
Abstract
Presenilin-1 (PSEN1) is the catalytic subunit of the γ-secretase complex, and pathogenic mutations in the PSEN1 gene account for the majority cases of familial AD (FAD). FAD-associated mutant PSEN1 proteins have been shown to affect APP processing and Aβ generation and inhibit Notch1 cleavage and Notch signaling. In this report, we found that a PSEN1 mutation (S169del) altered APP processing and Aβ generation, and promoted neuritic plaque formation as well as learning and memory deficits in AD model mice. However, this mutation did not affect Notch1 cleavage and Notch signaling in vitro and in vivo. Taken together, we demonstrated that PSEN1S169del has distinct effects on APP processing and Notch1 cleavage, suggesting that Notch signaling may not be critical for AD pathogenesis and serine169 could be a critical site as a potential target for the development of novel γ-secretase modulators without affecting Notch1 cleavage to treat AD.
Similar content being viewed by others

Introduction
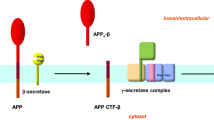
Deposition of amyloid-beta protein (Aβ) to form neuritic plaques is the unique neuropathological hallmark of Alzheimer’s disease (AD). Aβ is derived from the amyloid precursor protein (APP) by sequential cleavages of β- and γ-secretase in the amyloidogenic pathway. However, the non-amyloidogenic pathway is the predominant pathway in vivo, and APP is cleaved within Aβ domain at the Aβ Leu17 site by α-secretase, the Glu11β-secretase site by BACE1, or the Phe20 θ-secretase site by BACE2, thus precluding Aβ production [1,2,3]. In the amyloidogenic pathway, BACE1 first cleaves APP at the Aβ Asp1 site to produce sAPPβ and C99 (CTFβ99). Subsequently, γ-secretase cleaves C99 in the transmembrane domain to release Aβ and CTFγ. Among all the species of Aβ, Aβ40 is the predominant form of Aβ, but Aβ42 is the central component of neuritic plaques and the most toxic species promoting neurodegeneration. Thus, targeting β- and γ-secretase to reduce Aβ production or reduce the aggregation of Aβ has been one of the major strategies for AD drug development [4,5,6].
There have been numerous clinical trials of β- and γ-secretase inhibitors/modulators for AD treatment. Verubecestat (prodromal AD, Phase III), Elenbecestat (early AD, Phase III), AZD3293 (early and mild AD, Phase III), JNJ-54861911 (asymptomatic at-risk patients, Phase II/III), and CNP520 (asymptomatic at-risk patients, Phase III) are the representatives of BACE1 inhibitors in mid- and late-stage trials [7]. Verubecestat trial was halted in 2017 for lack of evidence of positive clinical effect, while the effect of others remained inconclusive [7]. Clinical trials of γ-secretase inhibitors (GSIs) have been unsuccessful due to the lack of efficacy or severe adverse effects at least partially associated with the inhibition of Notch signaling, such as the termination of the phase III trial of semagascestat (LY450139) [8]. Later on, γ-secretase modulators (GSMs) have attracted interest due to its Notch-sparing ability. One typical category of GSMs is NSAIDs (non-steroid anti-inflammatory drug), including ibuprofen, sulindac sulfide, and indomethacin, which selectively lower Aβ42 without affecting NICD generation [9]. Another category of potential GSMs, specifically regulating APP processing but not Notch (Gleeve), includes nucleotides and γ-secretase activating protein (GSAP) [10,11,12]. However, no γ-secretase inhibitor/modulator has been proven safe for clinical use.
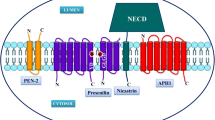
γ-Secretase cleaves many type-I transmembrane proteins including APP and Notch1 [13,14,15,16]. The γ-secretase complex consists of several key protein members including presenilins (PSEN1/PSEN2), Nicastrin, Aph-1, and Pen-2 [17]. The human PSEN1 gene (PSEN1) spans 87257 bp of genomic DNA on chromosome 14q24.3 and consists of 12 exons and 11 introns. PSEN1 protein undergoes auto-endoproteolysis at heterogeneous sites from amino acid 292 to 299, generating an 28-kD N-terminal fragment (NTF) and a 19-kD C-terminal fragment (CTF) [18]. However, it is not clear whether the endoproteolysis is an absolute requirement for γ-secretase activity since some presenilin mutants such as FAD-associated mutations PSEN1ΔE9 and PSEN2M292D are enzymatically active in the absence of endoproteolysis [19, 20]. PSEN1 mutations account for the majority of familial AD (FAD) cases. Pathogenic mutations significantly dysregulate γ-secretase activity, affecting APP processing and Aβ generation. Most PSEN1 mutants increase the relative amount of Aβ42 versus Aβ40 and promote neuritic plaque formation [21,22,23,24]. PSEN1 mutants also reduce Notch1 cleavage and consequently inhibit Notch signaling [16]. However, the role of Notch signaling in PSEN1 mutations-induced FAD pathogenesis remains elusive.
Notch signaling plays an important role in cell fate determination through local cell interactions [25]. The activation of Notch signaling is mediated by ectodomain shedding followed by the proteolytic cleavage of Notch1 at ε site by γ-secretase, releasing an intracellular domain of Notch1 (NICD) [15, 16]. NICD translocates to the nucleus to repress or activate the transcription of target genes [26]. Disruption of the PSEN1 gene in mice results in embryonic lethality and Notch1 deficient phenotypes [27, 28]. C. elegans deficient in SEL-12, the presenilin homolog, also have a Notch signaling deficiency phenotype with egg-laying defects (Egl phenotype) which can be rescued by the expression of wild-type human PSEN1 [29]. However, FAD-associated PSEN1 mutants show a reduced ability to rescue the Egl phenotype [29]. This indicates that PSEN1 is crucial for maintaining Notch signaling, while FAD-associated PSEN1 mutants display the loss-of-function phenotype of Notch signaling.
In this study, we discovered that a PSEN1S169del mutation facilitated Alzheimer’s pathogenesis by altering APP processing and promoting neuritic plaque formation and learning and memory deficits. However, this mutation had no effect on Notch1 cleavage and Notch signaling.
Results
PSEN1S169del promoted Aβ generation and neuritic plaque formation
Previously we identified a PSEN1 mutation (507-509delATC) in patients suffering from FAD with an onset age in the early 40s [30]. This deletion mutation is located in PSEN1 gene exon 6, resulting in expression of mutant PSEN1S169del protein lacking a serine in residue 169 in the transmembrane domain 3 (Supplemental Fig. 1).To investigate the effect of PSEN1S169del on AD pathogenesis, we generated heterozygous transgenic mice carrying the human PSEN1S169del mutation and APP23/PSEN1S169del double transgenic mice carrying two copies of the human PSEN1S169del gene (Supplemental Fig. 2). Compared with APP23 mice, both PSEN1 and PSEN1-NTF were significantly increased to 4.12 ± 0.18 and 1.96 ± 0.08-fold in APP23/PSEN1S169del mice (Fig. 1a). Aβ40 and Aβ42 were significantly increased in APP23/PSEN1S169del mice by 50.67 ± 3.24-fold and 349.65 ± 14.44-fold, respectively (p < 0.05) (Fig. 1b, c). More importantly, the ratio of Aβ42/40 was significantly increased to 6.96 ± 0.65-fold in APP23/PSEN1S169del mice compared with APP23 mice (p < 0.05) (Fig. 1d). These data demonstrated that PSEN1S169del altered APP processing, leading to the increase of Aβ42/40 ratio in vivo.

PSEN1S169del promotes APP processing, Aβ generation and neuritic plaque formation. a Cortical tissue was collected from APP23 (Control) and APP23/PSEN1S169del (S169del) mice at the age of 6 months and homogenized in RIPA-DOC lysis buffer. Full-length APP were detected with C20 antibody. Human PSEN1 and its NTF were detected with PSEN1N antibody. The PSEN1 protein levels were normalized to that of PSEN1WT. The values represent mean ± SEM. n = 3, *p < 0.05 by one-way ANOVA. At the age of 6 months, soluble Aβ40 (b) and Aβ4 2(c) from cortical tissue of APP23 and APP23/PSEN1S169del mice was measured according to Aβ ELISA Kit instructions (Invitrogen). d Aβ42/40 ratios were calculated. The values represent mean ± SEM, n ≥ 3, *p < 0.05 by Student's t test. e APP23/PSEN1S169del(S169del) and APP23 (Control) transgenic mice were euthanized at 6 and 12 months of age, and the brains were dissected, fixed, and sectioned. Amyloid plaques were detected with Aβ-specific monoclonal antibody 4G8 with the DAB method. Plaques were visualized by microscopy with ×40 magnification. Black arrows point to plaques. f Quantification of amyloid plaques at 6 months of age and 12 months of age. The values represent mean ± SEM; n = 10 each group, *p < 0.05 by Student’s t test. g Amyloid plaques were further confirmed using thioflavin S fluorescent staining and visualized by microscopy at ×40 magnifications. White arrows point to green fluorescent amyloid plaques.
Although APP23 mice developed amyloid plaques in the hippocampus and neocortex as early as 6 months of age, significant plaque deposition cannot be observed until 12 months of age [31]. To test whether PSEN1S169del promotes plaque deposition, APP23/PSEN1S169del and age-matched APP23 mice were euthanized at 6 and 12 months. Amyloid plaques were detected by 4G8 immunostaining (Fig. 1e). At 6 months, amyloid plaques were detected in APP23/PSEN1S169del mice, but not in APP23 mice by 4G8 (Fig. 1e, top panel) and the number of neuritic plaques was significantly increased in APP23/PSEN1S169del mice compared with APP23 mice at 6 months, 26.5 ± 1.09/slice vs. 0/slice (p < 0.05) (Fig. 1f), suggesting that PSEN1S169del significantly accelerated plaque formation in APP23/PSEN1S169del mice. At 12 months amyloid plaques could be easily detected in both APP23 mice and APP23/PSEN1S169del mice (Fig. 1e, bottom panel), and APP23/PSEN1 S169del mice exhibited significantly more plaques in the hippocampal region, 71.1 ± 4.79/slice vs. 32.6 ± 2.29/slice in APP23 mice (p < 0.05) (Fig. 1f). The number of plaques in APP23/PSEN1S169del mice was also significantly increased at 12 months compared with 6 months. The plaque results were further confirmed by thioflavin-S fluorescent staining (Fig. 1g). These data indicated that the PSEN1S169del mutation promoted deposition of Aβ to form neuritic plaque in vivo.
PSEN1S169del exacerbated learning and memory deficits
To investigate whether PSEN1S169del affects learning and memory, Morris water maze and fear conditioning tests were performed on the transgenic mice at 6 months of age. APP23/PSEN1S169del mice had similar body weight and performance in the rotor test as APP23 mice (data not shown). In the visible platform test, APP23/PSEN1S169del and APP23 mice had similar escape latency (29.48 ± 1.92 s vs.26.48 ± 1.88 s, p > 0.05) (Fig. 2a) and path length (5.88 ± 0.44 m vs. 5.57 ± 0.44 m, p > 0.05) (Fig. 2b), indicating that S169del mutation did not affect the mice’s motility and vision. In the hidden platform tests, APP23/PSEN1S169del mice showed significant deficits. APP23/PSEN1S169del mice had longer escape latency than APP23 mice (32.45 ± 3.51 s vs. 23.42 ± 2.71 s, p < 0.05) (Fig. 2c), and swam significantly longer distances than APP23 mice (6.03 ± 0.78 m vs. 4.39 ± 0.51 m, p < 0.05) (Fig. 2d) to reach the platform on the fourth day. In the probe trial, APP23/PSEN1S169del mice made significantly fewer hidden platform passes than APP23 mice (0.92 ± 0.23 vs.2.07 ± 0.50, p < 0.05) (Fig. 2e). A contextual fear conditioning test was also performed on the mice. The freezing time was similar between APP23/PSEN1S169del and APP23 mice on the first day. However, on the second day, APP23/PSEN1S169del mice froze significantly less than APP23 mice (23.52 ± 5.07% vs. 36.40 ± 3.92%, p < 0.05) (Fig. 2f). Taken together, the impaired performance of APP23/PSEN1S169del mice in the Morris water maze and fear conditioning tests indicated that PSEN1S169del exacerbated learning and memory deficits in vivo.

PSEN1S169del exacerbates learning and memory deficits. APP23 transgenic mice (Control, n = 14) and APP23/PSEN1S169del mice (S169del, n = 12) were subjected to Morris water maze test at 6 months of age. In the visible platform tests, APP23/ PSEN1S169del and APP23 mice exhibited a similar (a) escape latency and (b) swimming path length to escape onto the visible platform. The values represent mean ± SEM, p > 0.05 by Student’s t test. In the 4 days of hidden platform tests, APP23/ PSEN1S169del mice showed a longer (c) escape latency and (d) swimming path length to escape onto the hidden platform on the fourth day. The values represent mean ± SEM, *p < 0.05 by two-way ANOVA. e In the probe trial, APP23/ PSEN1S169del mice traveled into the target quadrant, significantly less platform crossing times than the controls. The values represent mean ± SEM, *p < 0.05 by Student’s t test. f On day 1 of the contextual fear conditioning test, APP23/ PSEN1S169del and APP23 mice demonstrated similar freezing levels immediately after foot-shock. Twenty-four hours later, APP23/ PSEN1S169del mice showed significantly reduced freezing levels compared with APP23 mice in the same contextual chamber without shock. The values represent mean ± SEM, * p < 0.05 by Student’s t test
PSEN1S169del affected APP processing and Aβ generation in vitro
To examine how the deletion mutation facilitates Alzheimer’s pathogenesis, we first investigated whether the S169del mutation affected auto-endoproteolytic processing of the mutant PSEN1 protein to generate NTF and CTF. NN cells, a PSEN1 and PSEN2 double knockout cell line, were transfected with the PSEN1S169del plasmid. We found that S169del did not affect the auto-endoproteolysis of this PSEN1 mutant (Figs. 3a, 4a,b). To investigate the effect of PSEN1S169del on APP processing and Aβ generation, PSEN1S169del, PSEN1 Y115H, PSEN1C410Y, and PSEN1WT plasmid were transfected into HAW cells, a cell line that stably expresses the wild-type human APP (Fig. 3a). Compared to wild-type PSEN1, PSEN1S169del significantly increased C83 level to 139.94 ± 14.6% (p < 0.05), similar to the effect of PSEN1Y115H and PSEN1C410Y, 155.93 ± 9.93% and 176.62 ± 28.80%, respectively (Fig. 3b). When γ-secretase activity was blocked by an inhibitor L685458, C83 levels were decreased to the same level in PSEN1WT, PSEN1S169del, PSEN1C410Y, and PSEN1Y115H-transfected cells (Fig. 3a, b). Compared with PSEN1WT, PSEN1S169del markedly decreased the production of both Aβ40 and Aβ42 to 24.71% ± 3.80% and 67.13% ± 4.02%, respectively (p < 0.01) (Fig. 3c, d). PSEN1C410Y and PSEN1Y115H reduced the production of Aβ40 to 32.11% ± 4.63% and 30.62% ± 5.28% of PSEN1WT, respectively (p < 0.01), whereas the production of Aβ42 was not affected compared with PSEN1WT (Fig. 3c, d). More importantly, PSEN1S169del, as well as PSEN1C410Y and PSEN1Y115H, significantly increased the ratio of Aβ42/40 to 280.31% ± 26.25%, 274.12% ± 14.15% and 286.13% ± 25.56% compared with PSEN1WT, respectively (p < 0.01) (Fig. 3e). These results demonstrated that PSEN1S169del altered APP processing and dramatically increased the ratio of Aβ42/40.

PSEN1S169del disrupts APP processing and Aβ generation in vitro. a HAW cells were transfected with pcDNA4-PSEN1WT, pcDNA4-PSEN1S169del, pcDNA4-PSEN1C410Y or pcDNA4-PSEN1Y115H. Forty-eight hours after transfection, half transfected cells of each line were treated with specific γ-secretase inhibitor (GSI), L685,458. Cell lysates were resolved in a 10−16% SDS-PAGE gel. PSEN1 and PSEN1 NTF were detected by rabbit anti-PSEN1 antibody PSEN1N. APP and C83 were detected by rabbit anti-APP antibody C20. Monoclonal anti-β-actin antibody (AC-15) was used to detect β–actin. b Quantification of C83 expression. N = 3. The values represent mean ± SEM, n ≥ 3, *p < 0.05 by one-way ANOVA. Conditioned media from the cells were collected and Aβ ELISA was performed to detect (c) Aβ40 and (d) Aβ42. e Aβ42/40 ratios were calculated. f NN cells were transfected with APPWT followed by the transfection of PSEN1 variants. PSEN1 and NTF were detected by PSEN1N. g Quantification of C83 expression in (f). h NN cells were transfected with C99 followed by the transfection of PSEN1 variants. C99 were detected by C20. i Quantification of C99 expression in (h). j Aβ40 and Aβ42 from conditioned media from cell lines in (h) were assayed by Aβ ELISA. k Aβ42/40 ratios were calculated. The values represent mean ± SEM, n ≥ 3, *p < 0.05 by one-way ANOVA

PSEN1S169del did not affect proteolytic release of NICD and Notch signaling. a pcDNA4-PSEN1WT, pcDNA4-PSEN1S169del or pcDNA4-PSEN1C410Y were transfected into V2 cells stably expressing mNotch1ΔE (NΔE). Cell lysates were analyzed by western blot. NΔE and NICD were detected by 9E10. The levels of NICD and NΔE were quantified by Licor image system and the ratios of NICD/NΔE were calculated. b NΔE and PSEN1 variants were transfected into NN cells. NΔE and NICD were detected by 9E10. c NΔE-SNAP and PSEN1 variants were overexpressed in NN cells and pulse labeled. After indicated chasing time, cells lysates were blotted for streptoavidinpulse-labeled NΔE (green) and NΔE (red). The levels of NICD and NΔE were quantified by Licor image system and the ratios of NICD/NΔE were calculated. d Hes-1 promoter activity was assessed in V2 cells co-transfected with Hes-1 promoter construct and PSEN1 variants. The promoter activity of PSEN1 variants transfected cells was normalized to PSEN1WT-transfected cells. e Hes-1 promoter activity was assessed in NN cells co-transfected with Hes-1 promoter construct, PSEN1 variants and NΔE. Forty-eight hours after transfection, cells were harvested and luciferase assay was performed. The promoter activity of PSEN1 variants transfected cells was normalized to PSEN1WT-transfected cells. The values represent mean ± SEM, n ≥ 3, *p < 0.05 by one-way ANOVA. f Egg retention assay of PSEN1 variants transgenic C. elegans. The transgenic lines were generated by microinjection of SEL-12 (SEL-12W) and human PSEN1 variants expression plasmids into SEL-12 deficient (SEL-12D) C. elegans. Wild-type C. elegans (N2) served as normal controls. The number of retained eggs was counted for ten worms for each line and the three lines for each variant averaged (n = 30 worms per group). The values represent mean ± SEM, *p < 0.05 compared to N2 and #p < 0.05 by one-way ANOVA
To eliminate the effect of endogenous presenilins, similar experiments were performed in NN cells. Compared with PSEN1WT-transfected NN cells, C83 was significant increased to 180.12 ± 11.55%, 163.33 ± 6.67%, and 170.14 ± 5.77% (p < 0.05) in NN-PSEN1S169del, NN-PSEN1Y115H, and NN-PSEN1C410Y cells, respectively (Fig. 3f, g). To further confirm the effect of PSEN1S169del on APP processing, we transfected the C99 construct into NN cells followed by the transfection of PSEN1 variants. We found that PSEN1S169del, PSEN1Y115H, and PSEN1C410Y significantly reduced C99 processing and increased C99 to 135.033 ± 1.41%, 130.93 ± 10.44%, and 150.18 ± 15.36% relative to PSEN1WT (p < 0.05), respectively (Fig. 3h, i). Consistently, the PSEN1S169del significantly reduced Aβ40 to 38.02 ± 3.15% (p < 0.05), while slightly increasing Aβ42 production to 114.18 ± 2.32% (p < 0.05) (Fig. 3j). The ratio of Aβ42/ Aβ40 was significantly increased to 305.81 ± 31.75% (p < 0.05) (Fig. 3k). PSEN1Y115H and PSEN1C410Y also showed markedly reduced Aβ40 to 39.50 ± 2.55% and 44.10 ± 3.85% (p < 0.05), while PSEN1Y115H increased Aβ42 production to 130.95 ± 1.95% (p < 0.05) and PSEN1C410Y had no significant effect on Aβ42 generation (94.02 ± 1.65%), leading to a significant increase of Aβ42/ Aβ40 ratios to 335.12 ± 26.55% and 217.12 ± 22.91% (Fig. 3j, k). These results clearly demonstrated that PSEN1S169del shifted the preference of γ-site cleavage, resulting in a dramatic increase of Aβ42/Aβ40 ratio.
PSEN1S169del had no effect on the Notch1 cleavage and Notch signaling
APP and Notch1 are two major substrates of the γ-secretase complex. While Aβ is believed to initiate the pathogenic cascade, impaired Notch1 cleavage and Notch signaling by pathogenic PSEN1 mutations might also contribute to AD pathogenesis [16]. To investigate the role of PSEN1S169del in Notch1 cleavage, PSEN1S169del was introduced into V2 cells stably expressing NΔE in HEK293 cells. NΔE is a truncated form of Notch1 that undergoes constitutive γ-secretase proteolysis to release NICD [16]. Consistent with previous findings, PSEN1Y115H and PSEN1C410Y significantly reduced NICD production to 67.61% ± 7.40% and 52.73% ± 5.21%, respectively (p < 0.05). However, there was no significant difference in NICD generation and NICD/NΔE ratios between PSEN1S169del and PSEN1WT (Fig. 4a). To avoid the interference of endogenous PSEN1/PSEN2, a similar experiment was performed in PSEN1/2 double knockout NN cells and the result showed that PSEN1S169del did not inhibit the proteolytic release of NICD from NΔE (Fig. 4b). To confirm the results, NΔE-SNAP and PSEN1 variants were overexpressed in NN cells. Cells lysates were blotted for streptavidin pulse-labeled NΔE (green) and non-labeled NΔE (red). PSEN1S169del displayed similar cleavage activity compared with PSEN1WT (p > 0.05) (Fig. 4c), whereas PSEN1Y115H and PSEN1C410Y significantly reduced the Notch1 cleavage and NICD generation to 56.2% ± 3.7% and 36.33% ± 4.10%, respectively (p < 0.05) (Fig. 4c).
To examine the mutant PSEN1’s effect on Notch signaling, Hes-1 promoter activity was assessed using luciferase assay. Hes-1 is a target gene of Notch signaling and its transcription is activated by NICD. Hes-1 promoter plasmid was co-transfected with PSEN1 plasmids into V2 cells. The Luciferase assay showed that Hes-1 promoter activity was similar between PSEN1S169del and PSEN1WT-transfected cells, while PSEN1Y115H and PSEN1C410Y significantly reduced the promoter activity to 75.30 ± 4.16% and 72.51 ± 0.93%, respectively (p < 0.05) (Fig. 4d). Similar experiments were performed using NN cells. Consistently, PSEN1S169del had no significant effect on Hes-1 promoter activity compared with PSEN1WT (p> 0.05) (Fig. 4e). However, PSEN1Y115H or PSEN1C410Y significantly reduced Hes-1 promoter activity to 67.44 ± 1.81% and 62.90 ± 1.38%, respectively (p < 0.05) (Fig. 4e). These results demonstrated that PSEN1S169del did not affect Notch1 cleavage and Notch signaling.
Next we examined the effect of PSEN1S169del on Notch signaling in vivo using SEL-12-deficient C. elegans model system. SEL-12 is one of the three orthologs of human presenilins. An SEL-12 deficiency causes an Egl phenotype, the phenotype of Notch signaling deficiency in C.elegans, i.e., increased eggs retained in gonads, which can be rescued by expression of wild-type human PSEN1. A number of FAD-associated PSEN1 mutants display reduced ability to rescue the SEL-12 deficiency-caused Egl phenotype [29, 32]. To determine the rescue ability of PSEN1S169del on SEL-12-deficient C. elegans and compare it to other PSEN1 constructs, three independent extrachromosomal array transgenic lines for each variant were established by microinjection of PSEN1wt, PSEN1S169del, PSEN1Y115H, or PSEN1C410Y plasmids into the sel-12 hermaphrodite germ line and the number of eggs in the gonads was counted. The other two FAD-associated PSEN1 mutants, PSEN1Y115H and PSEN1C410Y, served as negative controls. We found that the number of eggs retained in the gonads of C. elegans was significantly increased to 32.03 ± 0.58 in SEL-12-deficient line (SEL-12D) compared with 12.37 ± 0.51 in wild-type line (N2) (p < 0.05) (Fig. 4f). Both SEL-12 and human wild-type PSEN1 fully rescued the sel-12 egg-laying defect, reducing the number of retained eggs to 13.43 ± 0.56 and 13.20 ± 0.52, similar to the wild-type line (p > 0.05). The number of eggs in the gonads of PSEN1S169del transgenic line was not statistically different from that of PSEN1WT transgenic line, 15.20 ± 0.65 vs. 13.20 ± 0.52 (p > 0.05), while the other two FAD-associated mutants, PSEN1Y115H and PSEN1C410Y, had significantly more eggs in the gonad than PSEN1WT worms, 28.47 ± 1.04 and 29.60 ± 0.65, (Fig. 4f). These data indicated that PSEN1S169del did not affect Notch signaling in vivo.
Discussion
We have reported a novel PSEN1 S169del mutation in early-onset AD patients [30]. The mutation produced a clinical phenotype similar to that of sporadic AD [30]. Compared with the other two mutations at same amino acid, Ser169Pro and Ser169Leu, the mean age of onset was much older in patients with PSEN1S169del, 43y vs. 32y and 38y [33, 34]. In addition, the patient carrying PSEN1S169del mutation did not present some phenotypes that patients with Ser169Pro and Ser169Leu mutations had, such as myoclonic jerks and seizures, suggesting that PSEN1S169del mutation may have different functional role in the pathogenesis of AD. Although more than 200 PSEN1 mutations have been genetically linked to FAD, it remains elusive how PSEN1 mutations affect γ-secretase function on APP processing and Notch signaling pathway in AD pathogenesis. A recent report indicated that around 90% of 138 pathogenic FAD-associated PSEN1 mutations reduced the production of Aβ40 and Aβ42 in vitro assay [24]. Our results showed that the PSEN1S169del mutation reduced both Aβ40 and Aβ42 production in HAW cells. In NN cells overexpressing C99, PSEN1S169del mutation reduced Aβ40 production but slightly increased Aβ42 production. The inconsistent alteration of Aβ42 might be caused by the following reasons. First, it might be caused by the species or cell-specific effect, i.e., HEK293 (human embryonic kidney cells) and NN cells (mouse embryonic fibroblast). Moreover, both endogenous PSEN1/PSEN2 and exogenous PSEN1S169del were responsible for the γ-cleavage and Aβ generation in transfected HEK293 cells, while only exogenous PSEN1S169del was responsible for the γ-cleavage and Aβ generation in transfected NN cells (PSEN1/PSEN2 double knockout cells). In addition, the overexpression of different APP species, full-length APP and C99, may contribute to the difference of Aβ42 production. Furthermore, C99, the substrate for Aβ42 generation, is almost undetectable in HAW cells, suggesting the level of C99 in two cell lines may also contribute to the difference of Aβ42 production. However, more importantly, PSEN1S169del dramatically increased the ratio of Aβ42/40 in both cell lines, which is more relevant to the pathogenesis of AD. Consistently, S169del mutation also increased the ratio of Aβ42/40 in APP23/PSEN1S169del double transgenic mice, facilitating the deposition of Aβ to form neuritic plaques and exacerbating learning and memory deficits.
FAD-associated PSEN1 mutations not only alter APP processing and Aβ generation but also impair the cleavage of Notch1. Notch signaling is critical for embryo development, cells proliferation, and neurodegeneration. Notch1 is one of the major substrates of the γ-secretase complex. However, the importance of Notch signaling impairment in the development of AD remains elusive. Pathogenic PSEN1 mutations have been shown to markedly reduce γ-secretase cleavage of Notch1, leading to the reduction of NICD generation and impaired Notch signaling [16, 35]. Chávez-Gutiérrez et al. reported that the FAD-associated PSEN1 mutations might differentially regulated ε-site and γ-site cleavage in APP and Notch processing [36]. Notch is cleaved by γ-secretase at ε-site to release NICD, while APP is cleaved by γ-secretase at ε-site to release AICD. The ε-site cleavage was differentially impaired by FAD-associated PSEN1 mutations [37]. Among them, M139V and I213T mutations did not affect the ε-site cleavage of both Notch and APP. However, both of them affected γ-site cleavage of APP to generate relatively high level of longer Aβ, including Aβ42. Although this study suggested that some PSEN1 mutations may have no effect on Notch cleavage, this experiment was performed in test tubes by using purified substrates with approximate 99 amino acids. Therefore, the effect of these mutations on Notch1 cleavage needs to be confirmed in cultured cells or animals. In this study, we clearly showed that PSEN1S169del maintains normal Notch1 cleavage, NICD generation and Notch signaling in both wild-type and PSEN1/2-deficient cells. PSEN1S169del did not inhibit Notch signaling assessed by Hes-1 promoter activity in vitro as other pathogenic PSEN1 mutants did. More importantly, compared with PSEN1WT, PSEN1S169del completely rescued the egg-laying defective phenotype in SEL-12-deficient C. elegans. This is the first study that demonstrates a PSEN1 mutation that neither disrupts Notch1 cleavage and NICD generation nor impairs Notch signaling in vitro and in vivo. Our data highly suggest that Notch signaling impairment may not play a critical role in PSEN1 mutation-induced FAD.
PSEN1 forms the catalytic core of the γ-secretase complex. Parallel substituted cysteine accessible method (SCAM) and cross-link experiments confirmed that transmembrane domain 6 (TM6), TM7 and TM9 of PSEN1 formed the intramembrane chamber with two catalytic aspartates residing oppositely on TM6 and TM7 (Asp257 in TM6 and Asp385 in TM7), respectively [38,39,40,41,42]. The structure of PSEN1 is critical for its function as an intramembrane protease. Although the interaction between the GSIs/GSMs and the functional structures of γ-secretase/presenilin remains elusive, a recent study reported that a phenylimidazole-type GSMs (E2012) might modulate γ-secretase activity by interacting with TM1 of PSEN1 and triggering the piston movement [43]. Recent crystal structure analysis shed light on the elucidation of the structure of PSEN1 based on the conserved sequence of its homolog, presenilin/SPP, which indicated that TM2-4 served as a conserved core domain [44]. The serine 169 residue is located in the TM3 of PSEN1. Moreover, multiple residues in TM3 specifically affected the potency of some class of GSIs, such as ELN318463, altering APP processing and Aβ generation [45]. PS1S169A and PS1L173A significantly affected the potency of sulfonamides and DAPT but not L-685,458 in APP processing, suggesting that TM3 is a key domain and S169 may be the key residue in regulating γ-cleavage of APP [45]. Currently, the effect of this mutation on PSEN1 structure has not been reported. The exact role of this mutation and even TM3 on PSEN1 structure needs to be further investigated. In addition, the role of TM3 and S169 in Notch signaling has not been reported. Our study demonstrates that the pathogenic PSEN1 Ser169del functions separately in APP processing and the Notch signaling pathway, i.e. the mutation causes FAD but has no effect on Notch1 cleavage and Notch signaling. Serine169 in the TM3 of PSEN1 might be a critical site for separating PSEN1’s effect on APP processing from Notch cleavage, and TMD3 could be an allosteric domain regulating the specificity of γ-secretase. It could serve as a potential target for the development of novel GSMs for AD treatment by specifically modulating APP processing, but not affecting Notch1 cleavage to avoid the severe side effects. APP23/PSEN1S169del mice are the first AD model mice that only disrupt APP processing and Aβ generation resulting in neuritic plaque formation and memory deficits without an effect on Notch signaling. This new line of mice provides a new tool to investigate the mechanism underlying AD pathogenesis, and furthermore, it could be a valuable model to screen γ-secretase inhibitors/modulators for AD drug development.
Materials and methods
Plasmid constructs
PSEN1 wild-type, and mutant S169del, Y115H and C410Y cDNA were cloned into pcDNA4-MycHis (A) (Invitrogen). mNotch1ΔE m/v (NΔE) expresses the truncated form of mouse Notch1 that lacks the ectodomain and undergoes constitutive γ-secretase proteolysis. Plasmid ICV expresses NICD [16]. The plasmids pBY140-PSEN1WT (gifted from Dr. Baumeister of Albert-Ludwig University), -PSEN1S169del, PSEN1C410Y, PSEN1Y115H and pBY895 contain human PSEN1 and sel-12 coding sequences, respectively, which are driven by the sel-12 promoter. For pulse-chase assay, NΔE were cloned into pSNAP (New England Biolabs).
Cell culture and transfection
HEK293 cells were cultured in DMEM containing 10% FBS (Invitrogen). NN (PSEN1−/−/PSEN2−/−) cells were cultured in DMEM media supplemented with 15% FBS, 1 mM non-essential amino acids, 1 μM β-mecapitolethenoland, 50 U/mL ESGRO® Leukemia Inhibitory Factor (Millipore). Stable cell lines were maintained in media containing Zeocin or G418. All cells were maintained at 37 °C in an incubator containing 5% CO2. Transfection was performed with Lipofectamine 2000 Reagent (Invitrogen).
Immunoblot analysis and luciferase assay
Cells were lysed in RIPA-Doc lysis buffer and were resolved on 10% Tris-glycine or 16% Tris-tricine SDS-PAGE, and transferred to ImmobilonTM–FL phlyvnylidene fluoride membranes. Rabbit antibody C20 (1:2000) was used to detect APP and its CTFs. Rabbit PS1N (1:2000) was used to detect full-length PSEN1 and its NTF. 9E10 and M2 antibody were used to detect myc-tagged or Flag-tagged proteins, respectively. IRDyeTM 680-labeled or IRDyeTM 800CM-labeled secondary antibodies were used to detect the proteins and the membranes were visualized on the Odyssey system (LI-COR Biosciences). Hes-1 promoter assay was performed as described [16]. A Luciferase assay was performed following the manufacturer’s instruction and the luminescent signal was detected by a Luminometer (Turner Designs, TD20/20).
Aβ40/42 enzyme-associated immunosorbent assay (ELISA)
Conditioned medium was collected and protease inhibitors and AEBSF (ROCHE Diagnostics) were added to prevent degradation of Aβ. Lysis buffer (5 M guanidine HCl/50 mM TrisHCl) was added to homogenize brain tissues of mice at the age of 6 months. The soluble Aβ40 and Aβ42 were detected using β-amyloid 1-40 or β-amyloid 1-42 Colorimetric ELISA kit (Invitrogen).
Transgenic mouse generation
All animal experiment protocols were approved by the University of British Columbia Animal Care and Use Committee. APP23 mice carry human Swedish mutant APP 751. Human PSEN1S169del was cloned into Thy1-promotor-containing vector and the linearized DNA was microinjected into fertilized oocytes (CBA X C57Bl/6 F2) and survivors were transferred into pseudo-pregnant recipient female mice (CD-1/ICR). The expression of human PSEN1S169del was identified by RT-PCR and western blot. There was no significant difference in the body weights between the PSEN1S169del transgenic mice and control mice. APP23/PSEN1S169del mice were generated by crossing heterozygous APP23 mice with heterozygous Thy1-PSEN1S169del mice. The presence of both APPswe and PSEN1S169del transgenes was confirmed by genotyping with primers 5′-caccacagaatccaagtcgg and 5′-ggtatcttctgtgaatgggg for PSEN1S169del, and primers 5′-caccacagaatccaagtcgg and 5′-cttgacgttctgcctcttcc for APP.
Neuritic plaque staining
Brain was fixed and sectioned with a Leica cryostat to 30 μm thickness. Every 12th slice with the same reference position was mounted onto slides for staining with biotinylated 4G8 antibody (Biolegend, San Diego, CA). Approximately 24 slices were stained for each mouse. Plaques were visualized by the ABC and DAB method, and counted under microscopy with ×40 magnification. Thioflavin S staining was also performed to detect the green fluorescence-stained plaques.
Behavioral tests
The water maze test was performed in a pool 1.5 m in diameter and the platform was 10 cm in diameter. The procedure consists of 1 day of visible platform tests and 4 days of hidden platform tests, plus a probe trial 24 h after the last hidden platform test. The distance traveled, escape latency, and the number of passes through the platform were recorded by automated video tracking system (ANY-maze, Stoelting). The contextual fear conditioning test was previously described [46]. On day 1, mice were placed in the conditioning chamber for 5 min. After 180 s, the mice received an unconditioned foot-shock stimulus (1 mA, 50 Hz) for 3 s. Twenty-four hours later, mice were placed into the same chamber for 4 min without the foot-shock. In both trails, freezing behavior was recorded on a second-by-second basis with Freeze FrameTM (ActiMetrics Software).
Transgenic C. elegans and rescue assay
C. elegans were cultured on nematode growth medium seeded with Escherichia coli (E. coli; OP50). N2 Bristol strain (wild-type) and SEL-12-deficient strain, RB1672 sel-12(ok2078), were obtained from the Caenorhabditis Genetic Center (University of Minnesota). Transgenic lines of C. elegans were generated by DNA microinjection of SEL-12, PSEN1WT, PSEN1S169del, PSEN1C410Y, and PSEN1Y115H expression plasmids at a concentration of 10 µg/ml along with the co-injection marker (plasmid containing Pmyo-2::GFP) into the hermaphrodite germ line of the SEL-12-deficient strain, sel-12(ok2078), respectively. Stable F2 lines with transmission of Pmyo-2::GFP > 50% were chosen for the rescue assay. To overcome inherent variability in the expression levels from extrachromosomal arrays, three independent lines for each transgenic line were generated and assessed. The rescue ability was assessed by egg retention assay as described with a minor modification [47]. Briefly, worms were isolated at late L4 stage of larva and allowed to develop at 20 °C for 24 h. The resulting young adult worms were individually incubated for 30 min in 96-well cell culture plates containing 50 μl of 1% sodium hypochlorite which dissolves all tissues except eggs. Released eggs were then viewed and counted by using an inverted microscope (Tritec MINJ-1000).
References
Zhang S, Wang Z, Cai F, Zhang M, Wu Y, Zhang J, et al. BACE1 cleavage site selection critical for amyloidogenesis and Alzheimer’s pathogenesis. J Neurosci. 2017;37:6915–25.
Deng Y, Wang Z, Wang R, Zhang X, Zhang S, Wu Y, et al. Amyloid-beta protein (Abeta) Glu11 is the major beta-secretase site of beta-site amyloid-beta precursor protein-cleaving enzyme 1(BACE1), and shifting the cleavage site to Abeta Asp1 contributes to Alzheimer pathogenesis. Eur J Neurosci. 2013;37:1962–9.
Sun X, He G, Song W. BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 2006;20:1369–76.
Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, et al. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013;123:224–35.
Qing H, He G, Ly PT, Fox CJ, Staufenbiel M, Cai F, et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J Exp Med. 2008;205:2781–9.
Zeng J, Chen L, Wang Z, Chen Q, Fan Z, Jiang H, et al. Marginal vitamin A deficiency facilitates Alzheimer’s pathogenesis. Acta Neuropathol. 2017;133:967–82.
Mullard A. BACE inhibitor bust in Alzheimer trial. Nat Rev Drug Discov. 2017;16:155.
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369:341–50.
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–6.
Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, et al. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc Natl Acad Sci USA. 2003;100:12444–9.
He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, et al. Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature. 2010;467:95–98.
Fraering PC, Ye W, LaVoie MJ, Ostaszewski BL, Selkoe DJ, Wolfe MS. gamma-Secretase substrate selectivity can be modulated directly via interaction with a nucleotide-binding site. J Biol Chem. 2005;280:41987–96.
Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, et al. Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nat Cell Biol. 2000;2:463–5.
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–90.
De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–22.
Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci USA. 1999;96:6959–63.
Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, et al. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003;422:438–41.
Podlisny MB, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, et al. Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and Ala299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol Dis. 1997;3:325–37.
Jacobsen H, Reinhardt D, Brockhaus M, Bur D, Kocyba C, Kurt H, et al. The influence of endoproteolytic processing of familial Alzheimer’s disease presenilin 2 on abeta42 amyloid peptide formation. J Biol Chem. 1999;274:35233–9.
Steiner H, Romig H, Grim MG, Philipp U, Pesold B, Citron M, et al. The biological and pathological function of the presenilin-1 Deltaexon 9 mutation is independent of its defect to undergo proteolytic processing. J Biol Chem. 1999;274:7615–8.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–70.
Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–13.
Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–3.
Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc Natl Acad Sci USA. 2017;114:E476–E485.
Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6.
Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–8.
Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–39.
Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, et al. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–92.
Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, et al. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1996;93:14940–4.
Guo J, Wei J, Liao S, Wang L, Jiang H, Tang B. A novel presenilin 1 mutation (Ser169del) in a Chinese family with early-onset Alzheimer’s disease. Neurosci Lett. 2010;468:34–37.
Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, et al. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci USA. 2006;103:18727–32.
Baumeister R, Leimer U, Zweckbronner I, Jakubek C, Grunberg J, Haass C. Human presenilin-1, but not familial Alzheimer’s disease (FAD) mutants, facilitate Caenorhabditis elegans Notch signalling independently of proteolytic processing. Genes Funct. 1997;1:149–59.
Ezquerra M, Carnero C, Blesa R, Gelpi JL, Ballesta F, Oliva R. A presenilin 1 mutation (Ser169Pro) associated with early-onset AD and myoclonic seizures. Neurology. 1999;52:566–70.
Taddei K, Kwok JB, Kril JJ, Halliday GM, Creasey H, Hallupp M, et al. Two novel presenilin-1 mutations (Ser169Leu and Pro436Gln) associated with very early onset Alzheimer’s disease. Neuroreport. 1998;9:3335–9.
Chen F, Gu Y, Hasegawa H, Ruan X, Arawaka S, Fraser P, et al. Presenilin 1 mutations activate gamma 42-secretase but reciprocally inhibit epsilon-secretase cleavage of amyloid precursor protein (APP) and S3-cleavage of notch. J Biol Chem. 2002;277:36521–6.
Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–74.
Zhang S, Zhang M, Cai F, Song W. Biological function of Presenilin and its role in AD pathogenesis. Transl Neurodegener. 2013;2:15.
Sato C, Morohashi Y, Tomita T, Iwatsubo T. Structure of the catalytic pore of gamma-secretase probed by the accessibility of substituted cysteines. J Neurosci: Off J Soc Neurosci. 2006;26:12081–8.
Sato C, Takagi S, Tomita T, Iwatsubo T. TheC-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the gamma-secretase. J Neurosci: Off J Soc Neurosci. 2008;28:6264–71.
Takagi S, Tominaga A, Sato C, Tomita T, Iwatsubo T. Participation of transmembrane domain 1 of presenilin 1 in the catalytic pore structure of the gamma-secretase. J Neurosci: Off J Soc Neurosci. 2010;30:15943–50.
Watanabe N, Image I II, Takagi S, Tominaga A, Image Image I, Tomita T, et al. Functional analysis of the transmembrane domains of presenilin 1: participation of transmembrane domains 2 and 6 in the formation of initial substrate-binding site of gamma-secretase. J Biol Chem. 2010;285:19738–46.
Tolia A, Chavez-Gutierrez L, De Strooper B. Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the gamma-secretase complex. J Biol Chem. 2006;281:27633–42.
Cai T, Yonaga M, Tomita T. Activation of gamma-Secretase trimming activity by topological changes of transmembrane domain 1 of presenilin 1. J Neurosci. 2017;37:12272–80.
Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493:56–61.
Zhao B, Yu M, Neitzel M, Marugg J, Jagodzinski J, Lee M, et al. Identification of gamma-secretase inhibitor potency determinants on presenilin. J Biol Chem. 2008;283:2927–38.
Chen B, Bromley-Brits K, He G, Cai F, Zhang X, Song W. Effect of synthetic cannabinoid HU210 on memory deficits and neuropathology in Alzheimer’s disease mouse model. Curr Alzheimer Res. 2010;7:255–61.
Koelle MR, Horvitz HR. EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell. 1996;84:115–25.
Acknowledgements
This work was supported by Canadian Institutes of Health Research (CIHR) Operating Grant CCI-117952,TAD-117948, and MOP-142487to WS and CIHR Operating Grant #122216-2013 to CR. WS was the holder of the Tier 1 Canada Research Chair in Alzheimer’s disease. Sh.Z. was the recipient of the Chinese Scholarship Council award. Si.Z. is supported by UBC 4YF Scholarship. Assistance in C. elegans egg-counting was provided by Mahraz Parvand and Dawson Born.
Author contributions
WS conceived and designed the experiments; ShZ, FC, YW, TB, ZW, SiZ, DH and WS performed the experiments; ShZ, FC, YW, TB, ZW, SiZ, JG, LS, BT, CR and WS analyzed and contributed reagents /materials /analysis tools; ShZ, FC, YW, CR and WS wrote the paper. All authors reviewed the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Zhang, S., Cai, F., Wu, Y. et al. A presenilin-1 mutation causes Alzheimer disease without affecting Notch signaling. Mol Psychiatry 25, 603–613 (2020). https://doi.org/10.1038/s41380-018-0101-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-018-0101-x
- Springer Nature Limited
This article is cited by
-
Contactin-associated protein-like 2 (CNTNAP2) mutations impair the essential α-secretase cleavages, leading to autism-like phenotypes
Signal Transduction and Targeted Therapy (2024)
-
Amyloid β-based therapy for Alzheimer’s disease: challenges, successes and future
Signal Transduction and Targeted Therapy (2023)
-
CNTNAP2 intracellular domain (CICD) generated by γ-secretase cleavage improves autism-related behaviors
Signal Transduction and Targeted Therapy (2023)
-
Targeting NLRP3 inflammasome for neurodegenerative disorders
Molecular Psychiatry (2023)
-
Genetic Phenotypes of Alzheimer’s Disease: Mechanisms and Potential Therapy
Phenomics (2023)