FMRP. Then, we highlight several examples of missense mutations interfering with the PTM profile of specific proteins and leading to severe neurological conditions. Finally, we explore the possibility that FMR1 missense mutations could similarly interfere with the PTM homoeostasis of FMRP, thus participating in the aetiology of FXS.
The PTMs regulating the FMRP function
In neurons, proteins usually undergo PTMs in an activity-dependent manner. Among all signalling pathways modulating the FMRP function, the metabotropic glutamate receptors (mGluRs) play a critical role [51]. In hippocampal neurons, the activation of group 1 mGluRs (mGlu1R and mGlu5R) using the agonist dihydroxyphenylglycine (DHPG) triggers an mGluR-dependent long-term depression (mGluR-LTD) [61]. LTD is a form of synaptic plasticity generally described as a weakening of synapses resulting from the removal of AMPAR from the synapse [62, 63]. The mGluR-dependent LTD is a particular form of plasticity that requires new protein synthesis [61, 64]. The activation of type 1 mGluRs promotes the local translation of a subset of mRNAs that are essential to the expression of mGluR-LTD [64], including the one coding for FMRP itself [65, 66]. Given the role of FMRP as a translational repressor, it inhibits the synthesis of proteins required for mGluR-LTD and therefore acts as a brake for the expression of LTD. Accordingly, the lack of repression due to the absence of FMRP expression in Fmr1−/y neurons leads to an exaggerated local translation of pre-existent synaptic mRNAs and consequently, to an enhanced mGluR-LTD [50, 51].
Ubiquitination
Different studies reported that the initial phase of mGluR-LTD triggers the local synthesis of FMRP at synapses [65, 67]. Later, Hou et al. showed that the induction of the mGluR-LTD is associated with the rapid and transient increase in FMRP protein levels in the soma, nucleus and proximal dendrites [58]. The inhibition of protein synthesis completely abolishes the mGluR-induced increase in FMRP protein levels, indicating that this event requires new protein synthesis. The authors also applied DHPG to hippocampal slices in the presence of selective antagonists of either mGlu1R or mGlu5R and showed that the rapid increase in FMRP protein levels depends on the activation of mGlu5Rs [58]. In addition, the increased FMRP levels dropped back to basal levels after 10 min of DHPG washout, indicating that FMRP is also rapidly degraded upon the activation of mGluRs [58, 68]. Importantly, the decrease in FMRP protein levels after 10 min of DHPG stimulation is followed by new FMRP synthesis, resulting in the increased FMRP levels after 30 min of treatment [68]. The authors also demonstrated that the degradation of FMRP requires its poly-ubiquitination since it is abolished in the presence of the proteasome inhibitors MG132 and lactacystin. These results pointed out the role of the ubiquitin-proteasome system (UPS) in the dynamic regulation of FMRP levels during the mGluR-LTD [58]. Accordingly, MG132 pre-treatment also prevented the increase in PSD95 and CaMKII protein levels given that their mRNAs are targets of FMRP and their translation is up-regulated upon the activation of mGluRs. The fact that FMRP acts as a translational repressor on these two proteins also suggests that the rapid decrease in FMRP protein levels is sufficient to enable protein synthesis during mGluR activation [68]. Furthermore, blocking the proteasome activity impairs the late phase of mGluR-LTD, indicating that the rapid UPS-dependent degradation of FMRP is necessary to allow the translation of mRNA targets important for the expression of the mGluR-LTD [58]. Altogether, these data highlight the existence of a bidirectional mGluR-dependent regulation of the FMRP function by concomitantly promoting the synthesis and proteolysis of FMRP [58, 68].
Huang et al. identified the major ubiquitin ligase complex CdH1-APC as a critical enzyme for the ubiquitination of FMRP during mGluR-LTD. Knockout mice for a key component of the CdH1-APC complex display a strong impairment in both FMRP ubiquitination and degradation upon DHPG stimulation [69]. Accordingly, the loss of the mGluR-dependent FMRP ubiquitination in the CdH1 knockout mice impairs the mGluR-LTD in the hippocampus, thus confirming the role of the UPS system in this form of plasticity [69].
Phosphorylation
In 2002, Siomi et al. reported for the first time that dFMRP, the drosophila ortholog of FMRP, is phosphorylated in vivo within its C-terminal domain and identified the serine residue 406 (S406) as its major phosphorylation site [57]. They also demonstrated that dFMRP phosphorylation modulates its homomerization and ability to bind RNA molecules. However, Mazroui et al. later showed that FMRP phosphorylation is not required for its recruitment to polyribosomes [70]. Almost simultaneously, the serine 499 residue (S499, homologous to the S406 residue of dFMRP) was identified as the primary phosphorylation site of FMRP in the mouse brain [16] (Fig. 1). Unlike dFMRP, FMRP phosphorylation on S499 did not modify the total amount of mRNAs associated with FMRP. Nevertheless, ribosome run-off experiments in the presence of a non-specific inhibitor of translation revealed that the non-phosphorylated form of FMRP is almost absent from heavy polysomes (associated with efficient mRNA translation), whereas phosphorylated FMRP molecules are resistant to run-off and associate with stalled polyribosomes. These data therefore suggest that the phosphorylation of FMRP is required for its translational repressor role, while FMRP dephosphorylation rather triggers the translation of its associated mRNAs [16] (Fig. 2a).

a FMRP is constitutively phosphorylated on the S499 residue by CK2. FMRP-S499 phosphorylation may be required for secondary phosphorylation of nearby residues, potentially by S6K1 in an mGluR-dependent manner. Phosphorylated FMRP associates with stalled ribosomes, resulting in translational repression. Short mGluR activation (<1 min) triggers the PP2A-dependent dephosphorylation of FMRP, leading to granule disassembly and the translation of target mRNAs. Conversely, sustained mGluR activation triggers FMRP rephosphorylation by promoting the S6K1 activity and inhibiting PP2A in an mTOR-dependent manner, thus leading to translational repression. b Short mGluR activation (DHPG < 1 min) triggers FMRP dephosphorylation, leading to the translation of the mRNA targets of FMRP, including the Fmr1 mRNA. This results in a rapid and transient increase in FMRP protein levels. In parallel, dephosphorylation of FMRP leads to its ubiquitination and subsequent degradation via the proteasome, thus bringing the increased FMRP protein levels back to initial levels. Conversely, sustained mGluR activation (DHPG > 5 min) promotes FMRP rephosphorylation, resulting in translational repression to prevent an excessive protein synthesis.
This idea was further confirmed in a later study proposing that the rapid translation of FMRP targets is controlled by the mGluR-dependent phosphorylation state of FMRP [71]. In particular, the authors demonstrated that the DHPG-dependent activation of mGluRs triggers the rapid (<1 min) and transient PP2A-dependent dephosphorylation of FMRP along dendrites. According to the model in which the mGluR signalling bidirectionally regulates the phosphorylation and dephosphorylation of FMRP and consequently modulates the translation of the mRNAs bound to FMRP (including sapap3), the authors reported increased levels of the postsynaptic scaffolding protein SAPAP3 upon DHPG stimulation [71]. Similarly, while the phosphorylated form of FMRP is able to recruit the miR-125a/AGO2 inhibitory complex onto the 3′UTR of the psd95 mRNA to repress its translation, the mGluR-dependent dephosphorylation of FMRP leads to the release of AGO2 and allows the synthesis of PSD95 [23]. Accordingly, the dephosphorylation of FMRP promotes the translation of Arc in dendrites and the expression of the mGluR-LTD [72]. Narayanan et al. also showed that the sustained activation of mGluRs (1–5 min) promotes FMRP rephosphorylation in an mTOR-dependent manner through the suppression of the PP2A activity [71] (Fig. 2a). More recently, a similar mechanism has been described for dFMRP. In drosophila, light stimulation activates the calcium-dependent Mts protein, corresponding to the catalytic subunit of PP2A in mammals, which dephosphorylates dFMRP. The dephosphorylation of dFMRP results in the dissociation of dFMRP-containing ribosome complexes and leads to the translation of the dFMRP target ninaE mRNA that encodes the major fly rhodopsin Rh1, a G-protein coupled receptors that acts as light sensors in photoreceptors [73].
Interestingly, the ribosomal protein S6 kinase (S6K1) was identified as the major kinase for FMRP in hippocampal neurons [74]. This was validated using S6K1 knockout (S6K1−/−) brains, in which no phosphorylated FMRP was detected. S6K1−/− mice also displayed increased levels of SAPAP3 protein as observed in Fmr1−/y mice, further supporting that FMRP phosphorylation negatively regulates the translation of its target mRNAs [74]. The authors also showed higher PP2A-FMRP or S6K1-FMRP interaction upon 1 or 5 min of DHPG stimulation respectively, consistent with an earlier report of the same group [71, 74]. In addition, both mGluR-triggered mammalian target of rapamycin (mTOR) and ERK1/2 signalling pathways are required for the S6K1-dependent phosphorylation of FMRP. Noteworthy, PP2A inhibits S6K1 in mitogenic signalling [75, 76] and mTOR regulates both PP2A and S6K1 [71, 74]. In agreement with these reports, the blockade of PP2A activity with okaidic acid promotes an increased S6K1-dependent phosphorylation of FMRP at steady state that was potentiated upon DHPG stimulation. Altogether, these data revealed that a short mGluR activation (1 min) simultaneously triggers both FMRP dephosphorylation and S6K1 inhibition in a PP2A-dependent manner, while a sustained mGluR activation (>5 min) promotes the rephosphorylation of FMRP by releasing the inhibition of S6K1. Thus, the mGluR-driven dephosphorylation-phosphorylation PP2A-S6K1 signalling module appears to tightly regulate the translational repressor role of FMRP in a spatiotemporal manner [71, 74] (Fig. 2a).
Despite the work from Narayanan et al. [74], the S6K1-dependent phosphorylation of FMRP on the S499 residue remains controversial. As mentioned above, the S6K1 activity is regulated by the mTOR signalling [74]. Interestingly, the tuberous sclerosis complex (TSC) is a rare neurological disorder characterized by a hyperactive mTOR signalling [77]. Based on this evidence, Bartley et al. hypothesized that an S6K1-dependent FMRP-S499 hyperphosphorylation occurs in TSC mouse models [78]. However, the FMRP-S499 phosphorylation appeared unchanged in Tsc1+/− and conditional Tsc1−/− mice despite a significant increase in S6K1 activity. Furthermore, the pharmacological inhibition of mTOR or S6K1 activity did not affect the phosphorylation of the FMRP-S499 residue in vivo and the activation of mGluR in mouse neuroblastoma N2a cells did not change the ratio between the phosphorylated and total levels of FMRP. In addition, the phosphorylation of the S499 residue was unaltered in S6K1−/− mice. Altogether, these data strongly suggest that the FMRP-S499 phosphorylation is mTOR/S6K1-independent [78]. These findings are in contrast with previous reports supporting the role of mTOR/S6K1 signalling in controlling FMRP phosphorylation at the S499 residue [71, 74]. However, the authors suggested that this discrepancy probably depends on the experimental approaches used. Bartley et al. proposed that another kinase is responsible for the FMRP-S499 phosphorylation given that the sequence surrounding the S499 residue is not part of a phosphorylation consensus motif for S6 kinases [78]. Indeed, they later demonstrated that the casein kinase II (CK2) phosphorylates the mammalian FMRP-S499 residue [79] (Fig. 2a). CK2 was previously identified as the kinase phosphorylating FMRP in drosophila on a residue homologous to S499 (S406) [57]. Even if previously identified in drosophila, CK2 was not expected to phosphorylate the FMRP-S499 residue in mammals since it is a constitutively active kinase [80]. However, CK2 was recently shown to promote secondary hierarchical phosphorylation by other kinases, potentially in an activity-dependent manner [81]. Bartley et al. then used mass spectrometry to confirm that the FMRP region comprising the S499 residue is indeed phosphorylated but failed to precisely assign the secondary phosphorylated residues [79]. Finally, the authors found that both PP2A inhibition and mGluR stimulation increased the phosphorylation of the phosphomimetic FMRP-S499D mutant but not the one of the phosphodeficient FMRP-S499A, suggesting that a negative charge (phosphoS499) is required at this position to trigger the secondary phosphorylation of nearby residues regulated by PP2A and mGluRs [16, 79]. Whether S6K1 is the kinase responsible for such secondary activity-dependent phosphorylation remains to be elucidated (Fig. 2a).
While it is well-accepted that the mGluR-dependent phosphorylation of FMRP is required for its translational repressor role, some studies investigating FMRP phosphorylation have not addressed the contribution of mGluRs or other signalling pathways. However, it is likely that FMRP phosphorylation is regulated by neuronal activity in such cases. For instance, given that FMRP contributes to the assembly of Dicer-processed miRNAs onto target mRNAs through its KH2 domain [20], it has been hypothesized that miRNAs participate in the translational repressor role of FMRP. Because FMRP associates with stalled polyribosomes in its phosphorylated form [16], Cheever et al. performed immunoprecipitation experiments of total and phosphorylated FMRP and compared their RNA composition [82]. They reported a larger amount of 80-nucleotide-long RNAs associated with the phosphorylated form of FMRP corresponding to the size of precursor miRNAs. Interestingly, Dicer does not bind the phosphorylated form of FMRP, thus explaining why precursor miRNAs cannot be processed into mature ones. Indeed, the absence of phosphorylation within the residues 496–503 in FMRP is essential to allow its interaction with Dicer [82]. Altogether, given previous reports on the role of miRNAs as translational activators, the authors proposed that FMRP phosphorylation inhibits its association with Dicer, thus indirectly suppressing translation by reducing the production of mature miRNAs [82].
In the recent years, the in vivo function of the phosphorylation of human FMRP (hFMRP) has also been studied. hFMRP is transported along axons and localizes to axonal termini where its phosphorylation participates in the regulation of axonal complexity [83]. Both phosphomimetic S500D and phosphodeficient S500A mutants of hFMRP decreased the density of axonal GFP-FMRP puncta as well as the ability of the FMRP spliced variant isoform 7 to reduce axonal complexity [83]. Furthermore, Coffee et al. generated hFMRP-WT, phosphomimetic hFMRP-S500D and phosphodeficient hFMRP-S500A transgenes and specifically targeted their expression into neurons from dFmr1-null flies [84]. Both the wild type (WT) and S500D forms of hFMRP were able to fully rescue the neuronal FXS anomalies present in dFmr1-null flies, including increased protein levels, synaptic architecture defects at the neuromuscular junction and learning impairments. Conversely, the phosphodeficient hFMRP-S500A mutant failed to rescue any of these defects. Such results support a model in which FMRP phosphorylation is essential to regulate translation, axonal and synaptic architecture and consequently, the behaviour in flies [84].
Crosstalk between FMRP phosphorylation and ubiquitination
Several studies have provided evidence of a crosstalk between FMRP phosphorylation and ubiquitination. In line with previous findings [58, 68], Nalavadi et al. not only confirmed that DHPG stimulation induces the rapid ubiquitination and proteosomal-dependent degradation of FMRP, but also demonstrated that the PP2A-dependent dephosphorylation of FMRP at the serine 499 residue (S500 in human) contributes to this process [85]. Indeed, blocking PP2A activity abolished the degradation of FMRP following mGluR activation. Conversely, the expression of the phosphodeficient S499A mutant of FMRP in neuroblastoma cells led to increased levels of poly-ubiquitinated FMRP upon DHPG stimulation. These data indicate that FMRP dephosphorylation is required to allow its ubiquitination and subsequent degradation [85]. Furthermore, pre-incubation of neuroblastoma cells with the proteasome inhibitor MG132 blocks the mGluR-dependent degradation of FMRP without affecting its phosphorylation state, indicating that the UPS activity does not interfere with FMRP dephosphorylation [85]. Importantly, the authors showed that the translation of the FMRP target mRNA psd95 upon DHPG stimulation requires both the PP2A and UPS activity. Altogether, these findings confirm that the phosphorylation of FMRP is a prerequisite for its degradation, which eventually results in the translation of mRNAs that are usually repressed by FMRP [85]. To summarize, a short mGluR activation triggers FMRP dephosphorylation, leading to the translation of the mRNA targets of FMRP, including the Fmr1 mRNA. This results in a rapid and transient increase in FMRP protein levels. In parallel, dephosphorylation of FMRP leads to its ubiquitination and subsequent degradation, thus bringing increased FMRP protein levels back to initial levels. Conversely, sustained mGluR activation promotes FMRP rephosphorylation, resulting in translational repression to prevent excessive protein synthesis (Fig. 2b).
It has been recently reported that a mutated form of the polyglutamine binding protein 1 (PQBP1), the protein altered in the X-linked Renpenning syndrome, preferentially binds the non-phosphorylated form of FMRP. PQBP1 is a component of neuronal RNA granules, where it interacts with FMRP and participates in RNA granule transport and local translation in neurons. Such aberrant interaction between the mutant PQBP1 and FMRP likely impairs the rephosphorylation of FMRP, promoting its UPS-mediated degradation [86]. Consistently, the authors showed a dramatic increase in FMRP ubiquitination concomitant with a decrease in the total amount of FMRP in presence of the PQBP1 mutant. These results provided additional evidence of a functional interplay between phosphorylation and ubiquitination in controlling the stability and degradation of FMRP [86]. This work also suggests that alterations in the PTM profile of FMRP may interfere with its stability and degradation, thus impairing the FMRP function and ultimately leading to neuronal disorders.
Finally, recent data further supported the importance of the crosstalk between FMRP phosphorylation and ubiquitination for the mGluR-LTD. Choi et al. showed that CdH1-APC is not the only enzyme able to ubiquitinate FMRP. Indeed, they demonstrated that the chaperone-dependent E3 ubiquitin ligase Hsc70-interacting protein also participates in the expression of the mGluR-LTD in the rat hippocampus by ubiquitinating FMRP and controlling its degradation in a phosphorylation-dependent manner [87].
Sumoylation
Until 2018, the mGluR signalling pathway was only considered as a key player in FMRP ubiquitination and phosphorylation. Interestingly, our team recently demonstrated that FMRP is a target of sumoylation in the mammalian brain [60]. Sumoylation is a highly dynamic post-translational modification spatiotemporally regulated [88,89,90]. It consists in the covalent but reversible conjugation of the Small Ubiquitin-like MOdifier (SUMO) protein to specific lysine residues of target proteins. Initially, sumoylation was exclusively described as a nuclear protein modification. However, it is now clear that sumoylation also occurs outside the nucleus and acts as a key regulator of the neuronal function with essential roles in postsynaptic differentiation, neuronal excitability and synaptic transmission (for recent reviews, see refs. [91,92,93]). The balance between sumoylation and desumoylation is critical to the brain function and its disruption has been associated with several neurological disorders [92, 93]. Interestingly, this balance is governed by the activity of mGlu5Rs. A short mGlu5R activation promotes a transient trapping of the sole SUMO-conjugating enzyme UBC9 in dendritic spines in a protein kinase C (PKC)-dependent manner, leading to a rapid increase in the overall synaptic sumoylation [94]. Subsequently, the sustained activation of mGlu5Rs promotes the postsynaptic accumulation of the desumoylation enzyme SENP1, which results in the decrease of synaptic sumoylation back to initial levels [90].
We recently demonstrated that the activation of mGlu5Rs (<5 min) triggers the sumoylation of FMRP at the lysine 88 and 130 (K88 and K130) residues [60]. This event promotes the dissociation of FMRP from mRNA granules, allowing the release and local translation of its mRNA targets which in turn, regulate dendritic spine elimination and maturation (Fig. 3). According to the role of FMRP sumoylation in controlling spinogenesis, the expression of the SUMO-deficient form of FMRP (FMRP-K88,130R) in WT neurons impairs spine elimination and maturation [60], leading to a hyper-abundance of long dendritic protrusions similar to the one observed in Fmr1−/y neurons [47, 49] and FXS patients [43,44,45]. Given that mGlu5R activation promotes synaptic sumoylation in a PKC-dependent manner [94], FMRP may also be phosphorylated by PKC prior to its sumoylation. However, there is no evidence for such a phospho-dependent regulation of FMRP sumoylation in neurons to date. In addition, a crosstalk between FMRP sumoylation and ubiquitination may also exist given that both PTMs occur on lysine residues. FMRP sumoylation and the subsequent release of FMRP from mRNA granules may be a prerequisite for its ubiquitination on different acceptor lysine residues leading to protein degradation. Another possibility is that SUMO and ubiquitin directly compete to modify the same target lysine residues on FMRP, thus having antagonistic effects on the regulation of the FMRP function.

The mGlu5R-dependent sumoylation of FMRP promotes its dissociation from dendritic mRNA granules. This results in the release and local translation of its mRNA targets and thus, the regulation of spine elimination and maturation. Accordingly, the expression of a non-sumoylatable form FMRP in Fmr1−/y neurons leads to an aberrant granular shape due to an impaired dissociation of the FMRP mutant from dendritic mRNA granules, resulting in abnormal spine density and morphology (adapted from [60]).
Arginine methylation
Arginine methylation is a eukaryotic PTM that consists in the addition of methyl groups to arginine residues of target proteins. This modification participates in the modulation of RNA transcription and binding, protein–protein interactions and protein localization [95]. The addition of a methyl group alters the arginine side chain shape, increasing hydrophobicity and steric hindrance and/or removing hydrogen bond donors. Interestingly, arginine methylation usually occurs in arginine-glycine-rich domains of RNA-binding proteins and is mediated by protein arginine methyltransferases (PRMTs). While all PRMTs are capable of performing arginine monomethylation, type I PRMTs (PRMT1–4, 6 and 8) perform asymmetric dimethylation and type II (PRMT5, 7 and 9) catalyse symmetric dimethylation [95, 96]. Noteworthy, several PRMTs are crucial for neuronal differentiation and CNS myelination processes [97,98,99,100,101].
FMRP was first suggested to be a methylation substrate in 1995 [102]. However, it was only in 1999 when Ai et al. reported that a synthetic peptide comprised in the hFMRP RGG box was monomethylated by rat brain extract [103]. These data suggested that FMRP is substrate of an unknown PRMT present in brain homogenates [103]. Following studies demonstrated that FMRP is post-translationally mono- and dimethylated asymmetrically in its RGG box in mammalian cells [59, 104]. Furthermore, site-directed mutagenesis experiments identified the arginine residues R533, R538, R543 and R545 as the main sites of FMRP methylation [59, 105] (Fig. 1).
In the brain, FMRP binds and regulates the translation of several mRNAs critical to the brain function [10, 11]. Given that the RGG box is the primary RNA-binding domain of FMRP, several groups have addressed the role of arginine methylation on the ability of FMRP to bind its target mRNAs [59, 105, 106]. Interestingly, blocking arginine methylation with the indirect PRMT inhibitor adenosine-2′,3′-dialdehyde (AdOx) affects the binding of FMRP to homoribopolymer mimetic RNAs and some specific target mRNAs in vitro [106]. Further studies revealed that FMRP differentially binds its target mRNAs depending on the methylation state of specific arginine residue pairs [105]. In addition, methylation on R533 and R538 reduces FMRP association to polyribosomes, indicating that the presence of arginine residues at these positions is essential to regulate its polyribosomal association [105].
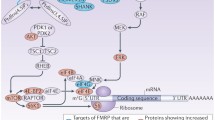
Arginine methylation is also involved in the modulation of FMRP protein–protein interactions. In particular, the inhibition of arginine methylation decreased the ability of FMRP to form heterodimers with its paralogue FXR1P, without affecting its ability to homomerize [107]. Indeed, the inhibition of arginine methylation leads to an accumulation of small FMRP-containing cytoplasmic granules in which FMRP is likely in the form of homomers. The authors speculated that FMRP associates with the translational machinery depending on its dimerization state, thus indicating that methylation would be controlling the ability of FMRP to specifically release the mRNA targets to be translated [107] (Fig. 4).

FMRP methylation in its RGG box regulates its homomerization within mRNA granules. FMRP methylation leads to the dissociation of FMRP-containing translational repressing complexes, resulting in the translation of specific mRNA targets. The methylation state of specific arginine residue pairs allows differential binding of FMRP to its mRNA targets.
Little is known about the signalling cues that trigger FMRP methylation. Denman et al. reported that FMRP methylation increases upon NGF stimulation in PC12 cells correlating with neurite outgrowth [108]. Interestingly, it was reported that GABAergic stimulation modulates protein arginine methylation [109]. The authors further showed that GABAergic stimulation decreases PRMT substrate methylation in Fmr1−/y hippocampal slices [109]. Indeed, both GABA signalling and protein arginine methylation are altered in Fmr1−/y mice [109, 110]. Thus, although speculative, the GABAergic signalling may be a regulator of FMRP methylation.
Noteworthy, while several studies have reported that PRMT1 methylates FMRP in vitro and in cell lines [59, 105, 111], PRMT3 and PRMT4 have also been proposed as potential candidates to accomplish FMRP methylation [104, 111]. It remains unknown whether PRMT1 is the only enzyme capable of methylating FMRP or whether a combination of PRMTs could be responsible for the methylation of the four specific arginine residues of FMRP in the brain. In addition, it still remains to be determined whether methylation occurs to inhibit RNA binding or rather to control the identity of the mRNAs bound to FMRP. There is no clear evidence whether FMRP can just switch from an unmethylated to a fully-methylated state or whether several forms of FMRP exist with one or more arginines methylated in vivo. Therefore, it will be of great interest to investigate whether different methylated forms of FMRP may confer distinct RNA-binding abilities.
Crosstalk between FMRP phosphorylation and arginine methylation
Interestingly, the methylatable arginine residues localize in close proximity to the primary phosphorylation site of FMRP (S499 in mouse, S500 in human). This evidence led to hypothesize that FMRP phosphorylation could modulate its methylation. However, the existence of a crosstalk between these PTMs to regulate the FMRP function was ruled out by two independent studies [59, 104]. More recently, Tsang et al. hypothesized an antagonistic effect of FMRP phosphorylation and methylation on RNA granule formation [112]. They showed that FMRP undergoes phase separation in vitro through its C-terminal low-complexity region (LCR). Phase separation is the creation of two distinct phases (a dense and a diluted one) from a homogenous mixture. In cells, phase separation leads to the formation of membraneless organelles, allowing the concentration of macromolecules (often RNA-binding proteins and RNA) and thus, underlying an intracellular organization to modulate complex biochemical reactions. More interestingly, the authors demonstrated that the phosphorylation of FMRP promotes phase separation indicating that FMRP phosphorylation plays a role in the formation of neuronal granules [112]. This is in agreement with previous reports showing that phosphorylated FMRP leads to translational repression [16, 71]. In particular, their data suggest that hierarchical phosphorylation regulates phase separation, thus linking granule formation to activity-dependent translation [112]. Conversely, FMRP methylation seems to decrease the propensity of phase separation in vitro, suggesting that FMRP methylation participates in granule disassembly [112]. These findings are consistent with the fact that FMRP forms homomers in granules and methylation prevents its homomerization [107]. However, it remains to be elucidated how FMRP C-terminal methylation impacts FMRP homomerization, which occurs through its N-terminal region.
Impact of FMRP PTMs on the pathophysiology of FXS
In the past decade, PTMs have been extensively linked to the development of neurological disorders, including Alzheimer’s [54] and Parkinson’s [113] diseases. However, alterations in the PTM homeostasis of FMRP have never been considered as a potential molecular mechanism underlying FXS given that FMRP is absent in patients presenting the typical CGG repeat expansion at the 5′ UTR of the FMR1 gene, which accounts for the majority of the cases [114]. Other mutagenic mechanisms leading to FXS have been reported, including deletions, promoter variants, splicing errors, missense and nonsense mutations [115,116,117,118]. To date, more than 120 sequence variants of unknown significance have been identified in the FMR1 gene [115]. Among them, only three missense mutations (I304N, G266E and R138Q) have been supported by functional studies showing association with the aetiology of FXS [115, 119,120,121,122]. Given that gene sequencing is not the standard care for individuals with developmental deficits, FMR1 missense mutations may be largely underdiagnosed and their frequency has not been well established. One large cohort study identified two novel FMR1 missense mutations in three unrelated individuals among 508 developmentally delayed males without the CGG repeat expansion. If proven pathogenic, the authors estimated that the frequency of newly discovered FMR1 missense mutations in their population would be a non-negligible 0.56% [123]. Noteworthy, the frequency of CGG repeat expansions is 2–3% in similar developmentally delayed populations [124,125,126,127]. The recurrence of some of these mutations, like the R138Q, further supports their pathogenicity [115, 117, 128]. Therefore, these missense mutations should be now considered as important contributors to the aetiology of FXS.
In this part of the review, we have explored the possibility that some FMR1 missense mutations may directly or indirectly alter the PTM profile of FMRP and consequently, its functional regulation, thus participating in the aetiology of FXS. This exciting hypothesis may lead to the development of novel therapeutic approaches for FXS patients carrying such mutations by targeting the altered PTM profile of FMRP. Importantly, while multiple efforts have been devoted to develop valid treatments for FXS patients, an effective therapy is still far from being discovered. Even if several promising preclinical studies succeeded in rescuing the cellular, synaptic and behavioural defects observed in animal models, such therapies failed to be translated into clinics due to the appearance of major side effects or lack of significant improvements (for recent comprehensive reviews, see refs. [11, 129,130,131]). Interestingly, it has already been reported that missense mutations interfere with the PTM profile of specific proteins, leading to severe neurological conditions. This is the case for spinocerebellar ataxia 13 (SCA13), a rare autosomal dominant disease resulting from missense mutations in the KCNC3 gene encoding for the Kv3.3 voltage-gated potassium channel. Over 20 affected individuals were identified carrying the same Kv3.3-R420H point mutation, which leads to the aberrant N-glycosylation of the channel, resulting in abnormal K+ currents due to the altered trafficking and reduced surface expression of the mutated channels [132].
Among the reported FMR1 missense mutations, only those encoding the I304N and G266E amino acid changes (Fig. 1) have been substantially studied [4, 9, 119,120,121, 133,134,135]. The FMRP protein levels are only reduced in a knock-in mouse model carrying the I304N mutation, probably due to an increased turnover of the mutant protein [120]. Both the I304N and G266E missense mutations lead to aberrant polyribosome association and mRNA binding, phenocopying the Fmr1−/y condition [9, 120, 121, 133]. Interestingly, polyribosome association and mRNA binding have been extensively linked to the phosphorylation and methylation status of FMRP. Therefore, it would be of interest to examine the phosphorylation and methylation status of the FMRP-I304N and FMRP-G266E mutants and assess whether these PTMs play a role in the aetiology of FXS in patients carrying these mutations.
More recently, another FMRP point mutation (F126S) was reported in a male patient presenting ID and autistic features [118]. This phenylalanine-to-serine mutation could potentially create a new phosphorylation site for serine-threonine kinases, such as PKC, as predicted by the online NetPhos3.1 and GPS5.0 software. Importantly, FMRP phosphorylation mediates ribosomal stalling, resulting in translational repression [16]. Thus, the presence of an additional phosphorylation site on FMRP may alter its phosphorylation homeostasis, leading to an excessive mRNA translation arrest and resulting in aberrant synapse elimination and/or maturation. In this case, the identification of the kinase responsible for such phosphorylation other than CK2 and S6K1 could represent an interesting therapeutic target without affecting the other phosphorylation sites of FMRP.
A similar strategy has been recently developed for the treatment of SCAs, a group of neurodegenerative diseases caused by polyglutamine expansions [136]. In particular, it is known that the PKA-dependent phosphorylation of the serine residue 766 of ataxin-1 (ATXN1), the protein affected in SCA1, decreases its degradation and increases its aggregation [137, 138]. Hearst et al. demonstrated that the inhibition of the PKA-dependent ATXN1 phosphorylation strongly reduced its aggregation in cultured cells and improved the morphology of Purkinje cells of cultured cerebellar slices. The authors also showed that this PKA inhibitory drug is able to cross the blood-brain barrier and localize to the cerebellum when intranasally delivered [136]. Such promising results encourage the possibility of therapeutically targeting one specific phosphorylated residue of a target protein in neurological disorders, including FXS.
Interestingly, the F126S mutation is also very close to the active K130 SUMO site of FMRP [60]. Since the phosphorylation of a target protein is often a prerequisite for its sumoylation on a nearby residue [92], the close proximity of the F126S mutation with the K130 SUMO site of FMRP raises the exciting possibility that phosphorylation of the mutated S126 residue may promote or hinder the sumoylation of FMRP and lead to aberrant synapse formation, which is characteristic of FXS. Future studies investigating the F126S mutation will need to address this possibility.
In the same line of evidence, the R138Q-FMRP missense mutation, identified in three unrelated individuals presenting developmental delays, ID and seizures [115, 117, 128], is also in close proximity to the active K130 SUMO site of FMRP [60]. This mutation may thus directly alter the mGlu5R-dependent sumoylation of FMRP and, consequently, its function, participating in the aetiology of FXS. The abnormal sumoylation of specific proteins has been previously associated with the pathogenesis of several neurological disorders. For instance, Tang et al. showed that the A548T mutation in synapsin Ia (SynIa), which has been linked to autism and epilepsy, leads to reduced levels of SynIa sumoylation and consequently, impairs neurotransmitter release. It is likely that such defects affect the balance between excitatory and inhibitory inputs and thus, underlie the pathology [139]. In addition, several MECP2 mutations frequently identified in Rett syndrome patients lead to reduced MeCP2 sumoylation. Interestingly, in vivo viral expression of MeCP2-WT or MeCP2 fused to SUMO rescued the social interaction, fear memory and LTP deficits displayed by the Mecp2 conditional knockout mice [140]. Collectively, these findings demonstrate the existence of a link between an altered protein sumoylation and the development of neurological disorders. Such correlation strongly supports the idea that aberrant FMRP sumoylation may underlie FXS in patients carrying the F126S and/or R138Q mutations, shedding light on sumoylation as a potential innovative therapeutic target for FXS.
Alterations in the sumoylation pathway have been implicated in the pathogenesis of cancer, cardiac diseases and neurodegenerative disorders (for recent reviews, see refs. [141,142,143]). Despite active drug discovery targeting the SUMO system, few compounds are actually being investigated in clinical studies [144,145,146] and only the FDA-approved pracinostat and topotecan are already used in the clinics to treat different cancer types [147,148,149]. These compounds are able to increase or decrease global sumoylation by directly or indirectly acting on the sole SUMO-conjugating enzyme UBC9 or the desumoylation enzymes SENPs [145, 146, 150,151,152,153,154,155,156,157,158]. Interestingly, topotecan has been successfully used to reduce sumoylation in neurons and glioblastoma cell lines [158]. Importantly, while the development of molecules targeting the sumoylation pathway is mainly at a preclinical stage, significant advances in drug discovery have been made in the analogous ubiquitination field. In 2003, the FDA approval of bortezomib, the first anticancer drug targeting the UPS [159], represented a milestone in the field since it demonstrated that targeting a complex network like ubiquitination or sumoylation is a feasible therapeutic approach.
Altered levels of FMRP have been identified in some neurological disorders other than FXS. In particular, levels of total and phosphorylated FMRP are reduced in the cerebellar vermis and superior frontal cortex of subjects with autism [160, 161], while mGlu5R expression is increased in the same brain regions in FXS [162] and autistic patients [160, 163]. Altogether, these findings suggest that the increased mGlu5R levels and its downstream signalling may promote FMRP dephosphorylation and consequently, its ubiquitination and degradation in autistic brains [161]. If so, blocking the mGlu5R-dependent dephosphorylation of FMRP should be considered as a potential innovative therapeutic option for autistic patients.
Reduced levels of FMRP have also been reported in carriers of premutated FMR1 alleles and associated with a significant degree of psychiatric disorders [164]. Low FMRP levels with normal FMR1 CGG triplet expansion and FMR1 mRNA levels have been detected in post-mortem brains from subjects with schizophrenia, bipolar disorder and major depressive disorders [165,166,167]. However, it remains unknown whether reduced FMRP levels are a cause or a consequence of the altered neuronal function in these disorders. Noteworthy, several mRNA targets of FMRP have been associated with autism, schizophrenia and mood disorder [30]. As above-mentioned, polyribosome association and mRNA binding depend on the phosphorylation and methylation status of FMRP [16, 59] and the release of mRNAs from dendritic granules relies on the mGlu5R-dependent sumoylation of FMRP [60]. Though uncertain, these PTMs may be dysregulated in pathological conditions and thus, become interesting innovative therapeutic targets.
Importantly, the therapeutic approaches targeting PTMs reviewed here have only been validated in preclinical studies using cells and animal models. However, their clinical safety and efficacy need to be further assessed to overcome the discrepancy between in vitro, in vivo and clinical trials. Special attention must be paid to the repositioning of FDA-approved compounds for other diseases, in particular those able to cross the blood-brain barrier given that their clinical safety has already been demonstrated. Furthermore, modulation of one specific PTM-modified residue remains a major challenge in the field given that a single PTM can affect several residues on the same target protein. The development of methods to ensure target accuracy is thus essential to establish the modulation of PTMs as a valid therapeutic option.
Concluding remarks and future directions
In this review, we summarized the current knowledge on the multiple PTMs that dynamically regulate the function of the RNA-binding protein FMRP, highlighting the complexity of its physiological regulation. We discussed how the interplay between these PTMs contributes to the functional regulation of FMRP in neurons. However, further work is still needed to determine whether FMRP is also the target of additional PTMs, such as acetylation, neddylation or glycosylation. The real challenge is now to get an integrated view of how these PTMs are activity-dependently interconnected to better understand the mechanisms governing the FMRP function in physiological conditions.
Here, we also explored the interesting possibility that the increasing number of FMR1 missense mutations recently identified in FXS patients may directly or indirectly disrupt the PTM homeostasis of FMRP, thus participating in the aetiology of the disease. While further work is still required to validate this possibility, the findings reviewed here suggest that targeting the PTMs of FMRP may be a promising and innovative therapeutic strategy for patients carrying FMR1 missense mutations. Finally, the link between pathological missense mutations and alterations in the post-translational regulation of specific proteins may be a common mechanism in multiple neurological disorders and thus, is worth examining in future research.
References
Davis JK, Broadie K. Multifarious functions of the fragile x mental retardation protein. Trends Genet. 2017;33:703–14.
Ashley CT Jr., Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–6.
Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74:291–8.
Darnell JC, Fraser CE, Mostovetsky O, Stefani G, Jones TA, Eddy SR, et al. Kissing complex RNAs mediate interaction between the fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 2005;19:903–18.
Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–99.
Myrick LK, Hashimoto H, Cheng X, Warren ST. Human FMRP contains an integral tandem Agenet (tudor) and KH motif in the amino terminal domain. Hum Mol Genet. 2015;24:1733–40.
Hu Y, Chen Z, Fu Y, He Q, Jiang L, Zheng J, et al. The amino-terminal structure of human fragile X mental retardation protein obtained using precipitant-immobilized imprinted polymers. Nat Commun. 2015;6:6634.
Alpatov R, Lesch BJ, Nakamoto-Kinoshita M, Blanco A, Chen S, Stutzer A, et al. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell. 2014;157:869–81.
Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1:109–18.
Bassell GJ. Fragile balance: RNA editing tunes the synapse. Nat Neurosci. 2011;14:1492–4.
Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci. 2013;16:1530–6.
Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–61.
Maurin T, Lebrigand K, Castagnola S, Paquet A, Jarjat M, Popa A, et al. HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic Acids Res. 2018;46:6344–55.
Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr., Moine H, Kooy RF, et al. Fragile X syndrome. Nat Rev Dis Prim. 2017;3:17065.
Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell. 2008;134:1042–54.
Ceman S, O'Donnell WT, Reed M, Patton S, Pohl J, Warren ST. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003;12:3295–305.
Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, et al. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–27.
Caudy AA, Myers M, Hannon GJ, Hammond SM. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 2002;16:2491–6.
Ishizuka A, Siomi MC, Siomi H. A drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 2002;16:2497–508.
Plante I, Davidovic L, Ouellet DL, Gobeil LA, Tremblay S, Khandjian EW, et al. Dicer-derived microRNAs are utilized by the fragile X mental retardation protein for assembly on target RNAs. J Biomed Biotechnol. 2006;2006:64347.
Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA, et al. Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci. 2004;7:113–7.
Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN, et al. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron. 2010;65:373–84.
Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O, et al. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol Cell. 2011;42:673–88.
Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P, et al. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 2009;7:e16.
Maurin T, Melko M, Abekhoukh S, Khalfallah O, Davidovic L, Jarjat M, et al. The FMRP/GRK4 mRNA interaction uncovers a new mode of binding of the fragile X mental retardation protein in cerebellum. Nucleic Acids Res. 2015;43:8540–50.
Eberhart DE, Malter HE, Feng Y, Warren ST. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet. 1996;5:1083–91.
Bardoni B, Sittler A, Shen Y, Mandel JL. Analysis of domains affecting intracellular localization of the FMRP protein. Neurobiol Dis. 1997;4:329–36.
Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci. 1997;17:1539–47.
Adinolfi S, Ramos A, Martin SR, Dal Piaz F, Pucci P, Bardoni B, et al. The N-terminus of the fragile X mental retardation protein contains a novel domain involved in dimerization and RNA binding. Biochemistry. 2003;42:10437–44.
Fernandez E, Rajan N, Bagni C. The FMRP regulon: from targets to disease convergence. Front Neurosci. 2013;7:191.
Siomi MC, Zhang Y, Siomi H, Dreyfuss G. Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them. Mol Cell Biol. 1996;16:3825–32.
Tamanini F, Van Unen L, Bakker C, Sacchi N, Galjaard H, Oostra BA, et al. Oligomerization properties of fragile-X mental-retardation protein (FMRP) and the fragile-X-related proteins FXR1P and FXR2P. Biochem J. 1999;343:517–23.
Bardoni B, Schenck A, Mandel JL. A novel RNA-binding nuclear protein that interacts with the fragile X mental retardation (FMR1) protein. Hum Mol Genet. 1999;8:2557–66.
Bardoni B, Castets M, Huot ME, Schenck A, Adinolfi S, Corbin F, et al. 82-FIP, a novel FMRP (fragile X mental retardation protein) interacting protein, shows a cell cycle-dependent intracellular localization. Hum Mol Genet. 2003;12:1689–98.
Schenck A, Bardoni B, Moro A, Bagni C, Mandel JL. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci USA. 2001;98:8844–9.
Bardoni B, Schenck A, Mandel JL. The fragile X mental retardation protein. Brain Res Bull. 2001;56:375–82.
Davidovic L, Jaglin XH, Lepagnol-Bestel AM, Tremblay S, Simonneau M, Bardoni B, et al. The fragile X mental retardation protein is a molecular adaptor between the neurospecific KIF3C kinesin and dendritic RNA granules. Hum Mol Genet. 2007;16:3047–58.
Ferron L, Nieto-Rostro M, Cassidy JS, Dolphin AC. Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat Commun. 2014;5:3628.
Castagnola S, Delhaye S, Folci A, Paquet A, Brau F, Duprat F, et al. New insights into the role of Cav2 protein family in calcium flux deregulation in Fmr1-KO neurons. Front Mol Neurosci. 2018;11:342.
Menon RP, Gibson TJ, Pastore A. The C terminus of fragile X mental retardation protein interacts with the multi-domain Ran-binding protein in the microtubule-organising centre. J Mol Biol. 2004;343:43–53.
Davidovic L, Bechara E, Gravel M, Jaglin XH, Tremblay S, Sik A, et al. The nuclear microspherule protein 58 is a novel RNA-binding protein that interacts with fragile X mental retardation protein in polyribosomal mRNPs from neurons. Hum Mol Genet. 2006;15:1525–38.
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14.
Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, et al. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol. 1985;67:289–95.
Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41:289–94.
Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98:161–7.
Yan Z, Kim E, Datta D, Lewis DA, Soderling SH. Synaptic actin dysregulation, a convergent mechanism of mental disorders? J Neurosci. 2016;36:11411–7.
Grossman AW, Elisseou NM, McKinney BC, Greenough WT. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084:158–64.
Bakker CE. Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian fragile X consortium. Cell. 1994;78:23–33.
Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, Zu T, Hoogeveen-Westerveld M, Severijnen L, et al. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol Dis. 2006;21:549–55.
Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99:7746–50.
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–7.
Venne AS, Kollipara L, Zahedi RP. The next level of complexity: crosstalk of posttranslational modifications. Proteomics. 2014;14:513–24.
Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805.
Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer's disease. Neurochem Int. 2011;58:458–71.
Xu H, Wang Y, Lin S, Deng W, Peng D, Cui Q, et al. PTMD: a database of human disease-associated post-translational modifications. Genomics, Proteom Bioinform. 2018;16:244–51.
Junqueira SC, Centeno EGZ, Wilkinson KA, Cimarosti H. Post-translational modifications of Parkinson's disease-related proteins: phosphorylation, sumoylation and ubiquitination. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2001–7.
Siomi MC, Higashijima K, Ishizuka A, Siomi H. Casein kinase II phosphorylates the fragile X mental retardation protein and modulates its biological properties. Mol Cell Biol. 2002;22:8438–47.
Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–54.
Stetler A, Winograd C, Sayegh J, Cheever A, Patton E, Zhang X, et al. Identification and characterization of the methyl arginines in the fragile X mental retardation protein Fmrp. Hum Mol Genet. 2006;15:87–96.
Khayachi A, Gwizdek C, Poupon G, Alcor D, Chafai M, Casse F, et al. Sumoylation regulates FMRP-mediated dendritic spine elimination and maturation. Nat Commun. 2018;9:757.
Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis–dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–5.
Henley JM, Barker EA, Glebov OO. Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34:258–68.
Chater TE, Goda Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front Cell Neurosci. 2014;8:401.
Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–7.
Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, et al. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci USA. 1997;94:5395–400.
Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24:2648–55.
Todd PK, Malter JS, Mack KJ. Whisker stimulation-dependent translation of FMRP in the barrel cortex requires activation of type I metabotropic glutamate receptors. Brain Res Mol Brain Res. 2003;110:267–78.
Zhao W, Chuang SC, Bianchi R, Wong RK. Dual regulation of fragile X mental retardation protein by group I metabotropic glutamate receptors controls translation-dependent epileptogenesis in the hippocampus. J Neurosci. 2011;31:725–34.
Huang J, Ikeuchi Y, Malumbres M, Bonni A. A Cdh1-APC/FMRP ubiquitin signaling link drives mGluR-dependent synaptic plasticity in the mammalian brain. Neuron. 2015;86:726–39.
Mazroui R, Huot ME, Tremblay S, Boilard N, Labelle Y, Khandjian EW. Fragile X mental retardation protein determinants required for its association with polyribosomal mRNPs. Hum Mol Genet. 2003;12:3087–96.
Narayanan U, Nalavadi V, Nakamoto M, Pallas DC, Ceman S, Bassell GJ, et al. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J Neurosci. 2007;27:14349–57.
Niere F, Wilkerson JR, Huber KM. Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J Neurosci. 2012;32:5924–36.
Wang X, Mu Y, Sun M, Han J. Bidirectional regulation of fragile X mental retardation protein phosphorylation controls rhodopsin homoeostasis. J Mol Cell Biol. 2017;9:104–16.
Narayanan U, Nalavadi V, Nakamoto M, Thomas G, Ceman S, Bassell GJ, et al. S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J Biol Chem. 2008;283:18478–82.
Novak-Hofer I, Thomas G. Epidermal growth factor-mediated activation of an S6 kinase in Swiss mouse 3T3 cells. J Biol Chem. 1985;260:10314–9.
Van Kanegan MJ, Adams DG, Wadzinski BE, Strack S. Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. J Biol Chem. 2005;280:36029–36.
Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372:657–68.
Bartley CM, O'Keefe RA, Bordey A. FMRP S499 is phosphorylated independent of mTORC1-S6K1 activity. PLoS ONE. 2014;9:e96956.
Bartley CM, O'Keefe RA, Blice-Baum A, Mihailescu MR, Gong X, Miyares L et al. Mammalian FMRP S499 is phosphorylated by CK2 and promotes secondary phosphorylation of FMRP. eNeuro. 2016;3:e0092-16.2016 1–16.
Ruzzene M, Di Maira G, Tosoni K, Pinna LA. Assessment of CK2 constitutive activity in cancer cells. Methods Enzymol. 2010;484:495–514.
St-Denis N, Gabriel M, Turowec JP, Gloor GB, Li SS, Gingras AC, et al. Systematic investigation of hierarchical phosphorylation by protein kinase CK2. J Proteom. 2015;118:49–62.
Cheever A, Ceman S. Phosphorylation of FMRP inhibits association with Dicer. RNA. 2009;15:362–6.
Zimmer SE, Doll SG, Garcia ADR, Akins MR. Splice form-dependent regulation of axonal arbor complexity by FMRP. Dev Neurobiol. 2017;77:738–52.
Coffee RL Jr., Williamson AJ, Adkins CM, Gray MC, Page TL, Broadie K. In vivo neuronal function of the fragile X mental retardation protein is regulated by phosphorylation. Hum Mol Genet. 2012;21:900–15.
Nalavadi VC, Muddashetty RS, Gross C, Bassell GJ. Dephosphorylation-induced ubiquitination and degradation of FMRP in dendrites: a role in immediate early mGluR-stimulated translation. J Neurosci. 2012;32:2582–7.
Zhang XY, Qi J, Shen YQ, Liu X, Liu A, Zhou Z, et al. Mutations of PQBP1 in Renpenning syndrome promote ubiquitin-mediated degradation of FMRP and cause synaptic dysfunction. Hum Mol Genet. 2017;26:955–68.
Choi YN, Jeong DH, Lee JS, Yoo SJ. Regulation of fragile X mental retardation 1 protein by C-terminus of Hsc70-interacting protein depends on its phosphorylation status. Biochem Biophys Res Commun. 2014;453:192–7.
Loriol C, Parisot J, Poupon G, Gwizdek C, Martin S. Developmental regulation and spatiotemporal redistribution of the sumoylation machinery in the rat central nervous system. PLoS ONE. 2012;7:e33757.
Loriol C, Khayachi A, Poupon G, Gwizdek C, Martin S. Activity-dependent regulation of the sumoylation machinery in rat hippocampal neurons. Biol Cell. 2013;105:30–45.
Schorova L, Pronot M, Poupon G, Prieto M, Folci A, Khayachi A, et al. The synaptic balance between sumoylation and desumoylation is maintained by the activation of metabotropic mGlu5 receptors. Cell Mol Life Sci. 2019;76:3019–31.
Gwizdek C, Casse F, Martin S. Protein sumoylation in brain development, neuronal morphology and spinogenesis. Neuromolecular Med. 2013;15:677–91.
Schorova L, Martin S. Sumoylation in synaptic function and dysfunction. Front Synaptic Neurosci. 2016;8:9.
Henley JM, Carmichael RE, Wilkinson KA. Extranuclear sumoylation in neurons. Trends Neurosci. 2018;41:198–210.
Loriol C, Casse F, Khayachi A, Poupon G, Chafai M, Deval E, et al. mGlu5 receptors regulate synaptic sumoylation via a transient PKC-dependent diffusional trapping of Ubc9 into spines. Nat Commun. 2014;5:5113.
Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13.
Blanc RS, Richard S. Arginine methylation: the coming of age. Mol Cell. 2017;65:8–24.
Huang J, Vogel G, Yu Z, Almazan G, Richard S. Type II arginine methyltransferase PRMT5 regulates gene expression of inhibitors of differentiation/DNA binding Id2 and Id4 during glial cell differentiation. J Biol Chem. 2011;286:44424–32.
Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, et al. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26:2749–62.
Simandi Z, Czipa E, Horvath A, Koszeghy A, Bordas C, Poliska S, et al. PRMT1 and PRMT8 regulate retinoic acid-dependent neuronal differentiation with implications to neuropathology. Stem Cells. 2015;33:726–41.
Hashimoto M, Murata K, Ishida J, Kanou A, Kasuya Y, Fukamizu A. Severe hypomyelination and developmental defects are caused in mice lacking protein arginine methyltransferase 1 (PRMT1) in the central nervous system. J Biol Chem. 2016;291:2237–45.
Scaglione A, Patzig J, Liang J, Frawley R, Bok J, Mela A, et al. PRMT5-mediated regulation of developmental myelination. Nat Commun. 2018;9:2840.
Liu Q, Dreyfuss G. In vivo and in vitro arginine methylation of RNA-binding proteins. Mol Cell Biol. 1995;15:2800–8.
Ai LS, Lin CH, Hsieh M, Li C. Arginine methylation of a glycine and arginine rich peptide derived from sequences of human FMRP and fibrillarin. Proc Natl Sci Counc, Repub China Part B, Life Sci. 1999;23:175–80.
Dolzhanskaya N, Merz G, Denman RB. Alternative splicing modulates protein arginine methyltransferase-dependent methylation of fragile X syndrome mental retardation protein. Biochemistry. 2006;45:10385–93.
Blackwell E, Zhang X, Ceman S. Arginines of the RGG box regulate FMRP association with polyribosomes and mRNA. Hum Mol Genet. 2010;19:1314–23.
Denman RB. Methylation of the arginine-glycine-rich region in the fragile X mental retardation protein FMRP differentially affects RNA binding. Cell Mol Biol Lett. 2002;7:877–83.
Dolzhanskaya N, Merz G, Aletta JM, Denman RB. Methylation regulates the intracellular protein-protein and protein-RNA interactions of FMRP. J Cell Sci. 2006;119:1933–46.
Denman RB, Dolzhanskaya N, Sung YJ. Regulating a translational regulator: mechanisms cells use to control the activity of the fragile X mental retardation protein. Cell Mol Life Sci. 2004;61:1714–28.
Denman RB, Xie W, Merz G, Sung YJ. GABAAergic stimulation modulates intracellular protein arginine methylation. Neurosci Lett. 2014;572:38–43.
Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci. 2010;30:9929–38.
Dolzhanskaya N, Bolton DC, Denman RB. Chemical and structural probing of the N-terminal residues encoded by FMR1 exon 15 and their effect on downstream arginine methylation. Biochemistry. 2008;47:8491–503.
Tsang B, Arsenault J, Vernon RM, Lin H, Sonenberg N, Wang LY et al. Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc Natl Acad Sci USA. 2019;116:4218–27.
Zhang J, Li X, Li JD. The roles of post-translational modifications on alpha-synuclein in the pathogenesis of Parkinson's diseases. Front Neurosci. 2019;13:381.
Monaghan KG, Lyon E, Spector EB. ACMG standards and guidelines for fragile X testing: a revision to the disease-specific supplements to the standards and guidelines for clinical genetics laboratories of the American College of Medical Genetics and Genomics. Genet Med. 2013;15:575–86.
Collins SC, Bray SM, Suhl JA, Cutler DJ, Coffee B, Zwick ME, et al. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A. 2010;152A:2512–20.
Collins SC, Coffee B, Benke PJ, Berry-Kravis E, Gilbert F, Oostra B, et al. Array-based FMR1 sequencing and deletion analysis in patients with a fragile X syndrome-like phenotype. PLoS ONE. 2010;5:e9476.
Sitzmann AF, Hagelstrom RT, Tassone F, Hagerman RJ, Butler MG. Rare FMR1 gene mutations causing fragile X syndrome: a review. Am J Med Genet A. 2018;176:11–8.
Quartier A, Poquet H, Gilbert-Dussardier B, Rossi M, Casteleyn AS, Portes VD, et al. Intragenic FMR1 disease-causing variants: a significant mutational mechanism leading to fragile-X syndrome. Eur J Hum Genet. 2017;25:423–31.
De Boulle K, Verkerk AJ, Reyniers E, Vits L, Hendrickx J, Van Roy B, et al. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet. 1993;3:31–5.
Zang JB, Nosyreva ED, Spencer CM, Volk LJ, Musunuru K, Zhong R, et al. A mouse model of the human fragile X syndrome I304N mutation. PLoS Genet. 2009;5:e1000758.
Myrick LK, Nakamoto-Kinoshita M, Lindor NM, Kirmani S, Cheng X, Warren ST. Fragile X syndrome due to a missense mutation. Eur J Hum Genet. 2014;22:1185–9.
Myrick LK, Deng PY, Hashimoto H, Oh YM, Cho Y, Poidevin MJ, et al. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc Natl Acad Sci USA. 2015;112:949–56.
Handt M, Epplen A, Hoffjan S, Mese K, Epplen JT, Dekomien G. Point mutation frequency in the FMR1 gene as revealed by fragile X syndrome screening. Mol Cell Probes. 2014;28:279–83.
Patsalis PC, Sismani C, Hettinger JA, Boumba I, Georgiou I, Stylianidou G, et al. Molecular screening of fragile X (FRAXA) and FRAXE mental retardation syndromes in the Hellenic population of Greece and Cyprus: incidence, genetic variation, and stability. Am J Med Genet. 1999;84:184–90.
Hecimovic S, Tarnik IP, Baric I, Cakarun Z, Pavelic K. Screening for fragile X syndrome: results from a school for mentally retarded children. Acta Paediatr. 2002;91:535–9.
Major T, Culjkovic B, Stojkovic O, Gucscekic M, Lakic A, Romac S. Prevalence of the fragile X syndrome in Yugoslav patients with non-specific mental retardation. J Neurogenet. 2003;17:223–30.
Biancalana V, Beldjord C, Taillandier A, Szpiro-Tapia S, Cusin V, Gerson F, et al. Five years of molecular diagnosis of Fragile X syndrome (1997–2001): a collaborative study reporting 95% of the activity in France. Am J Med Genet A. 2004;129A:218–24.
Diaz J, Scheiner C, Leon E. Presentation of a recurrent FMR1 missense mutation (R138Q) in an affected female. Transl Sci Rare Dis. 2018;3:139–44.
Sethna F, Moon C, Wang H. From FMRP function to potential therapies for fragile X syndrome. Neurochem Res. 2014;39:1016–31.
Gross C, Hoffmann A, Bassell GJ, Berry-Kravis EM. Therapeutic strategies in fragile X syndrome: from bench to bedside and back. Neurotherapeutics. 2015;12:584–608.
Castagnola S, Bardoni B, Maurin T. The search for an effective therapy to treat fragile X syndrome: dream or reality? Front Synaptic Neurosci. 2017;9:15.
Gallego-Iradi C, Bickford JS, Khare S, Hall A, Nick JA, Salmasinia D, et al. KCNC3(R420H), a K(+) channel mutation causative in spinocerebellar ataxia 13 displays aberrant intracellular trafficking. Neurobiol Dis. 2014;71:270–9.
Siomi H, Choi M, Siomi MC, Nussbaum RL, Dreyfuss G. Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell. 1994;77:33–9.
Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–38.
Ramos A, Hollingworth D, Pastore A. The role of a clinically important mutation in the fold and RNA-binding properties of KH motifs. RNA. 2003;9:293–8.
Hearst SM, Shao Q, Lopez M, Raucher D, Vig PJ. The design and delivery of a PKA inhibitory polypeptide to treat SCA1. J Neurochem. 2014;131:101–14.
Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, et al. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–68.
Jorgensen ND, Andresen JM, Lagalwar S, Armstrong B, Stevens S, Byam CE, et al. Phosphorylation of ATXN1 at Ser776 in the cerebellum. J Neurochem. 2009;110:675–86.
Tang LT, Craig TJ, Henley JM. SUMOylation of synapsin Ia maintains synaptic vesicle availability and is reduced in an autism mutation. Nat Commun. 2015;6:7728.
Tai DJ, Liu YC, Hsu WL, Ma YL, Cheng SJ, Liu SY, et al. MeCP2 SUMOylation rescues Mecp2-mutant-induced behavioural deficits in a mouse model of Rett syndrome. Nat Commun. 2016;7:10552.
Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17:184–97.
Anderson DB, Zanella CA, Henley JM, Cimarosti H. Sumoylation: implications for neurodegenerative diseases. Adv Exp Med Biol. 2017;963:261–81.
Zhang L, Yang TH, Li DW. Roles of sumoylation in heart development and cardiovascular diseases. Curr Mol Med. 2017;16:877–84.
Yang Y, Xia Z, Wang X, Zhao X, Sheng Z, Ye Y, et al. Small-molecule inhibitors targeting protein sumoylation as novel anticancer compounds. Mol Pharm. 2018;94:885–94.
Kho C, Lee A, Jeong D, Oh JG, Gorski PA, Fish K, et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat Commun. 2015;6:7229.
Bernstock JD, Ye DG, Lee YJ, Gessler F, Friedman GK, Zheng W, et al. Drugging sumoylation for neuroprotection and oncotherapy. Neural Regen Res. 2018;13:415–6.
Garcia-Manero G, Abaza Y, Takahashi K, Medeiros BC, Arellano M, Khaled SK, et al. Pracinostat plus azacitidine in older patients with newly diagnosed acute myeloid leukemia: results of a phase 2 study. Blood Adv. 2019;3:508–18.
Brave M, Dagher R, Farrell A, Abraham S, Ramchandani R, Gobburu J, et al. Topotecan in combination with cisplatin for the treatment of stage IVB, recurrent, or persistent cervical cancer. Oncology. 2006;20:1401–4, 1410; discussion 1410–11, 1415–6.
Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802.
Bernstock JD, Lee YJ, Peruzzotti-Jametti L, Southall N, Johnson KR, Maric D, et al. A novel quantitative high-throughput screen identifies drugs that both activate SUMO conjugation via the inhibition of microRNAs 182 and 183 and facilitate neuroprotection in a model of oxygen and glucose deprivation. J Cereb Blood Flow Metab. 2016;36:426–41.
Kumar A, Zhang KY. Advances in the development of SUMO specific protease (SENP) inhibitors. Comput Struct Biotechnol J. 2015;13:204–11.
Yang W, Sheng H, Wang H. Targeting the SUMO pathway for neuroprotection in brain ischaemia. Stroke Vasc Neurol. 2016;1:101–7.
Fukuda I, Ito A, Hirai G, Nishimura S, Kawasaki H, Saitoh H, et al. Ginkgolic acid inhibits protein sumoylation by blocking formation of the E1-SUMO intermediate. Chem Biol. 2009;16:133–40.
Fukuda I, Ito A, Uramoto M, Saitoh H, Kawasaki H, Osada H, et al. Kerriamycin B inhibits protein sumoylation. J Antibiot. 2009;62:221–4.
Takemoto M, Kawamura Y, Hirohama M, Yamaguchi Y, Handa H, Saitoh H, et al. Inhibition of protein sumoylation by davidiin, an ellagitannin from Davidia involucrata. J Antibiot. 2014;67:335–8.
Kim YS, Nagy K, Keyser S, Schneekloth JS Jr. An electrophoretic mobility shift assay identifies a mechanistically unique inhibitor of protein sumoylation. Chem Biol. 2013;20:604–13.
Zlotkowski K, Hewitt WM, Sinniah RS, Tropea JE, Needle D, Lountos GT, et al. A small-molecule microarray approach for the identification of E2 enzyme inhibitors in ubiquitin-like conjugation pathways. 2017;22:760–6.
Bernstock JD, Ye D, Gessler FA, Lee YJ, Peruzzotti-Jametti L, Baumgarten P, et al. Topotecan is a potent inhibitor of sumoylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci Rep. 2017;7:7425.
Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8:508–13.
Fatemi SH, Folsom TD. Dysregulation of fragile x mental retardation protein and metabotropic glutamate receptor 5 in superior frontal cortex of individuals with autism: a postmortem brain study. Mol Autism. 2011;2:6.
Rustan OG, Folsom TD, Yousefi MK, Fatemi SH. Phosphorylated fragile X mental retardation protein at serine 499, is reduced in cerebellar vermis and superior frontal cortex of subjects with autism: implications for fragile X mental retardation protein-metabotropic glutamate receptor 5 signaling. Mol Autism. 2013;4:41.
Lohith TG, Osterweil EK, Fujita M, Jenko KJ, Bear MF, Innis RB. Is metabotropic glutamate receptor 5 upregulated in prefrontal cortex in fragile X syndrome? Mol Autism. 2013;4:15.
Fatemi SH, Folsom TD, Kneeland RE, Liesch SB. Metabotropic glutamate receptor 5 upregulation in children with autism is associated with underexpression of both fragile X mental retardation protein and GABAA receptor beta 3 in adults with autism. Anat Rec (Hoboken). 2011;294:1635–45.
Bourgeois JA, Coffey SM, Rivera SM, Hessl D, Gane LW, Tassone F, et al. A review of fragile X premutation disorders: expanding the psychiatric perspective. J Clin Psychiatry. 2009;70:852–62.
Fatemi SH, Kneeland RE, Liesch SB, Folsom TD. Fragile X mental retardation protein levels are decreased in major psychiatric disorders. Schizophr Res. 2010;124:246–7.
Kelemen O, Kovacs T, Keri S. Contrast, motion, perceptual integration, and neurocognition in schizophrenia: the role of fragile-X related mechanisms. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:92–97.
Kovacs T, Kelemen O, Keri S. Decreased fragile X mental retardation protein (FMRP) is associated with lower IQ and earlier illness onset in patients with schizophrenia. Psychiatry Res. 2013;210:690–3.
Acknowledgements
We thank Franck Aguila for his excellent artwork. We gratefully acknowledge the ‘Jérôme Lejeune’ foundation and the ‘Agence Nationale de la Recherche’ (ANR-15-CE16-0015-01) for financial support. We also thank the French Government for the ‘Investments for the Future’ LabEx ‘SIGNALIFE’ (ANR-11-LABX-0028-01). MP is a PhD fellow from the international PhD ‘SIGNALIFE’ LabEx program.
Author information
Authors and Affiliations
Contributions
MP and AF: writing original draft and editing. SM: conceptualization, guidance, funding, writing and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Prieto, M., Folci, A. & Martin, S. Post-translational modifications of the Fragile X Mental Retardation Protein in neuronal function and dysfunction. Mol Psychiatry 25, 1688–1703 (2020). https://doi.org/10.1038/s41380-019-0629-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-019-0629-4
- Springer Nature Limited
This article is cited by
-
The multifaceted role of Fragile X-Related Protein 1 (FXR1) in cellular processes: an updated review on cancer and clinical applications
Cell Death & Disease (2024)
-
The Fragile X Protein Family in Amyotrophic Lateral Sclerosis
Molecular Neurobiology (2023)
-
Bidirectional regulation of synaptic SUMOylation by Group 1 metabotropic glutamate receptors
Cellular and Molecular Life Sciences (2022)
-
miR-142 downregulation alleviates rat PTSD-like behaviors, reduces the level of inflammatory cytokine expression and apoptosis in hippocampus, and upregulates the expression of fragile X mental retardation protein
Journal of Neuroinflammation (2021)
-
Proteomic insights into synaptic signaling in the brain: the past, present and future
Molecular Brain (2021)