
Abstract
Resistance to apoptosis in acute myeloid leukemia (AML) cells causes refractory or relapsed disease, associated with dismal clinical outcomes. Ferroptosis, a mode of non-apoptotic cell death triggered by iron-dependent lipid peroxidation, has been investigated as potential therapeutic modality against therapy-resistant cancers, but our knowledge of its role in AML is limited. We investigated ferroptosis in AML cells and identified its mitochondrial regulation as a therapeutic vulnerability. GPX4 knockdown induced ferroptosis in AML cells, accompanied with characteristic mitochondrial lipid peroxidation, exerting anti-AML effects in vitro and in vivo. Electron transport chains (ETC) are primary sources of coenzyme Q10 (CoQ) recycling for its function of anti–lipid peroxidation in mitochondria. We found that the mitochondria-specific CoQ potently inhibited GPX4 inhibition–mediated ferroptosis, suggesting that mitochondrial lipid redox regulates ferroptosis in AML cells. Consistently, Rho0 cells, which lack functional ETC, were more sensitive to GPX4 inhibition–mediated mitochondrial lipid peroxidation and ferroptosis than control cells. Furthermore, degradation of ETC through hyperactivation of a mitochondrial protease, caseinolytic protease P (ClpP), synergistically enhanced the anti-AML effects of GPX4 inhibition. Collectively, our findings indicate that in AML cells, GPX4 inhibition induces ferroptosis, which is regulated by mitochondrial lipid redox and ETC.

Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) is the most common acute leukemia in adults. Despite recent progress in molecularly targeted therapies, the 5-year overall survival rate is only about 30% [1]. This is mainly due to frequent relapse or refractory disease, which carries a dismal prognosis; these patients’ one-year survival rate is only 10% [2]. As a major cause of such therapy resistance, AML cells develop resistance to apoptosis induction through numerous mechanisms [3, 4], underlining the unmet need for the development of alternative therapeutic strategies that overcome apoptosis resistance.
Pathways of non-apoptotic regulated cell death are of growing interest in cancer research. One such form of regulated cell death is ferroptosis, which is characterized by iron dependency and triggered by the accumulation of lipid peroxidation on cellular membranes [5]. Increasing evidence from solid tumor studies supports the therapeutic potential of ferroptosis induction through its distinct mechanism of cell death independent of apoptosis. Major defense mechanisms against ferroptosis include the system xc–/glutathione/glutathione peroxidase 4 (GPX4) pathway [6, 7] and GPX4-independent pathways that utilize endogenous radical-trapping antioxidants such as coenzyme Q10 (CoQ) [8,9,10,11] or tetrahydrobiopterin [12, 13]. Among them, GPX4 is the only enzyme that can directly reduce lipid peroxides and its inhibition results in ferroptosis. GPX4 is a selenoprotein, whose translation is regulated by a unique protein synthesis mechanism that requires selenium. Because of its high rank in selenoprotein synthesis hierarchy, the expression of GPX4 is relatively retained even under selenium deficiency [14], suggesting its importance in cellular homeostasis.
AML cells exhibit increased reactive oxygen species production [15] and active iron uptake [16], both known determinants of ferroptosis susceptibility. We therefore hypothesize ferroptosis as a vulnerability in AML. While ferroptosis has been investigated recently in AML [17,18,19,20,21,22,23], the pathophysiological role of GPX4-mediated ferroptosis in AML remain largely unknown.
In the present study, we sought to elucidate the molecular mechanisms of GPX4 inhibition–induced ferroptosis in AML. We identified mitochondrial involvement in ferroptosis in AML cells, which provides a molecular rationale for targeting this disease-specific vulnerability to enhance the anti-leukemia effects of GPX4 inhibition.
Results
GPX4 is a potential therapeutic target in AML
To evaluate the therapeutic potential of targeting ferroptosis-regulating genes including GPX4 in a variety of cancer cell lines, we analyzed Cancer Dependency Map (DepMap) datasets using shinyDepMap [24], a tool that combines CRISPR and shRNA screening data of 423 cancer cell lines. This tool enables us to predict the essentiality of genes of interest across cancer cell lines as well as the selectivity of the gene essentiality among distinct cell types. In particular, we investigated ferroptosis suppressor genes annotated on FerrDb, a manually curated database of ferroptosis regulators and their disease associations [25, 26] (FerrDb V2, last access; 8/13/2022). GPX4 was the second-ranked gene with the high essentiality among the top 10 ferroptosis suppressors (efficacy score; X = −1.21), following MTOR (X = −1.454) (Fig. 1A). In addition, the selectivity of GPX4 (selectivity score; Y = 0.799) was much higher than that of MTOR (Y = 0.076), indicating that the essentiality of GPX4 is more cell line–specific. Importantly, when the cell lines were grouped by tumor types, AML was among those that are highly dependent on GPX4 (Fig. 1B). Meanwhile, consistently, MTOR exhibits high dependency not only in AML but more prominently in other tumor types (Fig. S1A). These analyses suggest that targeting GPX4 may have specific roles in AML.

A Efficacy/selectivity plot for the whole genes analyzed on shinyDepMap. The top 10 ferroptosis suppressor genes on FerrDb are annotated. B Dependency scores of GPX4 in all cell lines are clustered based on their lineages. A lower dependency score indicates a higher dependency on GPX4. Leukemia cell lines are subdivided into AML, B-cell ALL, and T-cell ALL, based on the original DepMap annotations. C GPX4 protein levels in 11 AML cell lines were determined by immunoblotting. The relative expression levels of GPX4 were assessed with densitometry as a ratio of GPX4 to α-tubulin (the loading control) and normalized to that of OCI-AML3. D, E GPX4 protein levels in peripheral blood mononuclear cells from 16 AML patients and bone marrow mononuclear cells from 4 healthy bone marrow donors (HD) were determined by immunoblotting (D). The relative expression levels of GPX4 were assessed with densitometry as a ratio of GPX4 to Histon H3 (the loading control), normalized to that of AML patient #17 that serves also as inter-membrane control, and plotted (E). High and low GPX4 bands suggest posttranslational modification, part of which appear as smears possibly due to partial degradation. For quantification, all the bands detected are included in the densitometry analysis. F GPX4 protein expression measured by mass spectrometry with tandem mass tag (TMT) labeling is plotted for AML samples (n = 44) and lineage-depleted normal bone marrow (NBM) samples (n = 3). Original data was obtained from a previous study [29].
We next examined GPX4 protein expression in AML cells. In AML cell lines, the expression levels were found variable among 11 lines tested (Fig. 1C). Primary AML cells also exhibit distinct expression levels of GPX4 protein across 16 samples tested (Fig. 1D). GPX4 expression in normal bone marrow bulk mononuclear cells are significantly higher than that in AML cells (Fig. 1D, E). Of note, distinct levels of GPX4 bands were detected in some of the primary AML samples which may suggest post-translational modification of the protein [27], and some of the smear pattern which may reflect partial degradation of GPX4 protein. However, the exact nature of these bands remains unclear in the present study. We thus performed further validation and analyzed the differential expression of GPX4 in AML cells and normal cells using publicly available datasets with larger sample size. First, we utilized GEPIA platform, a web server for cancer and normal gene expression profiling [28]. We found that GPX4 gene expression of AML samples from The Cancer Genome Atlas (TCGA) dataset (n = 173) were significantly lower than that of normal bone marrow samples from The Genotype-Tissue Expression (GTEx) dataset (n = 70) (Fig. S1B). We also analyzed a recent proteome dataset comparing primary AML (n = 44) and normal bone marrow (n = 3) cells [29]. Consistently, GPX4 protein expression is significantly lower in AML samples compared to normal bone marrow samples (Fig. 1F).
Next, based on previous studies indicating a negative impact of elevated GPX4 gene expression on AML patients survival [30, 31], we analyzed TCGA dataset (n = 160) [32] and Beat AML dataset (n = 350) [33] to study the correlation. While TCGA shows worse survival in patients with high GPX4 expression (Fig. S1C), Beat AML did not show significant survival difference (Fig. S1D). In addition, analysis of a publicly available proteome dataset [34] revealed no significant survival difference between AML patients with high and low GPX4 protein levels (Fig. S1E). These analyses do not fully contradict the previous studies that reported the survival impact of GPX4 expression but also suggest that the correlation of GPX4 and AML patient survival should be more carefully concluded. Multiple aspects including confounding factors likely cause these distinct results, warranting further investigations to determine whether there are specific subpopulations of AML where GPX4 expression has a survival impact.
GPX4 suppression induces ferroptosis in AML cells in vitro and in vivo
To test the anti-leukemia effects of the pharmacological inhibition of GPX4, we utilized ML210, to date a well-established, proteome-wide specific inhibitor of GPX4 [35, 36]. We determined GPX4 inhibition–induced cell death as the percentage of cells positive for annexin V and/or DAPI to capture both apoptotic and non-apoptotic cell death including ferroptosis. In OCI-AML3 cells, ML210 increased both, annexin V–positive and DAPI-positive fractions, which were effectively suppressed by the lipophilic antioxidants liproxstatin-1 (Lip1) and α-tocopherol (aToc) as well as the iron chelator deferoxamine (DFO) (Fig. 2A). These data indicate that, although annexin V is usually used as a marker of apoptosis, annexin V–positive AML cells that result from GPX4 inhibition are ferroptotic. This is consistent with previous findings demonstrating that lipid peroxidation upon ferroptosis can cause phosphatidylserine externalization without caspase activation [37]. Consequently, the pan-caspase inhibitor z-VAD-FMK did not inhibit ML210-induced cell death (Figs. 2B and S2A, B). Lip1, aToc, and DFO consistently suppressed ML210-induced lipid peroxidation, which was detected with C11-BODIPY (Fig. 2C). The iron-dependent lipid peroxidation and cell death induced by ML210 were confirmed in MOLM-13 and OCI-AML2 cells as well (Fig. S2C–F).

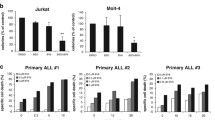
A, C OCI-AML3 cells were treated with 0.5 µM ML210 with or without 1 µM liproxstatin-1 (Lip1), 100 µM α-tocopherol (aToc), or 4 µM deferoxamine (DFO) for 48 hours. Cell death (A) and lipid peroxidation (C) were determined by annexin V (AnnV) and/or DAPI positivity or by C11-BODIPY581/591 staining, respectively, on flow cytometry. B OCI-AML3 cells were treated with 1 µM venetoclax (Ven) plus 0.2 µM AMG-176 (MCL-1 inhibitor) to induce apoptosis [65] or 0.2 µM ML210, with or without 25 µM z-VAD-FMK (zVAD), for 24 hours. D Parental AML cells were treated with ML210 for 24 hours, and specific cell death was calculated as described in Supplemental information. X axis represents ML210 concentration on a logarithmic scale. E The area under the cell killing curve (AUC) for ML210 (Fig. 2D) was correlated with the relative GPX4 expression level (Fig. 1C). Correlation coefficient (r) and statistical significance (p) on Pearson correlation analysis were indicated. F MOLM-13 cells with wildtype TP53 (WT), TP53 knockout (KO), or the indicated TP53 mutations were treated with ML210 for 72 hours. G Kasumi-1 cells transfected with shRNA targeting TP53 (shp53) or with scramble control shRNA (shC) were treated with ML210 for 72 hours. H Peripheral blood mononuclear cells from AML patients (n = 14) or bone marrow mononuclear cells from healthy bone marrow donors (HD) (n = 12) were treated with 10 µM ML210 for 48 hours. Cell death was determined for CD45+ bulk hematopoietic cell populations. I AUC for ML210 in primary AML cells (Fig. S2O) was correlated with the GPX4 expression level (Fig. 1D) for matching samples (n = 13).
Next, we evaluated the efficacy of ML210 across 11 AML cell lines. Sub-micromolar concentrations of ML210 induced cell death in most cell lines (Fig. 2D). When analyzed with the GPX4 protein expression on Fig. 1C, their sensitivities were negatively correlated with the expression levels (Pearson correlation analysis; r = −0.739, p = 0.009) (Fig. 2E). Among the cell lines tested, MV-4-11 is relatively sensitive to ML210 despite its high GPX4 protein level, indicating that other cell line-specific mechanisms may also play roles in the determination of the sensitivity. On immunoblot, well-established ferroptosis regulators tested did not show any unique feature that could potentially increase the sensitivity in MV-4-11 cells (Fig. S2G, H). Further investigations for other molecules as well as their functional assays are required to elucidate the mechanisms involved. To assess the effects of endogenous GPX4 upregulation on ferroptosis, we utilized selenium to enhance GPX4 biosynthesis. Supplementing culture media with seleno-L-methionine (SLM) increased GPX4 expression levels in AML cells (Fig. S2I). We further generated Doxycycline (Dox)-inducible short hairpin RNA (shRNA) constructs to knockdown eukaryotic elongation factor, selenocysteine-tRNA specific (EEFSEC), which is required for selenoprotein translation [38]. The Dox-induced knockdown of EEFSEC abrogated SLM-induced GPX4 upregulation (Fig. S2J), confirming that GPX4 upregulation occurs through increased protein synthesis of GPX4. As expected, AML cells cultured in SLM-supplemented media are resistant to ML210 (Fig. S2K). GPX4-mediated suppression of ferroptosis through selenoprotein synthesis is summarized in Fig. S2L.
We also assessed the anti-leukemia effect of GPX4 inhibition in TP53-mutant AML cell lines, which served as an in vitro model of one of the most clinically challenging genetic subtypes of AML. ML210 had similar efficacy among isogenic MOLM-13 cells with TP53 deletion or hotspot mutations (Fig. 2F). Another GPX4 inhibitor, RSL-3, also showed the similar cell death induction in these cells (Fig. S2M). Immunoblot revealed that xCT expression was slightly higher in TP53 knockout (KO) or mutant cells (Fig. S2N) compared to TP53 wild-type (WT) cells. This is consistent with the previous reports showing that TP53 is a negative regulator of SLC7A11 [39, 40] while xCT protein levels did not correlate the cell sensitivities to ferroptosis in these conditions. The knockdown of mutant TP53 in Kasumi-1 cells harboring the TP53R248H mutation also did not alter sensitivity to ML210 (Fig. 2G). Taken together, GPX4 inhibition exerts anti-leukemia effects independently of TP53 mutation status.
We then treated primary cells derived from AML patients (n = 14) or healthy bone marrow donors (n = 12) with ML210. AML patients included those who relapsed after or developed resistance to multiple regimens and/or those with TP53 mutations (Table S1). Most of the primary AML samples tested were sensitive to ML210 (Figs. 2H and S2O). Three samples showed resistance to ML210, but we could not identify any common clinical features explaining the resistance, including prior regimens, cytogenetics, or mutations. A correlation analysis of AML cell sensitivity to ML210 and GPX4 protein levels in primary AML samples (n = 13) showed negative correlation (Fig. 2I), which suggests that GPX4 protein expression is a potential predictor of AML cell sensitivity to GPX4 inhibition. Independently, cell death induced in CD45+ mononuclear hematopoietic cells from healthy bone marrow donors (HD) was significantly lower than that induced in AML patient samples (Fig. 2H). Besides, we found that the effective dose of ML210 was higher in primary cells (at the levels of micromolar) compared to cell lines, for which the mechanisms are yet to be investigated. Of note, cell death induced by higher dose of ML210 in primary cells was inhibited by Lip1 (Fig. S2P), consistent with the on-target effects of ferroptosis induction.
We next introduced Dox-inducible shRNAs targeting GPX4 (shGPX4) into OCI-AML3 cells (OCI-AML3-shGPX4-1 or -2 cells) (Fig. 3A). Treatment with Dox for 96 hours (shGPX4-1) or 72 hours (shGPX4-2) resulted in lipid peroxidation (Figs. 3B and S3A), followed by cell death induction at 120 hours (Fig. 3C), demonstrating that GPX4 downregulation induces lipid peroxidation prior to the cell death. The findings were confirmed in MOLM-13 cells and OCI-AML2 cells transfected with the same shRNAs (Figs. 3D, E and S3B, C). Cell death was induced already at 96 hours post Dox treatment in these two cell lines, perhaps partly due to lower baseline GPX4 expressions compared to that in OCI-AML3 cells. In addition, GPX4 knockdown sensitized OCI-AML3-shGPX4 cells to ML210 at a dose that was not effective by itself (Fig. 3F), consistent with the aforementioned negative correlation between GPX4 protein expression and sensitivity to GPX4 inhibition. Cell death induced by GPX4 knockdown was abrogated by Lip1 and aToc but not by DFO (Fig. S3D, E). Of note, AML cells did not tolerate the dose range of DFO used in solid cancer ferroptosis studies (up to 100 μM) [6, 41] due to its inherent anti-leukemia effects (Fig. S3F). Given that lipid peroxidation induced by shGPX4 (Figs. 3B and S3A) was more profound than that by ML210 treatment (Fig. 2C), the lower dose of DFO in AML cells was possibly insufficient to block the robust ferroptosis induced by GPX4 knockdown.

A OCI-AML3 cells transfected with shRNAs targeting GPX4 (shGPX4) or with control shRNA (shC) were treated with 1 µg/ml doxycycline (Dox) for 72 hours, and GPX4 protein levels were determined by immunoblotting. B OCI-AML3-shGPX4-1 cells were treated with 1 µg/ml Dox for 96 hours, and cell death (left panel) and lipid peroxidation (right panel) were determined by flow cytometry. C–E OCI-AML3-shGPX4 (C), MOLM-13-shGPX4 (D), and OCI-AML2-shGPX4 (E) cells were treated with Dox for 120 hours (C) or 96 hours (D, E). F OCI-AML3-shGPX4 cells were treated with 0.1 µg/ml Dox for 72 hours in combination with 0.1 µM ML210 for the last 24 hours. G Survival curves of the NSG mice transplanted with luciferase-labeled OCI-AML3-shGPX4-2 cells and treated with vehicle or tetracycline (n = 7 for each group) were plotted. H OCI AML3-shGPX4-2 cells were harvested from moribund mice after treatment with vehicle or tetracycline for 6–8 weeks (n = 3 for each group) and purified for AML cells as described in Supplemental information. GPX4 protein levels were determined by immunoblotting.
To determine the anti-leukemia effect of GPX4 inhibition in vivo, we labeled OCI-AML3-shGPX4-2 cells with luciferase and transplanted them into NSG mice. After engraftment was confirmed by bioluminescent imaging (BLI) (Fig. S3G), the mice were given regular water (vehicle) or tetracycline water to knockdown GPX4 in leukemia cells (n = 7/group). Tetracycline treatment significantly prolonged mouse survival (median survival 74 days vs 55 days) (Fig. 3G), confirming that GPX4 inhibition exerts anti-leukemia effects in vivo. Knockdown of GPX4 protein was verified for the leukemia cells harvested from mice on immunoblotting (Fig. 3H). The same tetracycline treatment did not extend survival of mice transplanted with OCI-AML3 cells with control shRNA (Fig. S3H).
Mitochondrial lipid peroxidation and electron transport chain complexes regulate AML cell ferroptosis
Because mitochondria are the centers of cellular redox and iron metabolism, we next investigated the involvement of mitochondria in AML cell ferroptosis. Morphologically, evaluation by transmission electron microscopy showed that, compared with control cells, ML210-treated OCI-AML3 cells had smaller mitochondria with an electron-dense mitochondrial matrix (Fig. 4A). This is consistent with previous ferroptosis studies in non-leukemia models [6, 39]. ML210-treated OCI-AML3 cells also had disorganized cristae with “vesicular” morphology, a phenomenon similar to the finding reported to occur during apoptosis induction in HeLa cells [42]. Functionally, ML210 increased mitochondrial superoxide production, which was largely blocked by Lip1, aToc, and DFO, in AML cells (Figs. 4B, C and S4A, B). This suggests that the mitochondrial superoxide induced by GPX4 inhibition is mostly iron-dependent lipid peroxidation. Indeed, flow cytometry with MitoPerOx staining, which specifically detects lipid peroxidation in mitochondria, revealed that ML210 consistently increased mitochondrial lipid peroxidation (Fig. 4D). Dox-induced GPX4 knockdown also increased mitochondrial superoxide production and lipid peroxidation in OCI-AML3 cells (Figs. 4E and S4C) and MOLM-13 cells (Fig. S4D).

A OCI-AML3 cells were treated with 0.5 µM ML210 or with DMSO as a control for 6 hours and then subjected to transmission electron microscopy to assess the mitochondrial ultrastructure. B, C OCI-AML3 cells were treated with 0.5 µM ML210 with or without 1 µM liproxstatin-1 (Lip1) (B), 100 µM α-tocopherol (aToc) (B), or 4 µM deferoxamine (DFO) (C) for 48 hours. Mitochondrial superoxide production was determined by MitoSOX staining and flow cytometry. The median fluorescence intensity (MFI) of the treated cells was normalized to that of the untreated cells. D OCI-AML3 cells were treated with 0.5 µM ML210 for 48 hours, and mitochondrial lipid peroxidation was determined by MitoPerOx staining and flow cytometry. E OCI-AML3-shGPX4-1 cells were treated with 1 µg/ml doxycycline (Dox) for 96 hours, and mitochondrial superoxide (left) and lipid peroxidation (right) were determined by flow cytometry. MEFs (F) or OCI-AML3 cells (G) were treated with 2.0 µM ML210 in combination with increasing concentrations of MitoQ or DecylQ as indicated on the x axis for 24 hours. Cell viability was determined as the percentage of annexin V– and DAPI-negative cells on flow cytometry. H OCI-AML3 cells were treated with 0.5 µM ML210 with or without 0.1 µM MitoQ or 10 µM MitoTEMPO for 48 hours. I OCI-AML3-shGPX4-1 cells were treated with 1 µg/ml Dox with or without 0.1 µM MitoQ or 10 µM MitoTEMPO for 120 hours. J Lysates from HL-60-RhoWT cells or -Rho0 cells were subjected to immunoblotting for the indicated proteins. HL-60-Rho0 cells or -RhoWT cells were treated with ML210 for 48 (K) or 24 (L) hours.
A previous study using mouse embryonic fibroblasts (MEFs) demonstrated that the mitochondria-targeting antioxidant mitoquinone mesylate (MitoQ) is far less potent than the non-targeting antioxidant decylubiquinone (DecylQ) in protecting cells from GPX4 inhibition–induced ferroptosis [43]. This suggests that ferroptosis usually arises from a redox imbalance in cytoplasm but not in mitochondria. To further investigate the involvement of mitochondria in leukemia cells, we compared the anti-ferroptosis effect of MitoQ in MEFs and OCI-AML3 cells. The expression of GPX4 protein in those cells were determined by immunoblotting (Fig. S4E). We confirmed that the concentration of MitoQ required in MEFs to block ML210-induced cell death was more than 5-fold that of DecylQ (Fig. 4F), which is consistent with the previous report [43]. In contrast, and unexpectedly, MitoQ was as potent as DecylQ in blocking ferroptosis in OCI-AML3 cells treated with ML210 (Fig. 4G), suggesting that AML cell ferroptosis is regulated predominantly by mitochondrial, rather than cytoplasmic, lipid peroxidation. In addition to MitoQ, MitoTEMPO, another mitochondria-targeted antioxidant, also blocked cell death caused by ML210 treatment or by Dox-induced GPX4 knockdown (Figs. 4H, I and S4F, G), further supporting the mitochondrial regulation of ferroptosis in AML cells.
Next, to investigate the role of mitochondrial respiration, the center of mitochondrial redox metabolism, in AML cell ferroptosis, we studied AML cells with deficient electron transport chain (ETC) complex through chronic exposure to ethidium bromide to specifically deplete mitochondrial DNA (Rho0 cells). Since OCI-AML3 cells were not tolerated to Rho0 status (data not shown), we utilized well-established HL-60-Rho0 cells [44]. Immunoblotting revealed that Rho0 cells had non-detectable levels of ETC complex proteins (subunits in complexes I, II, III, and IV) and lower level of GPX4 expression (Fig. 4J). Notably, HL-60-Rho0 cells are nearly 10 times as sensitive to ML210 as their wild type (WT) controls (HL-60-RhoWT) (Fig. 4K). Furthermore, prominent mitochondrial lipid peroxidation was induced in Rho0 cells by almost 10-fold lower concentration of ML210 as RhoWT cells, confirming that Rho0 cells are more sensitive to GPX4 inhibition–induced mitochondrial lipid peroxidation in the AML cell line (Fig. 4L). Although GPX4 downregulation could also contribute to higher sensitivity to GPX4 inhibition in Rho0 cells, we reasoned that the intact expressions of ETC complex proteins may protect AML cells from GPX4 inhibition–mediated ferroptosis, perhaps uniquely in AML cells.
GPX4 inhibition–mediated ferroptosis is synergistically enhanced by hyperactivation of mitochondrial caseinolytic protease P
Given Rho0 AML cells’ higher ferroptosis sensitivity, we investigated the therapeutic potential of a combination of ferroptosis induction and ETC downregulation in AML. To this end, we hyperactivated the mitochondrial protease caseinolytic protease P (ClpP) by imipridones (ONC201 or ONC212) to induce the degradation of ETC subunits and exert cancer-selective lethality [45]. Indeed, the combination of ML210 and ONC201 induced synergistic cell death in OCI-AML2 and MOLM-13 cells (Fig. 5A). The combinatorial anti-leukemia effect was associated with the cumulative induction of lipid peroxidation and mitochondrial superoxide production (Fig. 5B, C). Next, we studied if the synergistic anti-leukemia effect is associated with changes in GPX4 protein. Immunoblot of GPX4 showed a size shift for all the ML210-treated samples (Fig. S5A), which reflects covalent binding of ML210 or its metabolite, as reported previously [36]. Meanwhile, changes in total GPX4 protein expression levels are distinct among three AML cell lines, suggesting that alteration in GPX4 protein expression by the treatment is not the primary mechanisms responsible for the synergistic anti-leukemia effects.

A OCI-AML3, MOLM-13, and OCI-AML2 cells were treated with ONC201 and/or ML210 for 72 hours, and cell death was determined by flow cytometry. A combination index (C.I.) less than 1.0 indicates synergistic effects. OCI-AML3 cells were treated with ONC201 and/or ML210 for 48 hours, and lipid peroxidation (B) and mitochondrial superoxide production (C) were determined by flow cytometry. D Primary cells from AML patients (n = 8) were treated with ML210 and/or ONC201 for 48 hours. Combination indexes were calculated based on specific cell death in CD45+ population upon the combinatorial treatment for each sample and were plotted. One sample was excluded from C.I. assessment because ONC201 was completely ineffective and thus C.I. calculations were not applicable. The sample from patient #12, which was resistant to a single treatment with ML210 on Figs. 2H and S2O, is indicated by arrows. E Primary cells from AML patients (n = 8) or healthy bone marrow donors (HD) (n = 8) were treated with 10 μM ML210 and 5 μM ONC201 for 48 hours, and cell death was determined for CD45+ populations. The sample from AML patient #12 is indicated by an arrow. F OCI-AML3-CLPP-Y118A cells were treated with doxycycline (Dox) for 144 hours in combination with ML210 for the last 72 hours. OCI-AML3-shGPX4-1 cells were treated with Dox for 120 hours in combination with ONC201 (G) or ONC212 (H) for the last 72 hours. I A genome-wide CRISPR knockout screening performed on NALM6 cells treated with ClpP agonists [46] were re-analyzed and sgRNA frequency scores (RANKS scores) were plotted. J OCI-AML3, OCI-AML2, and MOLM-13 cells were treated with 2 µM ONC201 for up to 48 hours, and GPX4 expression was determined by immunoblotting. K HL-60-Rho0 cells or -RhoWT cells were treated with ML210 with or without 0.25 µM ONC212 for 48 hours.
The combinatorial treatment was also tested in primary samples. Among the 14 primary AML samples tested for ML210 single treatment (in Fig. 2H), 8 samples (including one sample exhibiting resistance to ML210 alone) were tested for the combinatorial treatment as well. Combination indexes were below 1.0 for most of the treatment conditions, indicating synergistic anti-leukemia effects (Fig. 5D). The synergism was also observed for the sample that exhibited resistance to ML210 alone (indicated by an arrow in Fig. 5D). Importantly, primary cells from healthy bone marrow donors (n = 8) exhibit significantly less cell death upon the combinatorial treatment compared to those from AML patients (Figs. 5E and S5B).
Furthermore, synergistic cell death was induced when the genetic activation of ClpP using a Dox-inducible CLPP-Y118A hyperactivated mutant was combined with ML210 and when Dox-inducible GPX4 knockdown was combined with ONC201 and ONC212 (Figs. 5F–H and S5C, D). We also tested the combinatorial anti-leukemia effect of GPX4 knockdown and ONC212 in vivo. A tendency of extended survival in the combination arm was observed, however, the difference compared to the other arms was not statistically significant (Fig. S5E).
Supporting the combinatorial effects of GPX4 inhibition and ClpP hyperactivation, our re-analysis of a previously published CRISPR screening data of NALM6 leukemia cells treated with imipridones [46] revealed GPX4 to be one of the top hits; knockdown of GPX4 sensitized cells to ClpP hyperactivation (Fig. 5I). In addition to GPX4, genes essential for selenoprotein synthesis (SEPSECS, EEFSEC, PSTK, SEPHS2) [47] were also top hits, suggesting that the inhibition of GPX4’s function as a selenoprotein is important for the synthetic lethality of GPX4 inhibition and ClpP hyperactivation. We also found that ClpP hyperactivation by ONC201 upregulates GPX4 protein expression (Fig. 5J), suggesting that GPX4 exerts a cell-protective response against mitochondrial proteotoxic stress in AML cells. ClpP hyperactivation sensitized ETC-intact RhoWT cells to GPX4 inhibition, but the synergism was not exerted in Rho0 cells (Fig. 5K). This indicates that the induced synergism depends on ETC, supporting the notion that the regulated protein expression of ETC subunits helps protect cells from ferroptosis. Of note, pharmacological inhibition of each ETC complex (complex I by IACS-010759 [48], complex III by Antimycin-A, or complex V by Oligomycin-A) did not enhance but rather inhibited ML210-induced cell death (Fig. S5F). These data suggest that functional inhibition of single ETC complex does not necessarily sensitize AML cells to ferroptosis. Since Rho0 cells are characterized not merely by the impaired ETC function but also structural abrogation of ETC involving multiple complexes, loss of the intact 3D structures and loss of functions of multiple ETC complexes by imipridone-mediated degradation may similarly matter for the induction of synergism.
Discussion
An increasing number of studies have been reported regarding the roles of mitochondria and ETC complex in ferroptosis across various cancer models, resulting in diverse conclusions [6, 49,50,51]. This is perhaps due to the highly context-dependent nature of ferroptosis [52, 53]. Findings of the present study suggest that intact ETC complexes regulate GPX4 inhibition–mediated ferroptosis in AML cells in a manner distinct from that of other tumor types. Specifically, HL-60-Rho0 cells with ETC defects were more sensitive to GPX4 inhibition compared to their parental cells. This is in clear contrast with previous studies demonstrating similar or impaired efficacy of the classical GPX4 inhibitor RSL3 in Rho0 status of 143B osteosarcoma cells [6] or SK-Hep1 liver sinusoidal endothelial cells [49], respectively. Similarly, another study reported that, in HT-1080 fibrosarcoma cells, mitophagy-mediated mitochondrial depletion blocked erastin-induced ferroptosis while had no effect on RSL3-induced ferroptosis [50]. A CRISPR knockout screening for K562 chronic myeloid leukemia cells revealed synthetic lethal interactions between GPX4 deletion and ETC inhibition [51]. In the meanwhile, our data suggest that inhibitions of a single ETC complex rather reduced AML cell sensitivity to ferroptosis. The sensitivity was increased only when several complexes of ETC were simultaneously and structurally abrogated (by Rho0 condition or ClpP hyperactivation). Of note, in the CRISPR screening study of K562 cells, they demonstrate that the synthetic lethal interaction was only observed when cells are cultured with spend media but not regular media, which may have led to the difference with our data. Taken together, our findings suggest that AML cells may be distinctively dependent on ETC to protect themselves from ferroptosis.
This mechanism of ETC-dependent protection from ferroptosis could be a unique vulnerability in AML in which GPX4 has been inhibited. Indeed, we observed that the combination of GPX4 inhibition and ClpP activation–mediated degradation of ETC proteins had synergistic anti-leukemia effects in AML cells. We acknowledge that ClpP targets not only ETC proteins but also other proteins, including those involved in mitochondrial translation or metabolic pathways [45], and the inhibition of such proteins could contribute to the cells’ increased vulnerability to GPX4 inhibition–mediated ferroptosis. We had previously reported that ClpP hyperactivation induces atypical integrated stress response (ISR) and ATF4 upregulation [45, 54] and a recent study shows that ISR-mediated ATF4 activation sensitizes cells to GPX4 inhibition through alteration of GSH metabolism [55]. Involvement of these pathways should also be evaluated in future studies. Nevertheless, the combinatorial effects of ClpP hyperactivation and GPX4 inhibition observed in parental AML cells but not in ETC-deficient Rho0 cells strongly suggest that this synergism and the ferroptosis protection mechanism both depend on intact ETC complexes.
Furthermore, we demonstrated that MitoQ had unexpectedly potent anti-ferroptotic effects in AML cells but, as reported previously, not in MEFs. The importance of mitochondria-localized CoQ is also in line with ETC’s protective effects against mitochondrial lipid peroxidation and resultant ferroptosis because ETC complexes reduce mitochondria-localized CoQ to CoQH2, which exerts anti-oxidative effects [56]. MitoQ has been reported to have similar protective effects in erastin- or RSL3-treated SK-Hep1 liver sinusoidal endothelial cells, in which induced ferroptosis was also associated with mitochondrial lipid peroxidation [49]. In addition, recent studies by Boyi Gan’s group demonstrated that DHODH and GPD2 on the mitochondrial membrane, both regulate the recycling of mitochondria-localized CoQ in collaboration with ETC, protect against ferroptosis [10, 11]. Although the functional relevance of these non-ETC CoQ-regulating enzymes in AML cell ferroptosis remains to be investigated, we speculate that the maintenance of a reduced form of mitochondrial CoQ primarily contributes to ETC’s protective role against ferroptosis in AML cells.
Lastly, here we have reported significant in vivo anti-leukemia activity of GPX4 inhibition using a genetic approach. At present, none of the publicly available GPX4 inhibitors are fully developed for use in in vivo studies due to their suboptimal bioavailability in animal models [36]. The development of specific GPX4 inhibitors with superior pharmacokinetic and pharmacodynamic profiles in vivo is necessary for the clinical implementation of GPX4-targeted therapies. In parallel, potential toxicity of GPX4 inhibitors in non-hematopoietic systems must be evaluated carefully, as our genetic model does not reflect systemic GPX4 inhibition. Our in vitro data suggests preferential cell death induction by GPX4 inhibition in primary AML cells compared to normal BM cells, possibly due to their differential expression levels of GPX4 protein. Multiple genetic models also support the tolerability of conditional GPX4 deletion in normal hematopoiesis [57, 58] and hematopoietic stem cells [59], while a recent paper reported sensitivity of human normal hematopoietic stem cells to ferroptosis [60], warranting further investigation especially in vivo. GPX4 has been reported not only as essential for early embryonic development in mice [61, 62] but also as linked to a rare neonatal lethal disorder, Sedaghatian-type spondylometaphyseal dysplasia, when loss-of-function mutations occur in humans [63]. These studies alert that inhibition of GPX4 is potentially toxic to other organs when systemically targeted. To avoid the potential toxicity, development of cancer-specific targeting strategy for GPX4 inhibition (e.g., antibody-drug conjugates to target a cancer-specific surface protein) would be one of the most effective ways. Developing combinatorial strategies to minimize the dose of GPX4 inhibitors could be another solution, particularly by targeting other cancer-specific molecular mechanisms. Together with a recent study suggesting the combinatorial inhibition of GPX4 and FLT3 in FLT3-mutant AML [23], the present study suggests that targeting the mitochondrial regulation of ferroptosis could potentiate the therapeutic efficacy of GPX4 inhibition in AML.
Materials and methods
Detailed materials and methods can be found in the Supplementary information.
Cell lines and cell culture
Parental AML cells as well as HEK293T cells and MEFs were obtained and cultured as described in Supplemental information. OCI-AML3-CLPP-Y118A cells were established by us and described previously [45]. CRISPR-engineered isogenic MOLM-13 cells with TP53 WT, TP53 KO, or TP53 R175H or R248Q mutants were provided by Steffen Boettcher (University of Zurich, Zurich, Switzerland) [64]. HL-60-Rho0 and -RhoWT cells were provided by Peng Huang (Department of Molecular Pathology, MD Anderson Cancer Center) [44].
Primary samples
The mononuclear cells were purified from peripheral blood of AML patients (blast >60%) and bone marrow of healthy hematopoietic stem cell donors by Ficoll density-gradient centrifugation and were cultured in RPMI 1640 culture media (#MT10040CV, Corning, Corning, NY) supplemented with 100 ng/ml human Flt3-ligand (#300-19, Peprotech, Cranbury, NJ), 100 ng/ml human SCF (#300-07, Peprotech), 20 ng/ml human TPO (#300-18, Peprotech), and 20 ng/ml human IL-3 (#200-03, Peprotech), as well as 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM L-glutamine, and 10% heat-inactivated fetal bovine serum.
Immunoblot analyses
For the immunoblot analyses, we used following antibodies; anti-GPX4 (#ab125066, Abcam, Cambridge, UK), anti-α-tubulin (#2125; Cell Signaling), anti-Histon H3 (#3638, Cell Signaling, Danvers, MA), anti-β-actin (#A5316, MilliporeSigma, Darmstadt, Germany, or #4970, Cell Signaling), anti-AIFM2 (FSP1) (#HPA042309, MilliporeSigma), anti-SCD1 (#PA5-19682, Thermo Fisher Scientific, Waltham, MA), anti-DHODH (#14877-1-AP, Proteintech, Rosemont, IL), anti-XCT/SLC7A11 (#12691, Cell Signaling), anti-ACSL4 (#sc-271800, Santa Cruz Biotechnology), anti-EEFSEC (#HPA035795, MilliporeSigma), anti-p53 (#sc-126, Santa Cruz Biotechnology, Dallas, TX), and total oxidative phosphorylation rodent antibody cocktail (#ab110413, Abcam).
Data availability
All data generated during this study are included in this published article and its supplementary files. Additional data are available from the corresponding author upon reasonable request.
Change history
08 March 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41375-024-02202-0
References
National Cancer Institute. Surveillance, Epidemiology, and End Results Program: Cancer Stat Facts: Leukemia — Acute Myeloid Leukemia (AML) 2022 [Available from: https://seer.cancer.gov/statfacts/html/amyl.html.
Ganzel C, Sun Z, Cripe LD, Fernandez HF, Douer D, Rowe JM, et al. Very poor long-term survival in past and more recent studies for relapsed AML patients: The ECOG-ACRIN experience. Am J Hematol. 2018;93:1074–81.
Kropp EM, Li Q. Mechanisms of resistance to targeted therapies for relapsed or refractory acute myeloid leukemia. Exp Hematol. 2022;111:13–24.
Ong F, Kim K, Konopleva MY. Venetoclax resistance: mechanistic insights and future strategies. Cancer Drug Resist. 2022;5:380–400.
Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381–96.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Wu S, Mao C, Kondiparthi L, Poyurovsky MV, Olszewski K, Gan B. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc Natl Acad Sci USA. 2022;119:e2121987119.
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci. 2020;6:41–53.
Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16:1351–60.
Weitzel F, Ursini F, Wendel A. Phospholipid hydroperoxide glutathione peroxidase in various mouse organs during selenium deficiency and repletion. Biochim Biophys Acta. 1990;1036:88–94.
Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood. 2011;117:5816–26.
Lopes M, Duarte TL, Teles MJ, Mosteo L, Chacim S, Aguiar E, et al. Loss of erythroblasts in acute myeloid leukemia causes iron redistribution with clinical implications. Blood Adv. 2021;5:3102–12.
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2:e1054549.
Ye F, Chai W, Xie M, Yang M, Yu Y. Cao L, et al. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am J Cancer Res. 2019;9:730–9.
Yusuf RZ, Saez B, Sharda A, van Gastel N, Yu VWC, Baryawno N, et al. Aldehyde dehydrogenase 3a2 protects AML cells from oxidative death and the synthetic lethality of ferroptosis inducers. Blood. 2020;136:1303–16.
Birsen R, Larrue C, Decroocq J, Johnson N, Guiraud N, Gotanegre M, et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica. 2022;107:403–16.
Pardieu B, Pasanisi J, Ling F, Dal Bello R, Penneroux J, Su A, et al. Cystine uptake inhibition potentiates front-line therapies in acute myeloid leukemia. Leukemia. 2022;36:1585–95.
Akiyama H, Carter BZ, Andreeff M, Ishizawa J. Molecular Mechanisms of Ferroptosis and Updates of Ferroptosis Studies in Cancers and Leukemia. Cells. 2023;12:1128.
Sabatier M, Birsen R, Lauture L, Mouche S, Angelino P, Dehairs J, et al. C/EBPa confers dependence to fatty acid anabolic pathways and vulnerability to lipid oxidative stress-induced ferroptosis in FLT3-mutant leukemia. Cancer Discov. 2023;13:1720–47.
Shimada K, Bachman JA, Muhlich JL, Mitchison TJ. shinyDepMap, a tool to identify targetable cancer genes and their functional connections from Cancer Dependency Map data. Elife. 2021;10:e57116.
Zhou N, Bao J. FerrDb: a manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database. 2020;2020:baaa021.
Zhou N, Yuan X, Du Q, Zhang Z, Shi X, Bao J, et al. FerrDb V2: update of the manually curated database of ferroptosis regulators and ferroptosis-disease associations. Nucleic Acids Res. 2023;51:D571–D82.
Cui C, Yang F, Li Q. Post-Translational Modification of GPX4 is a Promising Target for Treating Ferroptosis-Related Diseases. Front Mol Biosci. 2022;9:901565.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102.
Kramer MH, Zhang Q, Sprung R, Day RB, Erdmann-Gilmore P, Li Y, et al. Proteomic and phosphoproteomic landscapes of acute myeloid leukemia. Blood. 2022;140:1533–48.
Wei J, Xie Q, Liu X, Wan C, Wu W, Fang K, et al. Identification the prognostic value of glutathione peroxidases expression levels in acute myeloid leukemia. Ann Transl Med. 2020;8:678.
Shi ZZ, Tao H, Fan ZW, Song SJ, Bai J. Prognostic and Immunological Role of Key Genes of Ferroptosis in Pan-Cancer. Front Cell Dev Biol. 2021;9:748925.
National Cancer Institute. Genomic Data Commons Data Portal 2022. https://portal.gdc.cancer.gov/.
Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526–31.
Jayavelu AK, Wolf S, Buettner F, Alexe G, Haupl B, Comoglio F, et al. The proteogenomic subtypes of acute myeloid leukemia. Cancer Cell. 2022;40:301–17.e12.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7.
Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 2020;16:497–506.
Kloditz K, Fadeel B. Three cell deaths and a funeral: macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019;5:65.
Simonovic M, Puppala AK. On elongation factor eEFSec, its role and mechanism during selenium incorporation into nascent selenoproteins. Biochim Biophys Acta Gen Subj. 2018;1862:2463–72.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the system x(C)(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun. 2017;8:14844.
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50.
Sun MG, Williams J, Munoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, et al. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol. 2007;9:1057–65.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, et al. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem. 2003;278:37832–9.
Ishizawa J, Zarabi SF, Davis RE, Halgas O, Nii T, Jitkova Y, et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell. 2019;35:721–37.e9.
Jacques S, van der Sloot AM, Huard CC, Coulombe-Huntington J, Tsao S, Tollis S, et al. Imipridone Anticancer Compounds Ectopically Activate the ClpP Protease and Represent a New Scaffold for Antibiotic Development. Genetics. 2020;214:1103–20.
Conrad M, Proneth B. Selenium: Tracing Another Essential Element of Ferroptotic Cell Death. Cell Chem Biol. 2020;27:409–19.
Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24:1036–46.
Oh SJ, Ikeda M, Ide T, Hur KY, Lee MS. Mitochondrial event as an ultimate step in ferroptosis. Cell Death Discov. 2022;8:414.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of Mitochondria in Ferroptosis. Mol Cell. 2019;73:354–63.e3.
To TL, Cuadros AM, Shah H, Hung WHW, Li Y, Kim SH, et al. A Compendium of Genetic Modifiers of Mitochondrial Dysfunction Reveals Intra-organelle Buffering. Cell. 2019;179:1222–38.e17.
Magtanong L, Mueller GD, Williams KJ, Billmann M, Chan K, Armenta DA, et al. Context-dependent regulation of ferroptosis sensitivity. Cell Chem Biol. 2022;29:1409–18.e6.
Dixon SJ, Pratt DA. Ferroptosis: A flexible constellation of related biochemical mechanisms. Mol Cell. 2023;83:1030–42.
Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9:ra17.
Chen MS, Wang SF, Hsu CY, Yin PH, Yeh TS, Lee HC, et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget. 2017;8:114588–602.
Wang Y, Hekimi S. Understanding Ubiquinone. Trends Cell Biol. 2016;26:367–78.
Canli O, Alankus YB, Grootjans S, Vegi N, Hultner L, Hoppe PS, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–48.
Altamura S, Vegi NM, Hoppe PS, Schroeder T, Aichler M, Walch A, et al. Glutathione peroxidase 4 and vitamin E control reticulocyte maturation, stress erythropoiesis and iron homeostasis. Haematologica. 2020;105:937–50.
Hu Q, Zhang Y, Lou H, Ou Z, Liu J, Duan W, et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 2021;12:706.
Zhao J, Jia Y, Mahmut D, Deik AA, Jeanfavre S, Clish CB, et al. Human hematopoietic stem cell vulnerability to ferroptosis. Cell. 2023;186:732–47.
Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun. 2003;305:278–86.
Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502.
Liu H, Forouhar F, Seibt T, Saneto R, Wigby K, Friedman J, et al. Characterization of a patient-derived variant of GPX4 for precision therapy. Nat Chem Biol. 2022;18:91–100.
Boettcher S, Miller PG, Sharma R, McConkey M, Leventhal M, Krivtsov AV, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science. 2019;365:599–604.
Carter BZ, Mak PY, Tao W, Zhang Q, Ruvolo V, Kuruvilla VM, et al. Maximal Activation of Apoptosis Signaling by Cotargeting Antiapoptotic Proteins in BH3 Mimetic-Resistant AML and AML Stem Cells. Mol Cancer Ther. 2022;21:879–89.
Acknowledgements
The authors thank Kenneth Dunner Jr., (MD Anderson Cancer Center High Resolution Electron Microscopy Facility) for assisting with transmission electron microscopy experiments, Manu M Sebastian (MD Anderson Cancer Center Department of Veterinary Medicine and Surgery) for assisting with in vivo experiments, M.L.P. 2nd was supported by the 1R25CA240137 UPWARDS Training Program and the CPRIT Research Training Award CPRIT Training Program (RP210028), Steffen Boettcher (University of Zurich, Zurich, Switzerland) for providing cell lines, Joseph A Munch (MD Anderson Cancer Center Science Medical Library) for editing the manuscript, and Boyi Gan (MD Anderson Cancer Center Department of Experimental Radiation Oncology) for reviewing the manuscript. This work was supported in part by Japan Cancer Society Relay For Life My Oncology Dream Award (HA), The Mochida Memorial Foundation for Medical and Pharmaceutical Research (HA), The Uehara Memorial Foundation Research Fellowship (HA), TRIUMPH fellowship in the Cancer Prevention Research Institution of Texas (CPRIT) Training Program RP210028 (EA), Paul & Mary Haas Chair in Genetics (MA), NIH/NCI R41 CA275631 (MA), FDA R01 FD006118 (MA and JI) Leukemia SPORE P50 CA100632 Development Research Program (MA and JI), MD Anderson Multidisciplinary Research Program (MA and JI), MD Anderson Cancer Center Institutional Research Grant (JI), the Department of Defense CA220581 (JI), New Investigator Research Grant Program from Leukemia Research Foundation (JI), and various donor funds (MA). Part of the study was performed in the Flow Cytometry & Cellular Imaging Core Facility and High Resolution Electron Microscopy Facility at MD Anderson Cancer Center, which is supported in part by NCI Cancer Center Support Grant P30CA16672. The animal study was supported in part by the MD Anderson Cancer Center Support Grant CA016672 including animal housing and care in the Research Animal Support Facility (RASF). Imipridones ONC201 and ONC212 were kindly provided from Chimerix (Durham, NC).
Author information
Authors and Affiliations
Contributions
HA, MA, and JI designed and conceptualized the study. HA and JI designed experiments. HA, RZ, MT, SJY, AM, MLPII, and NB performed in vitro experiments and analyzed data. HA, RZ, LBO, SJY, AM, and SS performed in vivo experiments and analyzed data. HA, RZ, MT, SS, NB, ADS, MA, and JI interpreted the data and draw the conclusions. YN, PYM, and VRR provided technical and material supports of experiments. ZL and EA performed statistical analyses. HA and JI wrote the manuscript. MT, BZC, ADS, MA, and JI reviewed/revised the manuscript. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
MA is a stockholder of Chimerix. JI, MA, and ADS have filed invention disclosure forms related to the use of imipridones in cancers. ADS has received research funding from Takeda Pharmaceuticals, BMS and Medivir AB, and consulting fees/honorarium from Takeda, Novartis, Jazz, and Otsuka Pharmaceuticals. ADS is named on a patent application for the use of DNT cells to treat AML. ADS is a member of the Medical and Scientific Advisory Board of the Leukemia and Lymphoma Society of Canada.
Ethics approval and consent to participate
The mouse studies were performed following the guidelines approved by the Institutional Animal Care and Use Committees at MD Anderson Cancer Center. Primary samples were obtained from AML patients and hematopoietic stem cell donors after acquiring written informed consent following MD Anderson Cancer Center Institutional Review Board protocol and in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: In this article the funding from M.L.P. 2nd was supported by the 1R25CA240137 UPWARDS Training Program and the CPRIT Research Training Award CPRIT Training Program (RP210028) was omitted.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Akiyama, H., Zhao, R., Ostermann, L.B. et al. Mitochondrial regulation of GPX4 inhibition–mediated ferroptosis in acute myeloid leukemia. Leukemia 38, 729–740 (2024). https://doi.org/10.1038/s41375-023-02117-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-023-02117-2
- Springer Nature Limited
This article is cited by
-
Emerging role of glutathione peroxidase 4 in myeloid cell lineage development and acute myeloid leukemia
Cellular & Molecular Biology Letters (2024)