
Abstract
The diurnal regulation of dopamine is important for normal physiology and diseases such as addiction. Here we find a novel role for the CLOCK protein to antagonize CREB-mediated transcriptional activity at the tyrosine hydroxylase (TH) promoter, which is mediated by the interaction with the metabolic sensing protein, Sirtuin 1 (SIRT1). Additionally, we demonstrate that the transcriptional activity of TH is modulated by the cellular redox state, and daily rhythms of redox balance in the ventral tegmental area (VTA), along with TH transcription, are highly disrupted following chronic cocaine administration. Furthermore, CLOCK and SIRT1 are important for regulating cocaine reward and dopaminergic (DAergic) activity, with interesting differences depending on whether DAergic activity is in a heightened state and if there is a functional CLOCK protein. Taken together, we find that rhythms in cellular metabolism and circadian proteins work together to regulate dopamine synthesis and the reward value for drugs of abuse.
Similar content being viewed by others


Introduction
Many of the mechanisms underlying physiology and behavior are controlled by circadian rhythms. Studies from the liver find that organismal homeostasis is maintained by an interplay between circadian rhythms and metabolism [1, 2]. Few studies have investigated these mechanisms in the mammalian brain, despite evidence linking circadian and/or metabolic disruptions to psychiatric disorders, including bipolar disorder and drug addiction [3,4,5,6,7]. Circadian disruption, whether environmental or genetic, alters the behavioral responses to drugs of abuse [8,9,10,11]. For example, circadian gene mutant mice display differential locomotor sensitization and conditioned preference to cocaine [12, 13], and self-administration of cocaine [14], likely by modulating DA neurotransmission [15,16,17,18,19,20].
At the cellular level, rhythms are controlled by transcriptional–translational feedback loops [21], and ~40–60% of protein-coding genes are regulated by the molecular clock [22]. The core circadian transcription factors CLOCK and BMAL1 form heterodimers and bind to enhancer E-box promoter elements to directly control rhythmic gene transcription [21]. More recently, the molecular clock has been shown to couple with intracellular metabolic signaling pathways. One of the most well characterized is the interaction between CLOCK/BMAL1 and the energy-dependent histone and protein deacetylase SIRT1 to repress CLOCK-mediated transcription [23,24,25,26]. SIRT1 activity is modulated by the cofactor nicotinamide adenine dinucleotide (NAD+), and the reduced form, NADH, both of which respond to cellular energy demands and redox state [27]. Other studies have shown that NAD+ is important for maintaining energy homeostasis in the brain [28, 29], as well as calcium transport and mitochondrial respiration [29,30,31,32,33].
NAD+-dependent SIRT1 regulates exploratory behavior, anxiety, and depressive-like behaviors, and reward and motivated behaviors associated with drugs of abuse [34,35,36,37,38], suggesting a molecular link between circadian rhythms, metabolism, and psychiatric disorders. Moreover, drugs of abuse, including cocaine, alter cellular metabolism, redox state, and oxidative stress [39,40,41,42]. For example, fluctuations of NAD+ modulate calcium influx to neural cells during DA stimulation, reducing NAD+ bioavailability and attenuating further cellular activity [43, 44]. A functional relationship between NAD+ and DA in the brain may have implications in normal and pathological brain function.
Alterations in brain metabolism, circadian rhythms, and the mesolimbic DA system (ventral tegmental area (VTA) and nucleus accumbens (NAc)) contribute to the acute and prolonged effects on neural and behavioral plasticity of cocaine and other drugs of abuse [45,46,47,48,49,50]. Most neurons of the VTA are DAergic (~65%) [51,52,53], which send projections to the amygdala, medial prefrontal cortex (mPFC), NAc, and other striatal regions [54,55,56]. In the NAc, DA clearance follows a diurnal rhythm, which correlates with cocaine sensitization [57, 58]. The VTA is central to reward and motivational behaviors [59, 60], and circadian disruption within the VTA is implicated in anxiety, depressive, and drug-related behaviors [18, 61].
In the VTA, DAergic synthesis and release may be regulated by circadian rhythms. The rate-limiting step for DA synthesis is dependent on the availability and activity of tyrosine hydroxylase (TH) [62,63,64]. Interestingly, there are several putative circadian transcription factor-binding motifs within the TH promoter [15, 65,66,67], including E-boxes [68,69,70,71]. The location of these E-boxes to the transcriptional start site (TSS) and the cAMP response element (CRE) site [72, 73] suggests direct transcriptional regulation of TH by CLOCK/BMAL1.
Our previous studies demonstrated that CLOCK disrupted, ClockΔ19 mice, are hyper-DAergic [18, 74], with a propensity to self-administer drugs of abuse [14, 75]. The expression of TH in the VTA is also increased in ClockΔ19 mice [18, 76], suggesting that CLOCK may antagonize the transcriptional activity of TH, contrary to the typical transcriptional activation role of CLOCK. Here, we demonstrate that the diurnal rhythm of TH is controlled by the diurnal-dependent recruitment of CLOCK and CREB, whereby CLOCK and NAD+-dependent regulation of SIRT1, antagonize CREB-induced transcription during the day, which is relieved during the night, allowing for CREB-mediated TH transcription. Moreover, these mechanisms are disrupted in ClockΔ19 mice leading to elevated CREB-mediated TH transcription during the day. We also report these transcription factors within the VTA are important for regulating conditioned place preference (CPP) for cocaine.
Materials and methods
Mice
Experiments used adult (9–16 weeks old) male wild-type (Wt) and ClockΔ19 mutant littermates on a BALB/c background grouped housed (2–4 per cage). For electrophysiological experiments, mice were between 5 and 6 weeks old at an age where abnormal behaviors of the ClockΔ19 mice are exhibited [77]. Mice were maintained on a 12:12 light–dark cycle (lights on at 700, ZT0, and lights off at 1900, ZT12) with food and water provided ad libitum. Separate cohorts of mice were used for each of the experiments described below with the exception of the following groups: immunohistochemistry and gene expression was completed on subsets of mice following cocaine CPP. Animal use was conducted in accordance with the National Institute of Health guidelines and approved by the Institutional Animal Care and Use Committees of the University of Pittsburgh.
Drug administration
Cocaine hydrochloride was provided by the National Institute on Drug Abuse and dissolved with saline (0.9%, Fisher Scientific). Mice were injected intraperitoneally (i.p.) with cocaine (5 mg/kg for CPP), resveratrol (20 mg/kg, SelleckChem, Houston, TX, USA), or saline (0.9%) at a volume of 10 ml/kg. Systemic administration of resveratrol readily crosses the blood–brain barrier [78]. For gene, protein, and NAD+ expression studies (n = 6 per group per ZT), cocaine (20 mg/kg, i.p.) was administered acutely (single dose) or chronically (single dose per day for 14 days). Drugs were administered between ZT4-8.
RNA isolation, cDNA, and qPCR
Mice were killed throughout the LD cycle (n = 6 per genotype per ZT) and brains were rapidly extracted and VTA was micropunched, followed by isolation of total RNA and cDNA synthesis to measure gene expression using qPCR, as described previously [79] (Supplementary Methods). Relative gene expression was calculated using the 2−∆∆Ct method, normalized to reference gene 18S, and reported as mean ± SEM.
SDS-PAGE western blots
Mice were killed throughout the LD cycle (n = 6 per group or genotype per ZT) and brains were rapidly extracted, and VTA was micropunched, followed by homogenization and sonication. Protein lysates (10 μg) were boiled and then separated by SDS-PAGE and transferred to immunobilon-FL transfer membrane (Millipore), as described previously. The following primary antibodies were used: β-actin (1:2000; Sigma A2228), GAPDH (1:1000; Fitzgerald 10R-G109a), CREB (1:1000; Millipore MAB5432), phospho-CREB (1:1000; Millipore 06-519), and SIRT1 (1:1000; Millipore 07-131) (Supplementary Methods). Optical densities were quantified by NIH ImageJ software and normalized to the reference protein. Samples with values ±1.5 s.d. from group means were excluded.
Chromatin immunoprecipitation (ChIP) assays
Midbrain punches were removed from fresh sections then pooled (n = 6 pools (n = 2 mice per pool) per genotype per ZT) and immediately cross-linked (1% paraformaldehyde), then incubated with glycine followed by homogenization and chromatin sheering (400–800 bp fragments), as previously described [18]. Samples were incubated with antibodies for CLOCK, P-CREB, or SIRT1, coupled to magnetic beads. DNA-associated protein was recovered using PCR Purification Kit (Qiagen). qPCR measured binding enrichment at the distal or proximal promoter of TH. Enrichment levels were calculated relative to the IgG control, as previously described [18].
TH:luc reporter in vitro assays
Rat pheochromocytoma (PC12) or mouse neuroblastoma (N2A) cells (ATCC, Manassas, VA, USA) were grown and maintained in high-glucose DMEM supplemented (Invitrogen) with 10% FBS (Invitrogen). SIRT1 KO MEFs and CLOCK KO MEFs were grown and maintained in DMEM supplemented with 5% FBS and 5% newborn calf serum (Sigma-Aldrich). Semi-confluent cells (70–90%) were transfected with plasmids (5 or 10 µg) using Lipofectamine 2000 reagents (Invitrogen). Cells were lysed 48–72 h post-transfection with 200 µl of lysis buffer (Promega E1521, Fisher Scientific), then bioluminescence activity was measured. Luciferase activity was normalized to wells containing only TH:luc reporter plasmid and total protein. Plasmid construction of wild-type and E-box mutant TH:luc reporter gene constructs were prepared by inserting fragments (−1 kb or −250 bp) of the mouse promoter containing the putative TSS into a pGL3 luciferase vector. Mutagenic primers were designed to produce mutations at the putative E-box sites. CLOCK, CLOCKΔ19, and SIRT1 expression plasmids were cloned and modified. siRNA-mediated transfection was used to knockdown SIRT1 (Santa Cruz Biotech, SC-108043). After transfection, PC12 cells and MEFs were also treated with bath application of NAD+ (β-Nicotinamide adenine dinucleotide; Sigma, N8535), NMN (β-Nicotinamide mononucleotide; Sigma, N3501), NAM (Nicotinamide; Sigma, 73240), or the NAMPT inhibitor, FK866 (SelleckChem, S2799).
HPLC to measure NAD+ levels from mouse brain tissue
Brain samples were rapidly extracted and flash frozen using liquid nitrogen (n = 6 mice per ZT per genotype). Frozen tissue samples were rapidly homogenized in perchloric acid then neutralized in potassium carbonate. NAD+ concentrations were determined using an HPLC system (Prominence; Shimadzu Scientific Instruments, Columbia, MD) with a Supelco LC-18-T column (Sigma, 58970-U), as described previously [80, 81]. NAD+ concentrations were normalized to weights of frozen tissue samples.
Electrophysiology
Mice were anesthetized with isoflurane then decapitated to obtain horizontal brain sections (200 µm) of the VTA (single cell sampled per slice; n = 7 wild-type and n = 6 ClockΔ19 mice). Whole-cell current-clamp recordings were conducted with a MultiClamp 700 amplified (Molecular Devices, Sunnyvale, CA, USA) on VTA neurons selected under differential interference contrast (DIC) optics. Dopamine cells were chosen based on their relatively larger size and characteristic electrophysiological properties, including the HCN channel-mediated “sag” current at hyperpolarizing current steps and spontaneous firing of 1–3 Hz (Supplementary Information). Following stable baseline stable baseline whole-cell recordings, resveratrol (SIRT1 activator; 50 µM dissolved in 0.1% DMSO) was bath applied for ~20 min. One cell was sampled per slice. pClamp 10 software (Molecular Devices) was used for recordings and subsequent analyses. Spikes were counted manually and rheobase was measured as the minimum current required to elicit action potentials.
HPLC to measure catecholamine and metabolites
Mouse brains were sectioned and microdissected for PFC, NAc, dSTR, and VTA, then frozen on dry ice (n = 6 per genotype per ZT). Microdissected punches were homogenized. Supernatant was removed for biogenic monoamines analyses using HPLC. Using the HPLC solvent, the following biogenic amines elute in the following order: DOPAC, dopamine, and HVA. HPLC control and data acquisition are managed by Empower software. HPLC assays were conducted by the Neurochemistry Core at the Vanderbilt Brain Institute.
Stereotaxic surgery
Mice underwent stereotaxic brain surgery to bilaterally inject into the VTA (relative to bregma: angle 7°; AP, −3.2; ML, ±1.0; DV, −4.6), high titer viruses (1 µl) encoding either AAV5 GFP-Scrambled or GFP-mutant CREB (mCREB), or bicistronic (GFP driven by cytomegalovirus promoter and SIRT1 driven by IE4/5 promoter) p1005 herpes simplex virus (HSV) vectors with GFP alone or GFP with SIRT1 (SIRT-OX) [34] (Supplementary Methods). Mice injected with AAV5 constructs recovered for 3–4 weeks, while mice injected with HSV constructs recovered for 2–3 days prior to behavioral testing to allow for maximal viral expression.
Immunohistochemistry
Mice were deeply anesthetized and then perfused (4% paraformaldehyde). Sections (30 μM) were processed for GFP, TH, and DAPI, then secondary flurophore antibodies (Supplementary Methods). Sections were imaged with an epifluorescent microscope (×4, ×10 and ×40). Mice were excluded if viral spread was not localized to the VTA, with viral spread through the injection tract, or with asymmetrical infection between hemispheres (~5% of mice).
Conditioned place preference (CPP)
Mice underwent cocaine place conditioning as described previously [13]. A biased conditioning protocol was used since most mice (>75%) preferred one chamber or the other during the pre-test (day 1). On days 2–5, mice were either injected with saline or cocaine (5 mg/kg, i.p.) then placed into the white or black chamber for 20 min. On day 6, mice were given no injections and allowed to freely explore the apparatus, and the amount of time spent for each of the three chambers was measured. Stereotaxic virally injected mice recovered prior to cocaine CPP (n = 10–11 per group). Mice were injected on days 2–5 exactly 30 min prior to saline or cocaine with resveratrol (20 mg/kg, i.p., dissolved in 5% hydroxypropyl β-cyclodextrin, Sigma-Aldrich, n = 9 per group). Mice were injected between ZT9-11 on days 1–5 (~12–16 h prior to CPP) with saline or the NAD+ precursor, NMN (β-Nicotinamide mononucleotide, 500 mg/kg, i.p., dissolved in saline, Sigma-Aldrich, n = 9–10 per group). The CPP score was calculated as the amount of time spent within the conditioned chamber on the test day subtracted from the amount of time spent on the same side on the pre-test day.
Statistics
Sample sizes for each of the experiments were based on previous studies and/or power calculations from unpublished pilot experiments. Animals were assigned randomly to treatment groups to balance age, cage, and littermates across genotypes, where applicable. Diurnal variation of gene, protein, and NAD+ expression was analyzed using CircWave v1.4 software (courtesy of Dr. Roelof Hut, www.euclock.org). Sin–cosine regression was constrained to a period of 24 h, single harmonic, with significance at α = 0.05. The center of gravity for each fitted waveform was used to estimate acrophase, or peak, of diurnal variation. Unpaired Student’s t test was used to test significance between genotypes where noted. One-way ANOVA followed by Tukey’s multiple comparisons tests were used to test differences between groups for in vitro experiments where noted. Two-way ANOVA were used to examine main effects of genotype, treatment, condition, or time, and interactions for in vitro and in vivo experiments. Significant interactions (genotype × treatment or genotype × time) were followed by post hoc tests (Tukey’s, Dunnett’s, or Sidak’s, where appropriate). Data are expressed as mean ± SEM with a two-tailed α = 0.05 considered statistically significant. Data were processed using Microsoft Excel, CircWave, ImageJ, and GraphPad Prism software.
Results
The molecular clock regulates TH expression and DA levels in mouse midbrain and striatum
Mice with the ClockΔ19 mutation display disrupted DA rhythms [18, 76, 82], and elevated levels of TH [76], suggesting a link between CLOCK and DA synthesis. The Δ19 mutation renders a dominant-negative protein unable to promote gene transcription [83, 84]. TH is diurnally expressed with a peak (acrophase) during the night (or active) phase of light–dark (LD) cycle (Fig. 1a), which is increased during the day and early night with a shift in acrophase in ClockΔ19 mice with a disrupted molecular clock (Fig. 1a and Figure S1C–F, I). Protein expression of TH and the phosphorylated form (P-THser40) are increased during the day and night (Figure S1A, B), suggesting activated TH may be constitutively elevated in ClockΔ19.

CLOCK regulates the diurnal expression of TH in the VTA. a TH expression is significantly elevated during the day in the VTA of ClockΔ19 mice (ZT × genotype, F5,60 = 3.95, p < 0.01; Sidak’s post hoc tests, **p < 0.01). Peak expression is shifted earlier during the active phase (t = 6.4, p < 0.0001). Sine wave fit (p < 0.05) with multi-harmonic regression assuming 24 h period are superimposed on mean ± SEM (Wt, black; ClockΔ19, blue). n = 6 mice per ZT per genotype. b Mouse TH promoter with canonical-binding sites for CLOCK (E-boxes; white) and CREB (CRE; black) proximal to the ATG start site (arrowhead) and core promoter (gray box). c–e Diurnal variation of P-CREB, CLOCK, and SIRT1 binding to the proximal promoter site. c P-CREB-binding peaks during the night in Wt mice and remains constitutively elevated in ClockΔ19 mice (ZT, F1,16 = 6.5, #p = 0.02 and genotype, F1,16 = 13.12, p = 0.002). d CLOCK-binding peaks during the day in Wt mice (antiphase to P-CREB), with significantly reduced binding in ClockΔ19 mice (ZT × genotype, F1,16 = 32.84, p < 0.0001; Tukey’s post hoc tests, ****p < 0.0001, a Wt ZT4 vs. ZT16, p < 0.0001). e SIRT1-binding peaks during the day, similarly to CLOCK in Wt mice with no detectable binding in ClockΔ19 mice (ZT × genotype, F1,16 = 8.75, p < 0.01; Tukey’s post hoc tests, **p < 0.01, a Wt ZT4 vs. ZT16, p = 0.01). Red dotted line marks IgG background level. n = 6 mice per ZT per genotype. f CREB expression is unchanged in the VTA of ClockΔ19 mice. g P-CREB expression is reduced in the VTA of ClockΔ19 mice (ZT × genotype, F1,12 = 10.54, p < 0.01; Tukey’s post hoc tests, a ClockΔ19 ZT4 vs. ZT16, p = 0.02). h SIRT1 expression varies across day and night in Wt mice with reduced expression during the day in ClockΔ19 mice (ZT × genotype, F1,12 = 16.65, p < 0.01; Tukey’s post hoc, *p < 0.05, a ClockΔ19 ZT4 vs. ZT16, p < 0.01). i-l Brain regions were collected at ZT8-10 when TH expression is nearing the peak in ClockΔ19 mice then levels of DA and metabolites were measured using HPLC. Levels of DA (t = 13.33, ****p < 0.0001), DOPAC (t = 4.6, ***p < 0.001), and HVA (t = 5.96, ***p < 0.001), are increased, with unchanged DOPAC/DA ratio and reduced HVA/DA ratios (t = 6.25, ****p < 0.0001) in the dSTR of ClockΔ19 mice (i). In addition, DA levels are reduced in the mPFC of ClockΔ19 mice (t = 2.52, *p < 0.05), and DOPAC (t = 6.98, ****p < 0.0001), HVA (t = 5.1, ***p < 0.001), and DOPAC/DA (t = 6.13, ***p < 0.001) and HVA/DA (t = 3.58, **p < 0.01) ratios are also increased (j). Levels of DA (t = 5.01), DOPAC (t = 3.13), and HVA (t = 2.65) are increased in the VTA, with reduced DOPAC/DA (t = 2.23) and HVA/DA (t = 2.39) ratios (k). DA (t = 4.1), DOPAC (t = 3.31), and HVA (t = 3.4) are in the NAc of ClockΔ19 mice, with no changes in ratios (l). *p < 0.05, **p < 0.01, ***p < 0.001. All data are represented as mean ± SEM. n = 6 mice per ZT per genotype
The mouse TH promoter contains two canonical E-boxes (CACGTG)—~1341 bp and ~233 bp upstream from the TSS referred to as distal and proximal, respectively (Fig. 1b). The proximal E-box is located near the CRE site (~79 bp to TSS), where CREB binds to activate transcription [85, 86]. The proximity of these sites to the TSS, and to one another, suggest diurnal-dependent regulation by CLOCK and CREB. Chromatin immunoprecipitation (ChIP) assays reveals antiphasic P-CREB and CLOCK binding at the proximal E-box of TH promoter, whereby P-CREB and CLOCK preferentially bind during the night and day, respectively (Fig. 1c, d). CLOCK peaks at ZT4 (also at distal E-box, Figure S2C) when TH troughs, and P-CREB peaks at ZT16 when TH peaks (no binding of either P-CREB or SIRT1 at the distal promoter, Figure S2A, B; CLOCK IP verified using Clock knockout mice, Figure S2D). SIRT1 also binds with a diurnal pattern at the proximal promoter, peaking during the day (Fig. 1e).
In ClockΔ19 mice, diurnal binding at the proximal promoter of P-CREB, CLOCK, and SIRT1 is lost (Fig. 1c–e). SIRT1 occupancy is almost undetectable (Fig. 1e), potentially due to reduced SIRT1 at ZT4 (Fig. 1h). There are no diurnal differences in CREB (Fig. 1f), although P-CREB expression appears to increase at ZT4 and decrease at ZT16 (Fig. 1g), while SIRT1 shows the opposite pattern of expression in ClockΔ19 mice (Fig. 1h), suggesting disrupted diurnal variation of expression. Diurnal variation of Sirt1 gene (Figure S1H) and SIRT1 protein (Fig. 1h) are lost in ClockΔ19 mice, and the rhythm of the CLOCK target gene Nampt is completely reversed (Figure S1G, I), further indicative of disrupted SIRT1 signaling. Given the elevated TH levels during the day in ClockΔ19 mice and the diurnal patterns of promoter binding of these transcription factors, CLOCK may repress the transcriptional activity of TH at specific times of day when SIRT1 is also present.
As expected, levels of DA and the metabolites 3-4-Dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) of the VTA and areas downstream, the dSTR and mPFC, are altered in ClockΔ19 mice (Fig. 1i–l). DA levels are elevated in the dSTR (Fig. 1i), VTA (Fig. 1k), and NAc (Fig. 1l), but reduced in the mPFC (Fig. 1j), with higher DOPAC/DA and HVA/DA ratios in the VTA and lower ratios in the dSTR in ClockΔ19 mice. These findings indicate a dominant-negative CLOCK protein consequently impacts TH transcription, and levels of DA and metabolites, in mesocorticolimbic pathways, including the dSTR, mPFC, NAc, and VTA.
CLOCK represses CREB-induced TH transcription by interacting with SIRT1
We designed TH promoter constructs expressing either the distal and/or proximal regions containing the E-box and CRE sites driving a luciferase reporter (TH:luc). Cells were transfected with any of the following mouse expression constructs: CREB, CLOCK, CLOCK∆19, and/or SIRT1, and TH:luc. As expected, CREB reliably induces TH:luc activity (Fig. 2a, PC12 and Fig. 2f, MEFs). In PC12 cells, CREB-induced TH:luc activity is completely blocked by CLOCK, but not CLOCK∆19 (Fig. 2a). Mutation of the proximal E-box (CACGTG to CCCGGG), where CLOCK preferentially binds during the day, prevents CLOCK-mediated antagonism of CREB-induced activity (Fig. 2b). Similar effects are observed following mutation of the distal E-box (Figure S3A).

CLOCK, SIRT1, and metabolic cofactors modulate the transcriptional activity of TH. a PC12 cells were transfected and TH:luc promoter activity was measured. CREB induces activity of TH:luc, which is prevented by co-transfection with CLOCK, while CLOCKΔ19 fails to do so (one-way ANOVA, F4,30 = 35.1, p < 0.0001; Tukey’s post hoc tests, ****p < 0.0001). Amount of CLOCK plasmid transfected (small circle, 1 μg; large circle, 10 μg). n = 8–15 per condition, data represent an average across triplicate wells and multiple plates. b Mutation of the proximal E-box site of the TH promoter prevents the ability of CLOCK to repress transcriptional activity (one-way ANOVA, F7,24 = 250.3, p < 0.0001; Tukey’s post hoc tests, a TH vs. TH/CREB/CLOCK p < 0.05, ****p < 0.0001). n = 4 per condition, data represent an average across triplicate wells and multiple plates. c siRNA-mediated knockdown of SIRT1 prevents the ability of CLOCK to repress transcriptional activity, while SIRT1 further represses activity when co-transfected with CLOCK (one-way ANOVA, F3,32 = 3503, p < 0.0001; Tukey’s post hoc tests, *p < 0.05, ***p < 0.001, ****p < 0.0001). n = 4 per condition, data represent an average across triplicate wells and multiple plates. d SIRT1 restores the ability of CLOCKΔ19 to repress transcriptional activity (one-way ANOVA, F2,9 = 76.5, p < 0.0001; Tukey’s post hoc tests, **p < 0.01, ***p < 0.001). n = 4 per condition, data represent an average across triplicate wells and multiple plates. e TH:luc activity was measured in Wt, SIRT1 KO, and CLOCK KO MEFs. SIRT1 is required for CLOCK to prevent CREB-induced transcriptional activity (genotype × condition, F6,40 = 3503, p < 0.0001). CLOCK represses CREB-induced transcriptional activity in Wt (a TH vs. TH/CREB, p < 0.01; b TH/CREB vs. TH/CREB/CLOCK, p < 0.0001) and CLOCK KO (c TH/CREB vs. TH/CREB/CLOCK, p < 0.0001) MEFs, but fails to do so in SIRT1 KO MEFs. Co-transfection of CLOCK and SIRT1 in CLOCK KO MEFs prevents CREB-induced transcriptional activity (d TH/CREB/CLOCK vs. TH/CREB/CLOCK/SIRT1 p < 0.001). n = 4–5 per condition, data represent an average across triplicate wells and multiple plates. f CLOCK KO MEFs were transfected with either partial domains or full SIRT1: 1) GEX vector backbone control; 2) N terminus (required for CLOCK interaction [24], or 3) catalytic domain. Full SIRT1 and partial N terminus SIRT1 prevents CREB-induced TH:luc activity, with no effect of partial catalytic SIRT1 (one-way ANOVA, F4,20 = 68.14, p < 0.0001; Tukey’s post hoc tests, ***p < 0.001). n = 4–6 per condition, data represent an average across triplicate wells and multiple plates. Transfected construct (+). All data are represented as mean ± SEM. g PC12 cells were treated with NAD+ (1 μM), harvested 72 h later then measured TH:luc activity. CLOCK and NAD+ prevent CREB-induced TH:luc activity (one-way ANOVA, F8,27 = 16.23, p < 0.0001; Tukey’s post hoc tests, **p < 0.01, ****p < 0.0001). n = 4 per condition, data represent an average across triplicate wells and multiple plates. h PC12 cells were treated with NAM (10 mM), harvested 72 h later then measured TH:luc activity. NAM prevents CLOCK-mediated repression of TH:luc activity (one-way ANOVA, F8,27 = 16.23, p < 0.0001; Tukey’s post hoc tests, ****p < 0.0001). n = 4 per condition, data represent an average across triplicate wells and multiple plates. i PC12 cells were treated with varying concentrations of NAD+, harvested 48 h later then measured TH:luc activity. NAD+ further reduces TH:luc activity regardless of dose relative to CLOCK (one-way ANOVA, F6,21 = 118.1, p < 0.0001; Tukey’s post hoc tests, ****p < 0.0001, a TH vs. TH/CREB, p < 0.0001; b TH/CREB vs. TH/CREB/CLOCK, p < 0.05; c TH/CREB/CLOCK vs. TH/CREB/CLOCK + NAD+, p < 0.05). n = 4 per condition, data represent an average across triplicate wells and multiple plates. j siRNA-mediated knockdown of SIRT1 leads to increased TH:luc activity in the presence of NAD+ (one-way ANOVA, F5,18 = 67.56, p < 0.0001; Tukey’s post hoc tests, a TH/CREB/CLOCK/SIRT1siRNA vs. TH/CREB/CLOCK/SIRT1siRNA + NAD+ (100 μM), p < 0.05, b TH/CREB/CLOCK/SIRT1siRNA vs. TH/CREB/CLOCK/SIRT1siRNA + NAD+ (2000 μM), p < 0.05. n = 4 per condition, data represent an average across triplicate wells among multiple plates. n = 4 per condition, data represent an average across triplicate wells and multiple plates. k PC12 cells were transfected, treated with NMN (200 μM), then harvested after 72 h to measure TH:luc activity. One-way ANOVA followed by Tukey’s multiple comparisons tests, ****p < 0.0001. a TH/CREB + NMN vs. TH/CREB/SIRT1 + NMN, p < 0.0001. n = 6 per condition, data represent an average across triplicate wells among multiple plates. n = 6 per condition, data represent an average across triplicate wells and multiple plates. l Transfected Wt and SIRT1 KO MEFs were treated with NMN (400 or 800 μM), then harvested 48–72 h later to TH:luc activity. The repressive effects of NMN on TH:luc activity depended on SIRT1 and CLOCK (genotype × condition, F4,30 = 50.4, p < 0.0001, Tukey’s post hoc tests, **p < 0.01, ****p < 0.0001, a condition vs. TH/CREB Wt MEFs, p < 0.0001; b condition vs. TH/CREB SIRT1 KO MEFs, p < 0.05. n = 4 per condition, data represent an average across triplicate wells and multiple plates. Transfected construct (+). All data are represented as mean ± SEM
Based on our findings (see Fig. 1) and those from liver and other tissues [24, 25], we investigated whether CLOCK represses transcriptional activity via the recruitment of SIRT1 (Figure S4). CLOCK transfected with SIRT1 further reduces TH:luc activity, whereas siRNA-mediated knockdown of Sirt1 prevents the effects of CLOCK on CREB-induced transcriptional activity (Fig. 2c). SIRT1 restored the repressive effects of CLOCK∆19 (Fig. 2d). We then used Wt, CLOCK KO, and SIRT1 KO MEFs to investigate whether CLOCK and/or SIRT1 are necessary for reducing transcriptional activity. CLOCK fails to reduce activity in SIRT1 KO MEFs, yet when CLOCK and SIRT1 are co-transfected, activity is reduced (Fig. 2e). The interaction between CLOCK and SIRT1 requires the N terminus region of SIRT1 (aa1–231) [24]. In CLOCK KO MEFs, constructs expressing the catalytic domain or the C terminus of SIRT1 has no effect on TH:luc activity, while the N terminus region is sufficient for recapitulating SIRT1-mediated repression (Fig. 2f).
Cellular metabolic cofactors of SIRT1 modulate TH gene transcriptional activity
NAD+ and intermediate enzymes of the NAD+ salvage pathway controls the activity of SIRT1 (Figure S4) [24, 26, 87, 88]. NAD+ (1 μM) blocks CREB-induced TH:luc activity (Fig. 2g), consistent with the role of SIRT1 from our findings. Indeed, SIRT1 knockdown attenuates the effects of NAD+ on TH:luc activity (Fig. 2j). NAM is a byproduct of NAD+ metabolism and part of the biosynthesis pathway regulating SIRT1 [89]. As expected, CLOCK antagonism of CREB-induced TH:luc activity is prevented by NAM (Fig. 2h and Figure S3A). The precursor NMN is necessary for NAD+ biosynthesis and NAD+-dependent SIRT1 activity (Figure S4) [90, 91]. Similar to NAD+, NMN prevents CREB-induced TH:luc activity and further attenuates activity with CLOCK or SIRT1 (Fig. 2k). SIRT1 KO MEFs treated with NMN (400 or 800 μM) and transfected with CLOCK produces minimal changes on CREB-induced TH:luc activity unless SIRT1 is also present (Fig. 2l). NAD+ synthesis is controlled by the rate-limiting enzyme NAMPT. The NAMPT inhibitor FK866 (10 nM), which recapitulates the effects of SIRT1 inhibition via NAD+ depletion [24, 25, 92], attenuates the repressive effects of CLOCK, CLOCK/SIRT1, and CLOCKΔ19/SIRT1 on CREB-induced transcription (Figure S3C). These findings collectively demonstrate NAD+ bioavailability and biosynthesis contributes to the ability of CLOCK/SIRT1 to reduce the transcriptional potential of TH, thus linking CLOCK activity to the redox state. To further demonstrate these mechanisms are important for endogenous TH regulation in neural cells, we transfected mouse N2A cells. Similar to our findings in PC12 and MEF cells, CLOCK alone has no effect on basal TH:luc activity (Fig. 3a), while CLOCK or SIRT1 prevents CREB-induced activity, with CLOCK∆19 having no effect (Fig. 3b).

CLOCK and SIRT1 reduce TH expression in neural cells and in vivo. a Mouse N2A cells transfected with TH:luc and CLOCK has no effect on bioluminescence activity. b CLOCK and SIRT1 block CREB-induced TH:luc activity in mouse N2A cells (one-way ANOVA, F4,25 = 279.7, p < 0.0001; Tukey’s post hoc tests, ****p < 0.0001). n = 6 per condition, data represent an average across triplicate wells and multiple plates. c Separate cohorts of mice were injected with HSV-SIRT1 (or GFP, 2–3 days of expression) or AAV5-mCREB (or GFP, 3–4 weeks of expression) into the VTA. Triple-labeling immunohistochemistry shows robust viral expression in the VTA (green, GFP; red, TH; and blue, DAPI). d Resveratrol reduces TH expression in the VTA of Wt and ClockΔ19 mice (genotype, F1,20 = 25.67, p < 0.0001, treatment, F1,20 = 42.09, p < 0.0001, and genotype × treatment, F1,20 = 4.8, p = 0.04; Tukey’s post hoc tests, *p < 0.05, ****p < 0.0001, #VEH Wt vs. VEH ClockΔ19, p < 0.01). e Overexpression of SIRT1 reduces TH expression in the VTA of Wt and ClockΔ19 mice (genotype, F1,20 = 29.44, p < 0.0001, treatment, F1,20 = 213.4, p < 0.0001, and genotype × treatment, F1,20 = 40.88, p < 0.0001; Tukey’s post hoc tests, ****p < 0.0001, #VEH Wt vs. VEH ClockΔ19, p < 0.0001). f mCREB reduces TH expression only in ClockΔ19 mice (genotype × virus, F1,20 = 37.05, p < 0.0001; Tukey’s post hoc tests, *p < 0.05, ****p < 0.0001, #AAV5-GFP Wt vs. ClockΔ19, p = 0.004)
Mice treated with resveratrol (20 mg/kg, daily i.p., 4 days), a pharmacological activator of SIRT1 [93], display reduced TH expression in the VTA (Fig. 3d). In vivo manipulation of SIRT1 (HSV-SIRT1) specifically within the VTA of Wt and Clock∆19 mice (Fig. 3c) also reduces TH expression (Fig. 3e), whereas the effects of mCREB in VTA are genotype dependent (Fig. 3f). Thus, SIRT1 represses TH expression in vivo, while CREB may differentially regulate transcription depending on a functional CLOCK protein, and these results suggest overactive CREB activity in ClockΔ19 mice (Supplementary Table 2).
The effects of SIRT1 activation on VTA DA physiology and striatal DA levels depend on a functional CLOCK protein
We performed whole-cell current-clamp recordings from VTA DA cells (Figure S5). Using whole-cell recordings, slices were treated with vehicle (DMSO) followed by resveratrol. Resveratrol increases and decreases spike frequency in Wt and ClockΔ19 mice, respectively (Fig. 4a, c, d). VTA DA neurons are more depolarized in ClockΔ19 mice, indicating enhanced excitability (Fig. 4b); consistent with our previous findings, reporting increased spontaneous firing of DA neurons in slice and in vivo [76, 82], which is reduced by resveratrol (Fig. 4e, f). Consistent with our physiological findings, resveratrol increases DA levels in the NAc and dSTR of Wt mice, and reduces DA and DOPAC only in the NAc of ClockΔ19 mice (Fig. 4g, h and Figure S6), with no effects in the mPFC (Figure S7). Resveratrol modulates DA cell firing depending on a functional molecular clock and basal DAergic tone from the VTA—i.e., resveratrol enhances DA signaling in Wt mice with low to normal tone and reduces DA in ClockΔ19 mice with high tone (Fig. 4i).

Resveratrol modulation of DA cell excitability and striatal DA levels depends on a functional CLOCK protein. a Representative traces of action potentials during current injection of VTA DA neurons treated with DMSO or resveratrol from Wt and ClockΔ19 mice. Whole-cell traces from DMSO-treated slices are similar between genotypes (single trace pictured). b Resting membrane potential of VTA DA neurons in ClockΔ19 mice are depolarized, indicating enhanced intrinsic excitability (t = 2.2, p < 0.05). n = 7 cells per genotype. c Action potentials elicited by 200 pA injection into VTA DA neurons during DMSO or resveratrol (RES) application. RES increases and decreases spike frequency in Wt mice (t = 2.44, p < 0.05) and ClockΔ19 (t = 9.34, p < 0.001) mice, respectively. d RES increases and decreases spike frequency in Wt (t = 2.13, a p < 0.05) and ClockΔ19 (t = 7.1, b p < 0.001) mice relative to baseline. e Minimum current required to elicit an action potential indicates RES selectively (Wt, p = 0.11) enhances the rheobase (excitability) of VTA DA neurons from ClockΔ19 mice (t = 4.40, p < 0.01). f RES enhances the rheobase relative to baseline of VTA DA neurons from ClockΔ19 mice (t = 3.81, p < 0.01). g, h Levels of DA, DOPAC, and HVA were measured in the NAc and dSTR of Wt and ClockΔ19 mice at ZT8-10, when TH expression is approaching the peak in ClockΔ19 mice. Resveratrol increases DA in the NAc and dSTR of Wt mice and decreases DA in the NAc of ClockΔ19 mice ((g, left) NAc: genotype × treatment, F1,20 = 14.34, p < 0.01; Tukey’s post hoc tests, a Saline Wt vs. ClockΔ19 p < 0.001, *p < .05, ***p < 0.001; and (h, left) dSTR: genotype, F1,20 = 5.96, p < 0.05; Tukey’s post hoc tests, a Saline Wt vs. ClockΔ19 p < 0.05, *p < 0.05). Resveratrol decreases DOPAC in NAc of ClockΔ19 mice ((g, middle), genotype × treatment, F1,20 = 13.8, p < 0.01; Tukey’s post hoc tests, a Saline Wt vs. ClockΔ19 mice p < 0.05, *p < 0.05), with no overall effect on HVA ((g, right), genotype, F1,20 = 20.1, p < 0.001). All data are represented as mean ± SEM. i Schematic of diurnal regulation of TH by CLOCK/SIRT1 and CREB. During the day (inactive phase), CLOCK and SIRT1 bind to the proximal E-box within the TH promoter, when TH expression is low. Oscillating NAD+ (and NAM) may modulate the activity of SIRT1 and repression of transcriptional activity. During the night (active phase), CREB is recruited to the CRE site to promote the transcriptional activity of TH. In ClockΔ19 mice, recruitment of SIRT1 to the promoter is reduced potentially due to reduced expression during the day, while P-CREB binding is enhanced during the day and night. Elevated P-CREB activity constitutively increases TH protein expression in ClockΔ19 mice. CLOCK promoter binding during the day may prevent CREB from binding the CRE site. The truncated mutant CLOCK protein (missing 51 amino acids of the transactivational domain) or the inability to recruit other cofactors to the transcriptional complex permits CREB binding. Thus, CLOCK could block CREB from accessing the CRE site, suggesting steric hindrance
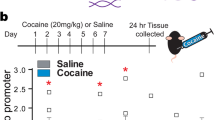
Diurnal variations of NAD+ and SIRT1 in VTA are affected by cocaine
To further extend these findings to the brain, we measured diurnal rhythms of NAD+ in the VTA using HPLC since NAD+ rhythms regulate SIRT1 in liver and other tissues. Intriguingly, NAD+ peaks during the middle of the day (Figure S8A), when SIRT1 binds the TH promoter (Fig. 1e); SIRT1 expression peaks (Figure S8B); and TH troughs (Fig. 1a and Figure S8E). Cocaine is known to alter redox state, SIRT1 activity, and the cellular response to DA [34, 35, 38, 44, 46, 94,95,96,97]. Thus, we investigated the effects of cocaine on diurnal rhythms of NAD+ and SIRT1 in the VTA. Acute cocaine (20 mg/kg, i.p., single injection) has no impact on NAD+; however, there is a complete loss of diurnal rhythm following chronic cocaine (20 mg/kg, i.p., 14 days) (Figure S8C and Figure S9A). SIRT1 diurnal rhythms are also lost following cocaine (Figure S8D), along with disruption in other circadian and metabolic genes (Figure S9) with minimal effects on CREB or P-CREB (Figure S9H, I). Acute cocaine completely flattens the diurnal rhythm of TH, which remains substantially dampened following chronic cocaine (Figure S8E). Diurnal rhythms of NAD+ and SIRT1 overlap, with peaks at the same times of day, and in antiphase to TH expression, which are altered by cocaine (Figure S8F).
The effects of SIRT1 activation on cocaine CPP depend on a functional CLOCK protein
Mice injected with AAV5-mCREB or HSV-SIRT1 into the VTA, or treated with resveratrol or NMN were subjected to cocaine CPP to determine whether these mechanisms are important for drug-conditioned reward (Fig. 5a). The effects of mCREB or SIRT1 activation led to opposite expression of TH in Wt and ClockΔ19 mice following cocaine CPP (Fig. 5b, resveratrol; Fig. 5c, NMN; Fig. 5d, SIRT1OX; and Fig. 5e, mCREB). Consistent with our previous findings [76], elevated cocaine CPP is robust in ClockΔ19 mice (Fig. 5f–i). Resveratrol (Fig. 5f), NMN (Fig. 5g), SIRT1 (Fig. 5h), or mCREB (Fig. 5i), almost completely prevents cocaine CPP in ClockΔ19 mice, yet enhances CPP in Wt mice, further supporting the differential regulation of VTA DAergic activity by NAD-dependent SIRT1 when DAergic cells are in a heightened or normal state. Taken together, our results demonstrate that CLOCK, SIRT1, and CREB are capable of bidirectionally modulating the in vivo transcriptional activity of TH and cocaine-conditioned reward (Supplementary Table 2).

In vivo regulation of TH expression and cocaine-conditioned place preference (CPP) by SIRT1 and CREB depends on a functional CLOCK protein. a Mice were administered resveratrol (red syringes; 20 mg/kg, i.p.) each conditioning day and saline (gray syringes) or cocaine (blue syringes; 5 mg/kg, i.p.) on alternating conditioning days. Another cohort of mice was administered NMN (brown syringes; 500 mg/kg, i.p.) at ZT9-11 on days 1–5. Separate cohorts of mice were injected with HSV-SIRT1 (or GFP, 2–3 days of expression) or AAV5-mCREB (or GFP, 3–4 weeks of expression) into the VTA prior to cocaine CPP. n = 9 per group RESV CPP; n = 10–11 per group SIRT1OX CPP; n = 10 per group mCREB CPP; and n = 9–10 per group NMN CPP. b–e Resveratrol, NMN, HSV-SIRT1, and AAV5-mCREB increases and decreases TH expression in Wt and ClockΔ19 mice, respectively (resveratrol, genotype, F1,20 = 4.78, p = 0.04 and genotype × treatment, F1,20 = 44.74, p < 0.0001; Tukey’s post hoc tests, a Vehicle Wt vs. ClockΔ19 p < 0.05, **p < 0.01, ***p < 0.001; NMN, genotype × treatment, F1,20 = 14.8, p < 0.001; Tukey’s post hoc tests, a Vehicle Wt vs. ClockΔ19 p < 0.05, *p < 0.05; HSV-SIRT1, genotype × virus, F1,20 = 25.75, p < 0.0001; Tukey’s post hoc tests, a HSV-GFP Wt vs. ClockΔ19 p < 0.05, **p < 0.01; and AAV5-mCREB, genotype, F1,20 = 14.48, p = 0.001, virus, F1,20 = 4.62, p = 0.04, and genotype × virus, F1,20 = 60.81, p < 0.0001; Tukey’s post hoc tests, a AAV5-GFP p < 0.05, **p < 0.001, ****p < 0.0001). f–i Resveratrol, NMN, HSV-SIRT1, and AAV5-GFP promote and attenuate cocaine CPP in Wt and ClockΔ19 mice, respectively (resveratrol, genotype × treatment, F1,32 = 20.28, p < 0.0001, Tukey’s post hoc tests, a Vehicle Wt and ClockΔ19 p < 0.05, *p < 0.05, **p < 0.01; NMN, genotype × treatment, F1, 34 = 27.34, p < 0.0001, Tukey’s post hoc tests, a Vehicle Wt vs. ClockΔ19 p < 0.01, *p < 0.05, ***p < 0.001; HSV-SIRT1, genotype × virus, F1,38 = 16.86, p = 0.0002; Tukey’s post hoc tests, a HSV-GFP Wt and ClockΔ19 p < 0.05, *p < 0.05; and AAV5-mCREB, genotype × virus, F1,36 = 10.4, p = 0.003; Tukey’s post hoc tests, a AAV5-GFP Wt vs. ClockΔ19, p < 0.01, *p < 0.05)
Discussion
Disruptions to the circadian system are linked to addiction and other psychiatric disorders. Metabolic oscillations are tightly coupled with the molecular clock [2, 25, 98]. Our results showing time-of-day-dependent expression of TH, transcriptional activity, and promoter occupancy of CLOCK, SIRT1, and CREB, together with our previous reports [14, 18, 74, 76, 77, 82], suggest circadian-dependent regulation of pathways involved in DA synthesis. Diurnal variation of TH transcription is regulated by time-of-day-dependent recruitment of CLOCK and SIRT1 to the proximal promoter in antiphase to CREB. During the day (inactive), CLOCK and SIRT1 reduce the transcriptional activity of TH, while CREB promotes transcription during the night (active). Furthermore, pharmacological activation of SIRT1 can modulate the excitability and firing of VTA DA neurons and DA levels downstream of the VTA. These mechanisms are disrupted by cocaine administration and this is important for the regulation of cocaine-conditioned reward. Together, our data support previous findings in peripheral tissues indicating coupling of metabolic and molecular clock pathways, which are important in the regulation of a number of physiological processes.
We establish here a functional role for NAD+-dependent regulation of SIRT1 signaling on CLOCK-mediated transcription of TH. NAM inhibits SIRT1 activity through the metabolizing of NAD+ [99,100,101,102]. Transcriptional activity of TH is attenuated by NAD+ and restored with NAM presumably by modulating SIRT1 activity, since these effects are less robust following SIRT1 knockdown or SIRT1 KO MEFs. The immediate precursor to NAD+, NMN, also reduces CREB-induced transcriptional activity, further supporting the role of NAD+ biosynthesis as a key regulator of SIRT1-mediated transcriptional repression.
The conversion of NAM to β-ΝΜΝ is catalyzed by the CLOCK target gene, NAMPT, positioning this enzyme as a pivotal factor coupling metabolic and circadian oscillators. The expression of Nampt and Sirt1 peaks near similar diurnal phases and antiphasic to SIRT1 and NAD+ levels, suggesting NAD+-dependent SIRT1 activity represses TH expression during the inactive phase. Daily rhythms of NAD+ directly modulates the diurnal activity of CLOCK/BMAL1/SIRT1 transcriptional complexes [24, 99]. Consistent with this, CLOCK and SIRT1 preferentially bind the TH promoter during the day when NAD+ peaks and TH expression is low. Moreover, the repression of transcriptional activity is dependent on the presence of the N terminus of SIRT1, which has previously been shown to be necessary for the interaction with CLOCK [87]. Together, these findings strongly suggest CLOCK-mediated repression of TH expression is dependent on NAD+ and the interaction with SIRT1.
Our findings suggest a functional CLOCK is able to antagonize CREB-mediated transcriptional activity of TH, where CLOCK promoter occupancy during the day, when TH expression is the lowest, prevents CREB occupancy. We previously reported significantly increased TH expression and altered DA synthesis in the VTA of ClockΔ19 mice [18, 76]. We extend these findings to demonstrate these changes in TH expression are likely due to impaired SIRT1 function or activity. While CLOCK∆19 protein is still capable of binding the promoter, SIRT1 binding (and expression) is markedly lower, potentially allowing for CREB recruitment and transcriptional activation. Another possibility is steric hindrance, where the CLOCK protein at the E-box site prevents CREB binding at certain times of day. Due to a splicing misread and the excision of exon 19, the mutant CLOCK protein is missing 51 amino acids of the transactivational domain [84]. In ClockΔ19 mice, CREB-induced transcriptional activity remains and P-CREB binding at the CRE site is constitutively enriched during day and night. Thus, the truncated CLOCK protein may permit CREB to bind and regulate TH transcription. Other genes are directly regulated by CLOCK/BMAL1 complexes and CREB, suggesting potential common mechanisms of circadian-dependent transcription at other gene promoters [103, 104]. Pleiotropic or “non-circadian” roles of these transcription factors may also be involved. The repression of transcriptional activity by CLOCK could be independent of circadian regulation, especially when considering the impact of cocaine on these mechanisms. For example, SIRT1 suppresses the transcriptional activity of TH under non-stimulated conditions, while overexpression or activation seems to increase TH expression if animals are also exposed to cocaine. Cocaine likely disrupts the typical action of these transcription factors or the activation of other cofactors and pathways. Interestingly, SIRT1 can both activate or inhibit transcription depending on the recruitment of additional cofactors or post-translational modifiers, and brain region [34, 87, 105].
The synthesis and release of DA is modulated by several pathways of the circadian system. For example, the dopamine transporter (DAT) has been shown to be necessary for diurnal variation of extracellular dopamine tone [16]. Moreover, midbrain TH expression seems to be also controlled by REV-ERBα and NURR1 to repress or promote transcription, respectively [15]. Interestingly, Rev-erbα and ClockΔ19 mutant mice are both hyper-DAergic and exhibit reduced anxiety-like and depressive-like behaviors [15, 18]. Our previous studies suggest altered DA synthesis and release in ClockΔ19 mice may be specific to the VTA and largely unaffected in the substantia nigra, suggesting complementary yet regionally distinct regulation of TH by molecular clock mechanisms, however, this has not been systematically investigated [15, 18]. CLOCK, in addition to other transcription factors, regulates the expression of Rev-erbα. Despite changes in liver of ClockΔ19 mice [106], we found diurnal expression of Rev-erbα was minimally affected, and may indicate these observed changes in TH of ClockΔ19 mice are independent of REV-ERBα.
The complete loss of CLOCK (e.g., CLOCK KO mice) may be compensated by NPAS2 [107,108,109,110]. NPAS2 and CLOCK independently dimerize with BMAL1 to maintain rhythms in various tissues [107, 111]. However, Clock∆19 mice gradually become arrhythmic in constant conditions [112,113,114], suggesting NPAS2 fails to compensate. Moreover, the CLOCK∆19 protein is capable of binding BMAL1, yet fails to recruit cofactors required for transcription [84], and leads to blunted molecular rhythms [115], consistent with our findings of flattened Per2 rhythms. In contrast, the diurnal variation of another CLOCK target gene Nampt was completely reversed in ClockΔ19 mice and TH expression appeared to phase-shifted with an overall increase in amplitude. Altered phase resetting, reduced circadian amplitudes, and phase shifts in ClockΔ19 mice have also been observed for other molecular and behavioral rhythms [116, 117]. Nevertheless, our results show ClockΔ19 mice have constitutively elevated activated TH in the VTA, further suggesting genetic disruption of the molecular clock disrupts DAergic transmission [18]. In addition, NPAS2 has little to no expression in the VTA [118], suggesting compensation by NPAS2 is unlikely to play a role in these studies.
Highly metabolic tissues, such as liver and brain, may rely on the coupling between metabolic and circadian oscillators in order to maintain homeostasis, energy utilization, and overall tissue health. An important cofactor with responsiveness to changes in cellular metabolism, mitochondrial biogenesis, and redox state is NAD+. NAD+ is almost completely depleted following DA stimulation, which is thought to be a consequence of the cells attempting to meet the energy demands of the mitochondria [43, 44]. Interestingly, chronic, but not acute, cocaine completely flattened the diurnal variations of NAD+ in the VTA, suggesting repeated administration of cocaine taxes mitochondrial function and impairs NAD+ signaling [119]. Chronic administration of drugs of abuse could impair neuronal metabolism, interfere with subsequent responses to stimuli, and promote oxidative stress.
The loss of rhythms of firing and release of DA from VTA neurons parallels loss of rhythms of locomotor activity, cocaine CPP, and self-administration in ClockΔ19 mice [14, 18, 74,75,76]. We replicate these findings of increased sensitivity to cocaine-conditioned reward in these mice, which may be due to elevated extracellular DA and DA turnover in the VTA and downstream striatal regions. As suggested, overactive CREB-mediated transcription of TH may be responsible for an overall increase of TH gene and protein expression in the VTA of ClockΔ19 mice. Indeed, VTA-specific viral overexpression of mutant CREB significantly reduces TH expression selectively in these mice, which is similar to overexpression of SIRT1, and pharmacological activation of SIRT1 by resveratrol. The reduction in TH expression corresponds with attenuated cocaine-conditioned reward. In addition, bath application of resveratrol to VTA slices from ClockΔ19 mice reduces the excitability and firing of DA neurons, further supporting the idea that SIRT1 activation antagonizes DA-driven hyperhedonia in ClockΔ19 mice. Other studies have shown that preventing SIRT1 in the NAc relieves stress-induced depressive-like behaviors [36] and attenuates drug self-administration [34, 35, 38]. Thus, targeting of SIRT1 or NAD+-dependent pathways may have therapeutic implications for those with comorbid mood, substance use, and metabolic disorders [120,121,122,123].
Repeated cocaine administration increases the expression of SIRT1 in the NAc, and SIRT1 activation either by overexpression or resveratrol activation can promote cocaine CPP and self-administration [34, 35, 38]. Similarly, systemically administered resveratrol, NMN, or VTA-specific overexpression of SIRT1 in wild-type mice increases cocaine CPP, potentially via enhanced DA neuronal excitability and extracellular DA in the NAc and dSTR. Expression of TH in the VTA is also increased following SIRT1 activation and cocaine administration in wild-type mice. These findings seem contrary to our initial results, demonstrating SIRT1 activation reduces the transcriptional activity of TH in vitro and in vivo. We speculate that suppressed TH expression following mCREB or SIRT1OX may lead to compensatory changes of DA neuronal activity or potentiated neural responses to cocaine. Cocaine may further perturb these molecular mechanisms which could underlie these discrepancies since cocaine altered the diurnal expression of TH, SIRT1, and NAD+. On the other hand, we observed the opposite direction of effects in ClockΔ19 mice, suggesting that when DA neuronal firing is constitutively elevated, mCREB or SIRT1 activation reduces DA activity and cocaine CPP. Future studies will need to further tease apart the mechanisms underlying these differences. Nevertheless, our findings collectively point toward NAD+-dependent SIRT1-mediated modulation of DA signaling involving CLOCK in the VTA.
Taken together, our results demonstrate diurnal variation of TH in the VTA is regulated by NAD+-dependent SIRT1 activity and CLOCK-mediated repression. These mechanisms may have important applications for substance use and implications for other psychiatric disorders. Targeting NAD+ or SIRT1 may be effective therapeutic approaches for comorbid mood and substance use disorders.
References
Cribbet MR, Logan RW, Edwards MD, Hanlon E, Bien Peek C, Stubblefield JJ, et al. Circadian rhythms and metabolism: from the brain to the gut and back again. Ann N Y Acad Sci. 2016;1385:21–40.
Eckel-Mahan K, Sassone-Corsi P. Metabolism and the circadian clock converge. Physiol Rev. 2013;93:107–35.
Vengeliene V, Cannella N, Takahashi T, Spanagel R. Metabolic shift of the kynurenine pathway impairs alcohol and cocaine seeking and relapse. Psychopharmacology. 2016;233:3449–59.
Barandas R, Landgraf D, McCarthy MJ, Welsh DK. Circadian clocks as modulators of metabolic comorbidity in psychiatric disorders. Curr Psychiatry Rep. 2015;17:98.
Kaidanovich-Beilin O, Cha DS, McIntyre RS. Crosstalk between metabolic and neuropsychiatric disorders. F1000 Biol Rep. 2012;4:14.
Karatsoreos IN. Links between circadian rhythms and psychiatric disease. Front Behav Neurosci. 2014;8:162.
Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–75.
Abarca C, Albrecht U, Spanagel R. Cocaine sensitization and reward are under the influence of circadian genes and rhythm. Proc Natl Acad Sci USA. 2002;99:9026–30.
Andretic R, Chaney S, Hirsh J. Requirement of circadian genes for cocaine sensitization in Drosophila. Science. 1999;285:1066–8.
Jansen HT, Sergeeva A, Stark G, Sorg BA. Circadian discrimination of reward: evidence for simultaneous yet separable food- and drug-entrained rhythms in the rat. Chronobiol Int. 2012;29:454–68.
Sleipness EP, Sorg BA, Jansen HT. Diurnal differences in dopamine transporter and tyrosine hydroxylase levels in rat brain: dependence on the suprachiasmatic nucleus. Brain Res. 2007;1129:34–42.
Halbout B, Perreau-Lenz S, Dixon CI, Stephens DN, Spanagel R. Per1(Brdm1) mice self-administer cocaine and reinstate cocaine-seeking behaviour following extinction. Behav Pharmacol. 2011;22:76–80.
Ozburn AR, Falcon E, Twaddle A, Nugent AL, Gillman AG, Spencer SM, et al. Direct regulation of diurnal Drd3 expression and cocaine reward by NPAS2. Biol Psychiatry. 2015;77:425–33.
Ozburn AR, Larson EB, Self DW, McClung CA. Cocaine self-administration behaviors in ClockDelta19 mice. Psychopharmacology. 2012;223:169–77.
Chung S, Lee EJ, Yun S, Choe HK, Park SB, Son HJ, et al. Impact of circadian nuclear receptor REV-ERBalpha on midbrain dopamine production and mood regulation. Cell. 2014;157:858–68.
Ferris MJ, Espana RA, Locke JL, Konstantopoulos JK, Rose JH, Chen R, et al. Dopamine transporters govern diurnal variation in extracellular dopamine tone. Proc Natl Acad Sci USA. 2014;111:E2751–2759.
Shumay E, Fowler JS, Wang GJ, Logan J, Alia-Klein N, Goldstein RZ, et al. Repeat variation in the human PER2 gene as a new genetic marker associated with cocaine addiction and brain dopamine D2 receptor availability. Transl Psychiatry. 2012;2:e86.
Sidor MM, Spencer SM, Dzirasa K, Parekh PK, Tye KM, Warden MR, et al. Daytime spikes in dopaminergic activity drive rapid mood-cycling in mice. Mol Psychiatry. 2015;20:1406–19.
Sorg BA, Chen SY, Kalivas PW. Time course of tyrosine hydroxylase expression after behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1993;266:424–30.
Spanagel R, Pendyala G, Abarca C, Zghoul T, Sanchis-Segura C, Magnone MC, et al. The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nat Med. 2005;11:35–42.
Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18:164–79.
Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci USA. 2014;111:16219–24.
Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–28.
Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–40.
Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–7.
Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+biosynthesis. Science. 2009;324:651–4.
Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223.
Hirrlinger J, Dringen R. The cytosolic redox state of astrocytes: maintenance, regulation and functional implications for metabolite trafficking. Brain Res Rev. 2010;63:177–88.
Ying W. NAD+ and NADH in brain functions, brain diseases and brain aging. Front Biosci. 2007;12:1863–88.
Henley J, Poo MM. Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 2004;14:320–30.
Konur S, Ghosh A. Calcium signaling and the control of dendritic development. Neuron. 2005;46:401–5.
Liang M, Yin XL, Wang LY, Yin WH, Song NY, Shi HB, et al. NAD+ attenuates bilirubin-induced hyperexcitation in the ventral cochlear nucleus by inhibiting excitatory neurotransmission and neuronal excitability. Front Cell Neurosci. 2017;11:21.
Tamsett TJ, Picchione KE, Bhattacharjee A. NAD+ activates KNa channels in dorsal root ganglion neurons. J Neurosci. 2009;29:5127–34.
Ferguson D, Koo JW, Feng J, Heller E, Rabkin J, Heshmati M, et al. Essential role of SIRT1 signaling in the nucleus accumbens in cocaine and morphine action. J Neurosci. 2013;33:16088–98.
Ferguson D, Shao N, Heller E, Feng J, Neve R, Kim HD, et al. SIRT1-FOXO3a regulate cocaine actions in the nucleus accumbens. J Neurosci. 2015;35:3100–11.
Kim HD, Hesterman J, Call T, Magazu S, Keeley E, Armenta K, et al. SIRT1 mediates depression-like behaviors in the nucleus accumbens. J Neurosci. 2016;36:8441–52.
Libert S, Pointer K, Bell EL, Das A, Cohen DE, Asara JM, et al. SIRT1 activates MAO-A in the brain to mediate anxiety and exploratory drive. Cell. 2011;147:1459–72.
Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE 3rd, Maze I, et al. Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron. 2009;62:335–48.
Aksenov MY, Aksenova MV, Nath A, Ray PD, Mactutus CF, Booze RM. Cocaine-mediated enhancement of Tat toxicity in rat hippocampal cell cultures: the role of oxidative stress and D1 dopamine receptor. Neurotoxicology. 2006;27:217–28.
Lopez-Pedrajas R, Ramirez-Lamelas DT, Muriach B, Sanchez-Villarejo MV, Almansa I, Vidal-Gil L, et al. Cocaine promotes oxidative stress and microglial-macrophage activation in rat cerebellum. Front Cell Neurosci. 2015;9:279.
Muriach M, Lopez-Pedrajas R, Barcia JM, Sanchez-Villarejo MV, Almansa I, Romero FJ. Cocaine causes memory and learning impairments in rats: involvement of nuclear factor kappa B and oxidative stress, and prevention by topiramate. J Neurochem. 2010;114:675–84.
Pomierny-Chamiolo L, Moniczewski A, Wydra K, Suder A, Filip M. Oxidative stress biomarkers in some rat brain structures and peripheral organs underwent cocaine. Neurotox Res. 2013;23:92–102.
Requardt RP, Wilhelm F, Rillich J, Winkler U, Hirrlinger J. The biphasic NAD(P)H fluorescence response of astrocytes to dopamine reflects the metabolic actions of oxidative phosphorylation and glycolysis. J Neurochem. 2010;115:483–92.
Requardt RP, Hirrlinger PG, Wilhelm F, Winkler U, Besser S, Hirrlinger J. Ca(2)(+) signals of astrocytes are modulated by the NAD(+)/NADH redox state. J Neurochem. 2012;120:1014–25.
Sordi AO, Pechansky F, Kessler FH, Kapczinski F, Pfaffenseller B, Gubert C, et al. Oxidative stress and BDNF as possible markers for the severity of crack cocaine use in early withdrawal. Psychopharmacology. 2014;231:4031–9.
Uys JD, Knackstedt L, Hurt P, Tew KD, Manevich Y, Hutchens S, et al. Cocaine-induced adaptations in cellular redox balance contributes to enduring behavioral plasticity. Neuropsychopharmacology. 2011;36:2551–60.
Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–32.
Nutt DJ, Lingford-Hughes A, Erritzoe D, Stokes PR. The dopamine theory of addiction: 40 years of highs and lows. Nat Rev Neurosci. 2015;16:305–12.
Volkow ND. Imaging the addicted brain: from molecules to behavior. J Nucl Med. 2004;45:13N–16N. 19N-20N, 22N passim
Volkow ND, Morales M. The brain on drugs: from reward to addiction. Cell. 2015;162:712–25.
Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577(Pt 3):907–24.
Nair-Roberts RG, Chatelain-Badie SD, Benson E, White-Cooper H, Bolam JP, Ungless MA. Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience. 2008;152:1024–31.
Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull. 1982;9:321–53.
Kabanova A, Pabst M, Lorkowski M, Braganza O, Boehlen A, Nikbakht N, et al. Function and developmental origin of a mesocortical inhibitory circuit. Nat Neurosci. 2015;18:872–82.
Morales M, Margolis EB. Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci. 2017;18:73–85.
Zhang JT, Ma SS, Yip SW, Wang LJ, Chen C, Yan CG, et al. Decreased functional connectivity between ventral tegmental area and nucleus accumbens in Internet gaming disorder: evidence from resting state functional magnetic resonance imaging. Behav Brain Funct. 2015;11:37.
Sleipness EP, Jansen HT, Schenk JO, Sorg BA. Time-of-day differences in dopamine clearance in the rat medial prefrontal cortex and nucleus accumbens. Synapse. 2008;62:877–85.
Sleipness EP, Sorg BA, Jansen HT. Time of day alters long-term sensitization to cocaine in rats. Brain Res. 2005;1065:132–7.
Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–63.
Nestler EJ, Carlezon WA Jr.. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–9.
Logan RW, McClung CA. Animal models of bipolar mania: the past, present and future. Neuroscience. 2016;321:163–88.
Biguet NF, Buda M, Lamouroux A, Samolyk D, Mallet J. Time course of the changes of TH mRNA in rat brain and adrenal medulla after a single injection of reserpine. EMBO J. 1986;5:287–91.
Fossom LH, Carlson CD, Tank AW. Stimulation of tyrosine hydroxylase gene transcription rate by nicotine in rat adrenal medulla. Mol Pharmacol. 1991;40:193–202.
Tank AW, Lewis EJ, Chikaraishi DM, Weiner N. Elevation of RNA coding for tyrosine hydroxylase in rat adrenal gland by reserpine treatment and exposure to cold. J Neurochem. 1985;45:1030–3.
Cambi F, Fung B, Chikaraishi D. 5’ flanking DNA sequences direct cell-specific expression of rat tyrosine hydroxylase. J Neurochem. 1989;53:1656–9.
Iwata N, Kobayashi K, Sasaoka T, Hidaka H, Nagatsu T. Structure of the mouse tyrosine hydroxylase gene. Biochem Biophys Res Commun. 1992;182:348–54.
Kobayashi K, Kaneda N, Ichinose H, Kishi F, Nakazawa A, Kurosawa Y, et al. Structure of the human tyrosine hydroxylase gene: alternative splicing from a single gene accounts for generation of four mRNA types. J Biochem. 1988;103:907–12.
Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–9.
Kumaki Y, Ukai-Tadenuma M, Uno KD, Nishio J, Masumoto KH, Nagano M, et al. Analysis and synthesis of high-amplitude Cis-elements in the mammalian circadian clock. Proc Natl Acad Sci USA. 2008;105:14946–51.
Yoo SH, Ko CH, Lowrey PL, Buhr ED, Song EJ, Chang S, et al. A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proc Natl Acad Sci USA. 2005;102:2608–13.
Yoshitane H, Ozaki H, Terajima H, Du NH, Suzuki Y, Fujimori T, et al. CLOCK-controlled polyphonic regulation of circadian rhythms through canonical and noncanonical E-boxes. Mol Cell Biol. 2014;34:1776–87.
Lazaroff M, Patankar S, Yoon SO, Chikaraishi DM. The cyclic AMP response element directs tyrosine hydroxylase expression in catecholaminergic central and peripheral nervous system cell lines from transgenic mice. J Biol Chem. 1995;270:21579–89.
Olson VG, Zabetian CP, Bolanos CA, Edwards S, Barrot M, Eisch AJ, et al. Regulation of drug reward by cAMP response element-binding protein: evidence for two functionally distinct subregions of the ventral tegmental area. J Neurosci. 2005;25:5553–62.
Roybal K, Theobold D, Graham A, DiNieri JA, Russo SJ, Krishnan V, et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci USA. 2007;104:6406–11.
Ozburn AR, Falcon E, Mukherjee S, Gillman A, Arey R, Spencer S, et al. The role of clock in ethanol-related behaviors. Neuropsychopharmacology. 2013;38:2393–2400.
McClung CA, Sidiropoulou K, Vitaterna M, Takahashi JS, White FJ, Cooper DC, et al. Regulation of dopaminergic transmission and cocaine reward by the Clock gene. Proc Natl Acad Sci USA. 2005;102:9377–81.
Parekh PK, Becker-Krail D, Sundaravelu P, Ishigaki S, Okado H, Sobue G, et al. Altered GluA1 (Gria1) function and accumbal synaptic plasticity in the ClockDelta19 model of bipolar mania. Biol Psychiatry. 2017. [Epub ahead of print].
Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506.
Spencer S, Falcon E, Kumar J, Krishnan V, Mukherjee S, Birnbaum SG, et al. Circadian genes period 1 and period 2 in the nucleus accumbens regulate anxiety-related behavior. Eur J Neurosci. 2013;37:242–50.
Stromsdorfer KL, Yamaguchi S, Yoon MJ, Moseley AC, Franczyk MP, Kelly SC, et al. NAMPT-mediated NAD(+) biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell Rep. 2016;16:1851–60.
Yoshino J, Imai S. Accurate measurement of nicotinamide adenine dinucleotide (NAD(+)) with high-performance liquid chromatography. Methods Mol Biol. 2013;1077:203–15.
Coque L, Mukherjee S, Cao JL, Spencer S, Marvin M, Falcon E, et al. Specific role of VTA dopamine neuronal firing rates and morphology in the reversal of anxiety-related, but not depression-related behavior in the ClockDelta19 mouse model of mania. Neuropsychopharmacology. 2011;36:1478–88.
King DP, Vitaterna MH, Chang AM, Dove WF, Pinto LH, Turek FW, et al. The mouse clock mutation behaves as an antimorph and maps within the W19H deletion, distal of Kit. Genetics. 1997;146:1049–60.
King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, et al. Positional cloning of the mouse circadian clock gene. Cell. 1997;89:641–53.
Lewis-Tuffin LJ, Quinn PG, Chikaraishi DM. Tyrosine hydroxylase transcription depends primarily on cAMP response element activity, regardless of the type of inducing stimulus. Mol Cell Neurosci. 2004;25:536–47.
Piech-Dumas KM, Tank AW. CREB mediates the cAMP-responsiveness of the tyrosine hydroxylase gene: use of an antisense RNA strategy to produce CREB-deficient PC12 cell lines. Brain Res Mol Brain Res. 1999;70:219–30.
Aguilar-Arnal L, Katada S, Orozco-Solis R, Sassone-Corsi P. NAD(+)-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nat Struct Mol Biol. 2015;22:312–8.
Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–60.
Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–107.
Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–41.
Koch-Nolte F, Fischer S, Haag F, Ziegler M. Compartmentation of NAD+-dependent signalling. FEBS Lett. 2011;585:1651–6.
Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–42.
Hu Y, Liu J, Wang J, Liu Q. The controversial links among calorie restriction, SIRT1, and resveratrol. Free Radic Biol Med. 2011;51:250–6.
Dietrich JB, Mangeol A, Revel MO, Burgun C, Aunis D, Zwiller J. Acute or repeated cocaine administration generates reactive oxygen species and induces antioxidant enzyme activity in dopaminergic rat brain structures. Neuropharmacology. 2005;48:965–74.
Gimenez-Xavier P, Gomez-Santos C, Castano E, Francisco R, Boada J, Unzeta M, et al. The decrease of NAD(P)H has a prominent role in dopamine toxicity. Biochim Biophys Acta. 2006;1762:564–74.
Macedo DS, de Vasconcelos SM, dos Santos RS, Aguiar LM, Lima VT, Viana GS, et al. Cocaine alters catalase activity in prefrontal cortex and striatum of mice. Neurosci Lett. 2005;387:53–56.
Numa R, Kohen R, Poltyrev T, Yaka R. Tempol diminishes cocaine-induced oxidative damage and attenuates the development and expression of behavioral sensitization. Neuroscience. 2008;155:649–58.
Bellet MM, Orozco-Solis R, Sahar S, Eckel-Mahan K, Sassone-Corsi P. The time of metabolism: NAD+, SIRT1, and the circadian clock. Cold Spring Harb Symp Quant Biol. 2011;76:31–38.
Aguilar-Arnal L, Ranjit S, Stringari C, Orozco-Solis R, Gratton E, Sassone-Corsi P. Spatial dynamics of SIRT1 and the subnuclear distribution of NADH species. Proc Natl Acad Sci USA. 2016;45:12715–20.
Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12:51–62.
Prozorovski T, Schulze-Topphoff U, Glumm R, Baumgart J, Schroter F, Ninnemann O, et al. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat Cell Biol. 2008;10:385–94.
Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–63.
Obrietan K, Impey S, Smith D, Athos J, Storm DR. Circadian regulation of cAMP response element-mediated gene expression in the suprachiasmatic nuclei. J Biol Chem. 1999;274:17748–56.
Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc Natl Acad Sci USA. 2002;99:7728–33.
Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013;153:1448–60.
Hughes ME, Hong HK, Chong JL, Indacochea AA, Lee SS, Han M, et al. Brain-specific rescue of Clock reveals system-driven transcriptional rhythms in peripheral tissue. PLoS Genet. 2012;8:e1002835.
DeBruyne JP, Weaver DR, Reppert SM. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat Neurosci. 2007;10:543–5.
Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–61.
Hogenesch JB, Chan WK, Jackiw VH, Brown RC, Gu YZ, Pray-Grant M, et al. Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. J Biol Chem. 1997;272:8581–93.
Reick M, Garcia JA, Dudley C, McKnight SL. NPAS2: an analog of clock operative in the mammalian forebrain. Science. 2001;293:506–9.
Landgraf D, Wang LL, Diemer T, Welsh DK. NPAS2 compensates for loss of clock in peripheral circadian oscillators. PLoS Genet. 2016;12:e1005882.
Kennaway DJ, Voultsios A, Varcoe TJ, Moyer RW. Melatonin and activity rhythm responses to light pulses in mice with the Clock mutation. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1231–1240.
Ochi M, Sono S, Sei H, Oishi K, Kobayashi H, Morita Y, et al. Sex difference in circadian period of body temperature in Clock mutant mice with Jcl/ICR background. Neurosci Lett. 2003;347:163–6.
Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–25.
Ripperger JA, Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat Genet. 2006;38:369–74.
Vitaterna MH, Ko CH, Chang AM, Buhr ED, Fruechte EM, Schook A, et al. The mouse Clock mutation reduces circadian pacemaker amplitude and enhances efficacy of resetting stimuli and phase-response curve amplitude. Proc Natl Acad Sci USA. 2006;103:9327–32.
Redlin U, Hattar S, Mrosovsky N. The circadian Clock mutant mouse: impaired masking response to light. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2005;191:51–59.
Garcia JA, Zhang D, Estill SJ, Michnoff C, Rutter J, Reick M, et al. Impaired cued and contextual memory in NPAS2-deficient mice. Science. 2000;288:2226–30.
Zhang Y, Xue Y, Meng S, Luo Y, Liang J, Li J, et al. Inhibition of lactate transport erases drug memory and prevents drug relapse. Biol Psychiatry. 2016;79:928–39.
Abe N, Uchida S, Otsuki K, Hobara T, Yamagata H, Higuchi F, et al. Altered sirtuin deacetylase gene expression in patients with a mood disorder. J Psychiatr Res. 2011;45:1106–12.
Kishi T, Fukuo Y, Kitajima T, Okochi T, Yamanouchi Y, Kinoshita Y, et al. SIRT1 gene, schizophrenia and bipolar disorder in the Japanese population: an association study. Genes Brain Behav. 2011;10:257–63.
Kishi T, Yoshimura R, Fukuo Y, Kitajima T, Okochi T, Matsunaga S, et al. The CLOCK gene and mood disorders: a case-control study and meta-analysis. Chronobiol Int. 2011;28:825–33.
Nivoli A, Porcelli S, Albani D, Forloni G, Fusco F, Colom F, et al. Association between sirtuin 1 gene rs10997870 polymorphism and suicide behaviors in bipolar disorder. Neuropsychobiology. 2016;74:1–7.
Acknowledgements
We thank Mark Brown and Mariah Hildebrand for animal care and genotyping. We also thank Eric Nestler, Deveroux Ferguson, and Rachel Neve for the mCREB and SIRT1 viruses, and Gary Thomas for the SIRT1 partial constructs. We thank Joe Takahashi for the ClockΔ19 mice and CLOCK expression constructs. This work was funded by DA039865, DA037636, MH106460, MH082876, DA023988, DA042886, NS058339, NARSAD, and IMHRO to CAM, and DA038564, DA041872, and NARSAD to RWL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Logan, R.W., Parekh, P.K., Kaplan, G.N. et al. NAD+ cellular redox and SIRT1 regulate the diurnal rhythms of tyrosine hydroxylase and conditioned cocaine reward. Mol Psychiatry 24, 1668–1684 (2019). https://doi.org/10.1038/s41380-018-0061-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-018-0061-1
- Springer Nature Limited
This article is cited by
-
Single nuclei transcriptomics in human and non-human primate striatum in opioid use disorder
Nature Communications (2024)
-
A Pattern to Link Adenosine Signaling, Circadian System, and Potential Final Common Pathway in the Pathogenesis of Major Depressive Disorder
Molecular Neurobiology (2022)
-
Valproate reverses mania-like behaviors in mice via preferential targeting of HDAC2
Molecular Psychiatry (2021)
-
Cocaine-mediated circadian reprogramming in the striatum through dopamine D2R and PPARγ activation
Nature Communications (2020)
-
Characterization of genetically complex Collaborative Cross mouse strains that model divergent locomotor activating and reinforcing properties of cocaine
Psychopharmacology (2020)