
Abstract
The FLT3-ITD mutation is associated with poor prognosis in acute myeloid leukemia (AML). FLT3 tyrosine kinase inhibitors (TKIs) demonstrate clinical efficacy but fail to target leukemia stem cells (LSC) and do not generate sustained responses. Autophagy is an important cellular stress response contributing to hematopoietic stem cells (HSC) maintenance and promoting leukemia development. Here we investigated the role of autophagy in regulating FLT3-ITD AML stem cell function and response to TKI treatment. We show that autophagy inhibition reduced quiescence and depleted repopulating potential of FLT3-ITD AML LSC, associated with mitochondrial accumulation and increased oxidative phosphorylation. However, TKI treatment reduced mitochondrial respiration and unexpectedly antagonized the effects of autophagy inhibition on LSC attrition. We further show that TKI-mediated targeting of AML LSC and committed progenitors was p53-dependent, and that autophagy inhibition enhanced p53 activity and increased TKI-mediated targeting of AML progenitors, but decreased p53 activity in LSC and reduced TKI-mediated LSC inhibition. These results provide new insights into the role of autophagy in differentially regulating AML stem and progenitor cells, reveal unexpected antagonistic effects of combined oncogenic tyrosine kinase inhibition and autophagy inhibition in AML LSC, and suggest an alternative approach to target AML LSC quiescence and regenerative potential.

Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) leukemic stem cells (LSC) reside at the apex of a cellular hierarchy that initiates and maintains disease [1]. LSC demonstrate properties of self-renewal, quiescence, and chemoresistance. LSC persistence after therapy and contribute to resistance and relapse. An internal tandem duplication (ITD) in the juxta-membrane domain of the FMS-like tyrosine kinase-3 (FLT3) is a common molecular abnormality in AML [2, 3]. FLT3-ITD mutations predict for increased risk of relapse and poor survival. FLT3 tyrosine kinase inhibitors (TKIs) have shown benefit in the treatment of FLT3-ITD AML. However, responses are not sustained and resistance remains a challenge [4,5,6,7]. There is considerable interest in understanding mechanisms of persistence of FLT3-ITD AML LSC after treatment.
Autophagy is a cellular stress response that maintains homeostasis by lysosomal degradation and recycling of damaged organelles and protein aggregates [8]. Autophagy plays a crucial role in aging, cancer and metabolic disease [9, 10]. Autophagic flux is enhanced in hematopoietic stem cells (HSC), and important for maintaining HSC function [11,12,13]. Lentiviral knockdown of autophagy genes Atg5 and Atg7 leads to impaired HSC engraftment. Atg7null or Atg5null mice develop anemia, loss of HSC function and myeloproliferation. Autophagy-deficient HSC show mitochondrial accumulation, metabolic activation, increased proliferation, myeloid differentiation, and impaired self-renewal [12, 14]. The tumor suppressor, p53, is also an important component of stress signaling, and has an important relationship between autophagy. Autophagy suppresses p53, and suppression of p53 by autophagy may contribute to tissue preservation and tumor promotion [15].
Autophagy participates in leukemia development, maintenance, and treatment resistance [16]. Autophagy inhibition has been explored as a treatment strategy in AML [17,18,19,20,21,22] and CML [23,24,25]. AML CD34 + cells were more sensitive to the autophagy inhibitor hydroxychloroquine (HCQ) than normal CD34 + cells [18]. Deletion of Atg5 or Atg7 in MLL-ENL leukemic mice prolonged survival, reduced LSC and enhanced chemosensitivity [26]. Autophagy was required for MLL-AF9 AML initiation [27]. However, the role of autophagy in modifying FLT3-ITD LSC function and treatment response is not clear.
Lys05 is a highly potent, second generation autophagy inhibitor [28]. Like HCQ, Lys05 accumulates in and deacidifies lysosomes, impairing autophagy flux [23, 28]. Here we studied the effect of Lys05 in FLT3-ITD AML, using patient samples and a genetic mouse model. We show that autophagy inhibition enhances mitochondrial respiration and activates and depletes FLT3-ITD AML LSC. However, TKI treatment unexpectedly antagonizes the effects of autophagy inhibition on LSC attrition by inhibiting mitochondrial respiration. We further show that TKI-mediated targeting of AML LSC and progenitors is p53-dependent. Autophagy inhibition enhances p53 activity and increases TKI-mediated inhibition of AML progenitors. In contrast, autophagy inhibition reduces p53 activity in LSC and reverses TKI-mediated inhibition of LSC. These findings provide new insights into the role of autophagy in modulating leukemic stem and progenitor cell function and response to tyrosine kinase inhibition.
Materials and methods
Mice
C57BL/6 mice were from Jackson Laboratory (Bar Harbor, ME), and C57BL/6.SJL mice from Charles River Laboratories (Frederick, MD). Mx1-Cre Tet2fl/fl mice and FLT-ITDki mice (Jackson Laboratory) were crossed to generate FLT3-ITDki/Mx1-Tet2f/f mice. Cre activity was induced by intraperitoneal (IP) injections of polyinosine-polycytosine (poly(I:C)) (12.5 mg/Kg) every other day for 7 doses. C57BL/6-Tg(CAG-RFP/EGFP/Map1lc3b) autophagy reporter mice were developed by Min Xie, and utilize differential pH sensitivities for red fluorescent protein (RFP; pKa 4.5) and enhanced green fluorescent protein (EGFP; pKa 5.9) [29]. NOD.Cg-Prkdcscid IL2rgtmlWjl/SzJ mice (NSG) and NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl Tg(CMV-IL3, CSF2, KITLG) (NRGS) mice were purchased from Jackson Laboratory. All experiments were performed using 8–10 weeks old mice of both sexes. Sample size was based on previous studies that established the number of mice per group required to detect significant differences. Mice were randomized between treatment groups based on sex and on levels of engraftment. Blinding was not used. Mouse care was in accordance with federal guidelines and protocols approved by the UAB Institutional Animal Care and Use Committee.
Human samples
Peripheral blood (PB) and bone marrow (BM) samples were obtained from FLT3-ITD AML and non FLT3-ITD AML patients seen at the University of Alabama at Birmingham (UAB). Patients characteristics are shown in Table S1. Normal peripheral blood stem cells were obtained from transplant donors. Mononuclear cells were isolated by Ficoll-Hypaque (Sigma Diagnostics, St. Louis, MO) centrifugation. CD34 + cells were isolated using immunomagnetic beads (Miltenyi Biotech, Auburn, CA). CD34 + CD38− and CD34 + CD38 + cells were selected by flow cytometry. MV4;11 cells were obtained from ATCC and MOLM-13 cells from the German Collection of Microorganisms and Cell Cultures. Human sample acquisition was approved by the UAB Institutional Review Board in accordance with assurances filed with the Department of Health and Human Services and met all requirements of the Declaration of Helsinki. Informed consent was obtained from AML patients and transplant donors.
Stem/progenitor cell culture
Human CD34+ cells were cultured in Stemspan serum-free enhanced medium (SFEM; Stem Cell Technologies) with human interleukin-3 (IL-3)(20 ng/ml), FLT3 ligand (FLT3L)(100 ng/ml); thrombopoietin (TPO)(50 ng/ml); stem cell factor (SCF) (100 ng/ml)(PeproTech, Cranbury, NJ) for 48 h [30]. Mouse C-kit+ cells were cultured in StemSpanTM SFEM medium supplemented with murine IL-3 (10 ng/ml); GM-CSF (10 ng/ml); SCF (25 ng/ml) and TPO (25 ng/ml)(PeproTech) for 24 h [11].
Mouse hematopoietic cell analyses
BM cells from leukemic FLT3-ITDki Mx1-cre Tet2f/f mice (CD45.2) were transplanted into irradiated (400 cGy × 2) CD45.1 mice. Following leukemia development, mice were treated with Lys05 (20 mg/kg/day, IP), AC220 (10 mg/kg/day, oral gavage) or vehicle for 2 weeks. Secondary repopulation was analyzed by transplanting pooled BM from treated mice (2 × 106 cells/mouse) with equal numbers of wild-type (WT) cells (CD45.1) into CD45.1 mice irradiated at 800 cGy. Blood, BM, and spleen cells were labeled with anti-CD45.1, CD45.2 and lineage-specific antibodies. Long-term HSC (LT-HSC; Lin−Sca1+cKit+Flt3−CD150 + CD48−), short-term HSC (ST-HSC; Lin− Sca1+cKit+ Flt3-CD150-CD48-), multipotent progenitors (MPP; Lin- Sca1+cKit+Flt3−CD48+) and granulocyte-macrophage progenitors (GMP; Lin-Sca1-c-Kit+CD34 + FcRII/IIIhi) were analyzed using a BD LSR Fortessa X-20 flow cytometer (BD, San Jose, CA) and FlowJo (Version 8.5.2) [31].
For xenograft experiments, 5 × 106 peripheral blood mononuclear cells from FLT3-ITD AML patients were incubated for 30 min with OKT3 antibody (Bio X Cell, BE0001-2) and transplanted into irradiated NSG or NRGS mice. BM and spleen cells were analyzed for human CD45 + cells and subpopulations by labeling with human antibodies (CD34, CD33, CD38, CD14,CD11b, CD3, CD19). Details of antibodies are shown in Table S2.
For cell cycle analysis, BM cells were labeled for surface markers, treated with Cytofix/Cytoperm™ (BD), labeled with Ki67 and DAPI, and analyzed by flow cytometry.
RNA interference
AML CD34+ cells were transfected with siRNAs to ATG5 (SMARTpool,Cat:M-004374-04-0005, Dharmacon-Horizon Discovery, Lafayette, CO) using an Amaxa cell Nucleofector IIb (Lonza, Basel, Switzerland) [32]. FITC-labeled siRNA (Silencer™ Select Negative Control No. 1 siRNA, Thermo Fisher, Waltham, MA) was used to evaluate transfection efficiency, with positivity above 90%. MV4-11 and Molm13 (MOI = 2) were transduced with PLKO lentivirus vectors expressing ATG5 or control shRNA with polybrene (4 µg/ml). Transduction efficiency exceed 90%. LSK cells were transduced with SI506A-1 vectors (System Bioscience, SBI) expressing EF1a-GFP-T2A-Puro and ATG5 (5′-TATGAAGAAAGTTATCTGGGTA-3′), p53 (5′-GTACATGTGTAATAGCTCC-3′) or control shRNA. LSK cells from FLT3-ITD leukemic mice were cultured overnight in SFEM medium with cytokines (10 ng/ml SCF; 10 ng/ml TPO) and exposed to lentivirus (MOI = 100) and TransDux (SBI) with centrifugation at 2500 rpm for 90 min. Transduction efficiency after 48 h of culture for control, ATG5 and P53 shRNA expressing vectors was 82.7%, 79.8% and 78.3% respectively. Transduced cells were injected into irradiated recipient CD45.1 mice (25,000 CD45.2 LSK cells plus 200,000 CD45.1 supportive cells per mouse).
Extracellular flux analysis (Mito Stress Test)
c-kit+ cells were isolated using immunomagnetic beads (Miltenyi Biotech). Oxygen consumption rate (OCR) was measured using the Seahorse XF96 analyzer (Agilent) as previously described [31].
Statistics
Results are shown as mean plus/minus standard error of the mean (SEM). Significance values were calculated using Prism version 9.0 software (GraphPad Prism, La Jolla, CA) using parametric and nonparametric t-tests (Mann–Whitney test), or analysis of variance as appropriate based on normality of distribution. Survival was analyzed using Kaplan–Meier analysis.
Results
Autophagy inhibition reduces human FLT3-ITD AML CD34+cell growth and enhances TKI sensitivity in vitro
MV4-11 and Molm13 FLT3-ITD AML cells were transduced with a GFP-LC3-RFP-LC3ΔG autophagy reporter plasmid (Fig. 1A). The expressed protein is cleaved by ATG4 proteases into equimolar amounts of GFP-LC3 and RFP-LC3ΔG. GFP-LC3 is recruited to the autophagosome and degraded, whereas RFP-LC3ΔG remains cytosolic and intact. The GFP:RFP signal ratio inversely correlates with autophagic activity [33]. Reporter function was validated by showing that autophagy inducers (Torin, Rapamycin, and PP242) reduced GFP:RFP ratios, and autophagy inhibitors (Lys05, CQ, and SAR405) increased GFP:RFP ratios (Fig. S1A). To study the relationship of autophagy to TKI and Lys05 sensitivity, cells with low autophagy flux (AT-low, top 20% of GFP-expressing cells) or high autophagy flux (AT-high, bottom 20% of GFP-expressing cells) were purified (Fig. 1A). Differences in GFP:RFP ratios between AT-high and AT-low cells were maintained after 72 h culture (Fig. 1A, Fig. S1B). AT-high cells showed reduced apoptosis after treatment with the Flt3 TKI AC220, and increased apoptosis after exposure to the autophagy inhibitor Lys05, compared to AT-low cells (Fig. 1A, Fig. S1C). Autophagy inhibition via shRNA-mediated knockdown of ATG5 enhanced sensitivity of FLT3-ITD + Molm13 and MV4-11 but not non-FLT3-ITD OCI-AML3 AML cells to the AC220 (Fig. 1B, Fig. S1D). Similarly treatment with the autophagy inhibitors Lys05 or SAR405 treatment enhanced sensitivity of Molm13 and MV4-11 but not OCI-AML3 AML cells to the FLT3 TKIs AC220 or Giltertinib (Fig. 1C, Fig. S1E). The combination of AC220 and Lys05 led to synergistic growth inhibition (Combination index 0.59 to 0.88) (Fig. S1F).

A GFP-LC3-RFP-LC3ΔG expressing plasmid used to measure autophagy in live cells (left). Flow cytometry sorting strategy and histogram for autophagy flux (AT) high versus AT low cells on Molm13 cell lines (middle). Apoptosis of AT high cells and AT low cells measured by Annexin V and DAPI after exposure to Lys05 (5 μM) and/or AC220 (2 nM) for 48 h (right). B Survival of Molm13, MV4-11 and OCI-AML3 cells transduced with ATG5-shRNA expressing lentivirus vectors and cultured for 48 h with/without AC220 treatment (20 nM). C Survival of Molm13, MV4-11 and OCI-AML3 cells after 48 h exposure to drugs at indicated concentration. Significance values: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant. Results represent mean ± SEM of multiple replicates.
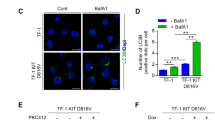
CD34+ progenitor cells and CD34− mature cells from FLT3-ITD AML patients showed similar levels of autophagy flux. Normal and FLT3-ITD AML CD34+ progenitors also showed similar autophagy flux (Fig. S2A). Lys05 inhibited autophagy flux in AML CD34+ cells, evidenced by increased LC3B expression, LC3B-II expression, and LC3B punctae formation (Fig. 2A, Fig. S2B). AC220 treatment did not affect autophagy flux in AML CD34+ cells, in contrast to prior reports for FLT3-ITD AML cell lines or unselected primary cells [34]. Lys05 reduced FLT3-ITD AML CD34+ and CD34− cell growth and survival, and the combination of Lys05 and AC220 synergistically increased inhibition of cell growth and survival compared to AC220 alone (Fig. 2B, Fig. S2C, D). In contrast, Lys05 did not affect survival and minimally affected growth of non-FLT3-ITD AML CD34+ and CD34− cells (Fig. 2B, Fig. S2C). ATG5 knockdown using siRNA also significantly reduced AML CD34+ cell growth and cell survival, and ATG5 knockdown cells showed increased inhibition with AC220 compared to control siRNA treated cells (Fig. 2C). Autophagy inhibition with Lys05 only modestly inhibited normal CD34+ cell proliferation and did not affect survival (Fig. S2E). We conclude that autophagy inhibition selectively targets FLT3-ITD AML compared with non-FLT3-ITD AML and normal CD34+ progenitor cells and enhances their sensitivity to TKI.

A Effect of Lys05 (5 μM) and AC220 (20 nM) on autophgy flux in FLT3-ITD AML CD34+ cells. LC3B-FITC MFI was measured in by flow cytometry after 24 h exposure (n = 4–5) (left). LC3B protein expression was measured by Western blotting after 16 h treatment from FLT3-ITD AML #178 (middle). LC3B puncta were measured by immunofluorescence microscopy after 4 h exposure from FLT3-ITD AML #178 (right). B FLT3-ITD AML CD34+ cells (n = 7–8) and non FLT3-ITD AML CD34+ cells (n = 4) were treated with Lys05 with/without AC220 for 48 h, cell survival was measured by Annexin V and DAPI labeling (left) and cell growth was meadured using CellTiter-Glo (right). C FLT3-ITD AML CD34+ cells (n = 4) were transfected with ATG5-siRNA and control-siRNA. Western blotting was performed to measure ATG5 protein levels (left). Cell survival (middle) and cell growth (right) and were measured after culture for 48 h with/without AC220(20 nM) treatment. Significance values: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant. Results represent mean ± SEM of multiple replicates.
Autophagy inhibition enhances TKI-mediated targeting of primary human FLT3-ITD cells engrafted in immunodeficient mice
We evaluated effects of autophagy inhibition in vivo on primary FLT3-ITD AML cells xenografts established in immunodeficient NSG or NRGS mice (Fig. 3A). Mice were injected with primary human cells from three FLT3-ITD AML patients. Following establishment of engraftment 8–12 weeks after transplantation, mice were treated with vehicle, AC220 (10 mg/kg/day, gavage), Lys05 (20 mg/kg/day, IP) or the combination for 3 weeks. We confirmed that in vivo treatment with Lys05 increased LC3b accumulation in engrafted human CD45+ cells (Fig. S3A). Lys05 treatment did not consistently impact AML CD45+ cells engraftment. AC220 reduced human CD45+ cells in 2 of the 3 samples (#173 and #395). Combined treatment with Lys05 and AC220 led to significantly increased inhibition of human CD45+ cells compared to AC220 alone in all samples (Fig. 3B, Fig. S3B). These results indicate that autophagy inhibition enhanced TKI-mediated targeting of primary human FLT3-ITD cells in vivo even when cells were insensitive to AC220 alone.

A Experimental design. Irradiated NSG mice (250 cGy) or NRGS mice (400 cGy) were transplanted with peripheral blood mononuclear cells from FLT3-ITD AML patients (5 × 106 cells per mouse, OKT3 antibody incubated). After successful human cell engraftment was confirmed after 8–12 weeks, mice were treated with vehicle, Lys05, AC220 or combination for 3 weeks, and human cells in PB, BM and spleen cells were analyzed (n = 3–8). B The percents of human CD45+ cells engrafted in BM of NRGS or NSG mice engrafted with FLT3-ITD AML samples #173, #395 and #591 are shown. C BM cells were labeled with Ki67 and DAPI, and cell cycle of human CD34 + CD38− cells from FLT3ITD AML #395 was measured by flow cytometry. Representative cell cycle flow plots and compiled results are shown. Significance values: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant. Results represent mean ± SEM of multiple replicates.
Quiescence is a key characteristic of LSC. Lys05 treatment reduced primitive CD34 + CD38− cells in G0 and increased cells in G1. Surprisingly, addition of AC220 to Lys05 increased CD34 + CD38− cells in G0 compared to Lys05 alone, indicating that AC220 inhibited Lys05-induced cell cycling (Fig. 3C). Lys05 did not significantly affect cell cycle in CD34 + CD38 + progenitor cells (Fig. S3C). These observations indicate that autophagy inhibition reduces quiescence of primitive FLT3-ITD AML cells, but that FLT3 kinase inhibition counters this effect.
Autophagy inhibition enhances TKI-mediated inhibition of FLT3 ITD AML progenitors and increases AML stem cell cycling in a genetic mouse model
We investigated the effect of autophagy inhibition on FLT3-ITD AML LSC using a genetic mouse model generated by crossing FLT3-ITDki and Mx1-Cre Tet2fl/fl mice. This model is similar to a previously reported FLT3-ITD AML model generated by crossing FLT3-ITDki and Vav-Cre Tet2fl/fl mice [35]. Mice develop a lethal AML-like disease (median survival 200 days), and LSC are present within primitive Lin− Sca-1+ Kit + (LSK) cells, which include ST-HSC and MPP, but are depleted of LT-HSC. LSK cells in this model are reported to show limited sensitivity to TKI treatment [35]. We found that primitive LSK cells from AML mice showed modestly enhanced autophagy flux compared to differentiated cells (Fig. S4A), but LSK cells from AML and normal mice showed similar autophagy flux (Fig. S4B).
AML BM cells (CD45.2) were transplanted into irradiated recipients (CD45.1) to generate cohorts with similar leukemia onset. Mice were treated with Lys05, AC220, or the combination for 2 weeks (Fig. 4A). We confirmed that Lys05 inhibited autophagy flux in vivo by showing that Lys05 treatment enhanced maintenance of EGFP puncta in BM cells of CAG-RFP/EGFP/Map1lc3b reporter mice (Fig. S4C) [36]. Circulating WBC and neutrophils (Fig. S4D) were reduced in all treatment arms in AML mice at time of completion of treatment. AC220 and combination treatment effectively reduced spleen size, splenic GMP, MPP and STHSC (Fig. S4E), compared to vehicle, consistent with reduction in disease burden. AC220 and combination treatment reduced BM GMP but not BM MPP and ST-HSC (Fig. 4B, Fig. S4F). In WT mice, Lys05 did not affect WBC, increased neutrophils slightly and reduced BM GMP without affecting MPP, ST-HSC and LT-HSC (Fig. S4G) after 2 weeks of treatment.

A Experimental plan. BM cells were obtained from FLT3-ITDki Mx1-cre Tet2f/f CD45.2 mice following leukemia induction and were transplanted into irradiated (400 cGy × 2) 8–10 weeks old CD45.1 mice. Blood draws were performed every 2 weeks to check for leukemia development. After 6–8 weeks, mice were treated with vehicle, Lys05 (20 mg/kg/day, IP), AC220 (10 mg/kg/day, oral gavage), or the combination of AC220 and Lys05 for 2 weeks (n = 10–12). B The flow gating strategy for stem/progenitor subsets is shown (left) and total number of GMP and ST-HSC per 2 femurs and 2 tibiae (4 bones) are shown (right). C BM cells were labelled with Ki67 and DAPI, and cell cycle analysis was performed (n = 6–7). A representative FACS plot and a graph of compiled data for LSK cells are shown. Significance values: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant. Results represent mean ± SEM of multiple replicates.
Lys05 treatment significantly reduced the proportion of BM LSK cells in G0 and increased cells in G1 compared to vehicle, indicating enhanced cell cycling. On the other hand, AC220 or the combination of Lys05 and AC220 did not significantly alter LSK cell cycling (Fig. 4C). Similar results were seen for ST-HSC (not shown). Lys05 and AC220 treatment did not affect cell cycle in GMP cells, but the combination of Lys05 and AC220 modestly increased GMP cycling (Fig. S4H). Treatment of wild type mice with Lys05 led to modest reduction in BM LSK cells in G0, but to a lesser extent than AML LSK cells (Fig. S4I).
Autophagy inhibition reduces repopulating capacity of FLT3-ITD AML stem cells in a genetic mouse model
The effect of treatment on LSC repopulation capacity was evaluated by transplanting BM cells from treated mice (CD45.2) into WT secondary recipient mice (CD45.1) with competitor BM cells (CD45.1) (Fig. 5A). Lys05-treated cells generated reduced total donor chimerism, donor myeloid chimerism, BM cellularity, donor myeloid cells, B cells, T cells, GMP, MPP and ST-HSC in BM of secondary recipients compared to vehicle treated cells. AC220-treated cells also generated reduced repopulation in secondary recipients. Surprisingly, cells from mice receiving combination treatment did not show reduced secondary repopulation compared to cells from vehicle treated mice, indicating that combination of AC220 with Lys05 has antagonistic effects on AML LSC repopulating capacity (Fig. 5B, Fig. S5A–D).

A Experimental design: FLT3-ITD AML mice were treated for 2 weeks with vehicle, Lys05, AC220 or the combination of Lys05 and AC220, as described for Fig. 4A, and 2 × 106 BM cells (CD45.2+) obtained from treated mice were injected into irradiated (400 cGy×2) recipient CD45.1 mice via tail vein together with 2 × 106 BM cells from wild type CD45.1 mice (n = 6–8). B Donor myeloid chimerism from peripheral blood were monitored every 4 weeks. The total number of donor GMP and ST-HSC per 2 femurs and 2 tibiae (4 bones) are shown. C LSK cells isolated from FLT3-ITDki /Mx1-Cre Tet2f/f leukemic BM were transduced with lentivirus vectors expressing ATG5-shRNA and GFP or ctrl-shRNA and GFP. Reduction in ATG5 protein levels in GFP+ cells was confirmed by Western blotting. Engrafted mice were treated with AC220 or vehicle for 2 weeks. Total number of GFP + GMP and GFP + ST-HSC in BM of recipient mice (2 femurs and 2 tibias) are shown (n = 4–7). Significance values: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant. Results represent mean ± SEM of multiple replicates.
We evaluated the effects of genetic targeting of autophagy in murine AML stem cells by transducing FLT3-ITD AML cells with lentivirus vectors expressing ATG5-shRNA or control-shRNA and GFP. We confirmed reduction in ATG5 mRNA and protein levels (Fig. 5C, Fig. S5E) in LSK cells. Mice engrafted with ATG5 or control shRNA expressing LSK cells were treated with AC220 or vehicle for 2 weeks. ATG5 knockdown resulted in increased numbers of BM GFP+ cells, myeloid cells, GMP and ST-HSC. AC220 treatment reduced ATG5-knockdown GFP+ populations but not control GFP+ populations (Fig. 5C, Fig. S5F). These results are consistent with increased cell cycling following autophagy inhibition. Differences between results of ATG5 shRNA and Lys05 treatment likely reflect differential effects of chronic, long-standing autophagy inhibition with ATG5 shRNA versus acute inhibition with Lys05 and targeting and upstream autophagy gene versus the lysosome.
These results indicate that autophagy inhibition enhances AML LSC cycling and reduces their repopulating potential. However, increased AML LSC attrition is antagonized by TKI treatment, indicating that it is FLT3 kinase dependent. In contrast, the combination of Lys05 and TKI synergistically inhibits AML progenitor cells.
Mitochondrial respiration is enhanced in AML stem cells following autophagy inhibition but is reduced in combination with tyrosine kinase inhibition
We performed RNA sequencing on AML LSC (LSK) and progenitor cells (GMP) from mice treated with Lys05, A220 and the combination of Lys05 and AC220. Gene set enrichment analysis (GSEA) confirmed enrichment of quiescence (Fig. S6A), AML stemness [37], and HSC signatures in AML LSK cells (Fig. S6B), and GMP and myeloid signatures [38] in AML GMP cells (Fig. S6C). Lys05-treated AML LSK cells showed enhanced cell cycle and reduced quiescence signatures compared to vehicle treated cells (Fig. 6A, B). Quiescence signatures were unchanged in LSK cells treated with AC220 or the combination of AC220 and Lys05 (Fig. S6D). These results were consistent with our cell cycle analysis (Fig. 4C).

RNASeq analysis was performed on FLT3-ITD AML LSK cells and GMP sorted from Lys05, AC220, combination and vehicle treated mice (4 replicates per group), and Gene-set Enrichment Analysis (GSEA) of differentially expressed genes was performed. A, B Representative GSEA plot showing downregulation of a quiescence gene set and upregulation of G2M checkpoint gene sets in LSK from Lys05 versus vehicle treated mice (A). Heatmap of expression of quiescence and cell cycling genes in Lys05 treated LSK cells (B). C Representative plots showing upregulation of oxidative phosphorylation gene sets in LSK from Lys05 versus vehicle treated mice. GSEA plots showing downsregulation of OXPHOS gene sets in LSK from mice treated with AC220 versus vehicle, combination of Lys05 and AC220 versus vehicle and combination of Lys05 and AC220 versus Lys05. Significance values: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant. Results represent mean ± SEM of multiple replicates.
Lys05-treated LSK cells were enriched for mitochondrial, oxidative phosphorylation (OXPHOS) (Fig. 6C, Fig. S6E), long chain fatty acid synthesis, fatty acid beta oxidation, TCA cycle and glutathione metabolism gene signatures (Fig. S6F). However, OXPHOS signatures were downregulated in LSK cells treated with AC220 compared to vehicle. OXPHOS signatures were also reduced in LSK cells treated with a combination of AC220 and Lys05 compared to vehicle or to Lys05 alone (Fig. 6C).
Lys05 treatment increased mtDNA copy number in LSK cells, indicating increased mitochondrial mass (Fig. 7A), but decreased mitochondrial membrane potential measured by TMRE labeling (Fig. 7B), suggesting that autophagy inhibition allows accumulation of stressed mitochondria. We evaluated mitochondrial respiration by extracellular flux analysis using the Seahorse apparatus to measure oxygen consumption rate (OCR). In view of the high cell number requirement, BM c-Kit+ progenitor cells rather than purified LTHSC were used for these assays. Lys05 modestly increased maximal mitochondrial respiratory capacity, but not basal or ATP-linked respiration. AC220 did not significantly affect OCR by itself, but markedly reduced basal and maximal OCR with Lys05 (Fig. 7C). Since increased mitochondrial respiration plays a critical role in promoting HSC activation and differentiation, reduced mitochondrial respiration may explain the antagonistic effects of TKI on Lys05-mediated LSC attrition.

A Mitochondrial DNA (mtDNA) from Lys05 and vehicle treated LSK cells (n = 5) was quantified based on ratio of mt-CoxII or mt-CytB to nuclear DNA. B Mitochondrial membrane potential was measured by TMRE labeling in LSK cells from Lys05 and vehicle treated mice (n = 5). C Extracellular flux analysis for oxygen consumption rate (OCR) was performed on c-Kit enriched cells obtained from FLT3-ITDki/Mx1-Cre Tet2f/f leukemic mice after 2 weeeks of indicated drug treatment (n = 4–5). A representative plot and a graph of compiled data are shown. Significance values: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant. Results represent mean ± SEM of multiple replicates.
Autophagy inhibition differentially modulates p53 activity in TKI-treated FLT3-ITD AML progenitor and stem cells
We evaluated the relationship between autophagy and p53 activation in FLT3-ITD AML cells. Autophagy induction (Torin, starvation) reduced p53 protein levels (Fig. S7A) whereas autophagy inhibitors (CQ, SAR405) increased p53 protein levels (Fig. S7B). Autophagy inhibition (Lys05) prolonged p53 protein half-life in Cycloheximide-treated cells whereas autophagy induction (Torin) enhanced p53 degradation (Fig. S7C). Moreover p53 protein levels were increased in AT-low cells compared to AT-high cells (Fig. S7D). These observations together indicate that p53 may be a substrate of the autophagosome. We further show that p53 knockdown abrogated effects of Lys05 on survival and growth of FLT3-ITD AML cells (Fig. S7E), and of AC220 and combined Lys05 and AC220 treatment on survival (Fig. S7F, G).
RNA sequencing showed that p53 gene signatures were enriched in GMP from Lys05 and AC220 treated mice compared with AC220 treated mice (Fig. 8A). RT-Q-PCR analysis confirmed that p53 target genes p21, Noxa and Puma were upregulated in GMP from combination compared to AC220 treated mice (Fig. 8A). p53 target genes p21, Bax, Noxa and Puma were also upregulated in human FLT3-ITD AML CD34+ progenitor cells treated with the combination compared to AC220 alone (Fig. 8A). In contrast, p53 gene signatures and p53 target genes were reduced in LSK from Lys05 and AC220 treated mice compared with AC220 treated mice (Fig. 8B). Therefore autophagy inhibition enhances p53 activity in TKI-treated progenitor cells, but paradoxically reduces p53 activity in TKI-treated LSC.

A Enrichment plot for combination of Lys05 and AC220 comapred with AC220 treatment for GMP cells (left). The expression of p53 target genes, p21, Bax, Noxa and Puma, measured by Q-RT-PCR in GMP cells treated with the Lys05 or/and AC220 for 2 weeks in vivo (n = 3–5) (middle). Expression of p53 target genes, p21, BAX, NOXA and PUMA, in FLT3-ITD AML CD34+ cells after treatment with Lys05 or/and AC220 for 48 h (n = 4) (right). B Enrichment plot for combination of Lys05 and AC220 comapred with AC220 treatment for LSK cells (left). The expression of p53 target genes, p21, Bax, Noxa and Puma, measured by Q-RT-PCR in LSK cells treated with the Lys05 or/and AC220 for 2 weeks in vivo (n = 3–5) (right). C LSK cells isolated from FLT3-ITDki /Mx1-Cre Tet2f/f leukemic BM were transduced with lentivirus vectors expressing P53-shRNA and GFP or ctrl-shRNA and GFP. Engrafted mice were treated with Lys05, AC220 or vehicle for 2 weeks. Reduction in P53 protein levels in GFP+ cells was confirmed by Western blotting. Total number of GFP+ myeloid cells and GFP + ST-HSC in BM of recipient mice (2 femurs and 2 tibias) are shown (n = 3–5). Significance values: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant. Results represent mean ± SEM of multiple replicates.
We evaluated the role of p53 in murine FLT3-ITD LSC using lentivirus vectors expressing p53-shRNA (Fig. S7H). We confirmed effective inhibition of p53 mRNA and protein expression in LSK cells (Fig. 8C, Fig. S7I). Mice engrafted with control-shRNA and p53-shRNA expressing LSK cells were treated with vehicle or Lys05 or AC220 for 2 weeks. p53 shRNA expressing GFP+ cells and GFP+ myeloid cells were increased compared to control shRNA expressing cells in BM of Lys05 treated mice, but GFP + LSK and ST-HSC were not increased (Fig. 8C, Fig. S7J). These results support a role for p53 in mediating effects of autophagy inhibition in mature leukemic cells but not in LSC. p53 knockdown led to increased GFP+ total cells, myeloid cells, LSK and ST-HSC in BM of AC220 treated mice, indicating that AC220-mediated inhibition of both mature leukemic cells and LSC is p53-dependent. Enhanced p53 activity seen with Lys05 given in combination with AC220 may explain increased inhibition of committed progenitors and mature cells, whereas reduced p53 activity seen with the combination may explain reduced LSC inhibition compared to AC220 alone.
Discussion
Autophagy is a cellular degradation process of adaptation to intracellular and environmental stresses. There is growing evidence that autophagy inhibition can limit growth of established tumors and enhance response to therapy. Our studies show that autophagy inhibition enhances the sensitivity of human FLT3-ITD AML progenitor cells to TKI and the combination of autophagy inhibition and TKI significantly reduces leukemia burden in FLT3-ITD AML mice. Importantly, autophagy inhibition increases FLT3-ITD AML LSC activation and cycling and depletes their secondary repopulating potential. However, TKI treatment unexpectedly antagonized the ability of autophagy inhibition to deplete repopulating capacity of AML LSC. These results indicate that FLT3-ITD kinase activity drives increased LSC attrition following autophagy inhibition, and autophagy protects FLT3-ITD AML LSC from oncogenic stress. They also indicate that combining TKI treatment with autophagy inhibition differentially affects AML stem cells versus progenitor cells. Finally, they reveal an unexpected, stem cell-specific, antagonistic interaction of autophagy inhibition with FLT3-ITD kinase inhibition.
Quiescent HSC exhibit low OXPHOS levels and switch to a high OXPHOS state following activation [39, 40]. Autophagy-deficient HSC demonstrate increased mitochondrial content accompanied by an activated metabolic state, impaired self-renewal and regenerative potential, and enhanced myeloid differentiation [12]. Here we show that autophagy inhibition leads to mitochondrial accumulation, perturbed mitochondrial function and increased mitochondrial respiratory capacity in AML stem cells. Altered mitochondrial metabolism may explain increased activation and reduced repopulating potential. Attenuation of mitophagy may also contribute to myeloid differentiation, cell cycle arrest, and reduced self-renewal [41, 42]. Other metabolic changes, including altered lipid metabolism, which has complex interplays with autophagy, may also play a role [43]. OXPHOS was markedly downregulated with combined TKI treatment and autophagy inhibition, indicating that increased mitochondrial respiration following autophagy inhibition is FLT3-ITD kinase activity driven, and potentially explaining antagonistic effects of TKI on autophagy inhibition-mediated activation and attrition of AML LSC.
Autophagy inhibition enhanced TKI-mediated targeting of committed progenitors, mature cells and FLT3-ITD cell lines. Autophagy upregulation can be a mechanism for escape from drug treatment. In CML, another tyrosine kinase-dependent leukemia, TKI treatment induces autophagic flux, and combined autophagy inhibition and TKIs treatment enhances eradication of LSC [23, 24, 44, 45]. FLT3-ITD expression can increase autophagy flux in AML cell lines [34]. We observed enhanced sensitivity of FLT3-ITD + AML progenitors to autophagy inhibition compared to FLT3-ITD negative cells. These results are consistent with a recent study found that identified autophagy as an important mechanism of TKI resistance in FLT3-ITD AML [46]. However, unlike this and previous studies, we did not observe differences in autophagy flux between FLT3-ITD AML and normal stem and progenitor cells, or increased autophagy flux with TKI treatment, suggesting that FLT3-ITD effects on autophagy may be cell context-dependent.
AML CD34+ cell sensitivity to autophagy inhibition has been associated with intact p53 protein [18]. p53 knockdown prevented TKI-mediated inhibition of both primitive and mature leukemic cells. Our studies suggest that p53 is a substrate for the autophagosome and that autophagy inhibition reduced p53 degradation and increased p53 levels in FLT3-ITD cell lines, and upregulated p53 activity in TKI-treated FLT3-ITD progenitor cells. p53 upregulation following autophagy inhibition may contribute to enhanced TKI-mediated targeting of committed progenitors. On the other hand, autophagy inhibition downregulated p53 activity in TKI-treated AML LSC, potentially contributing to reduced TKI-mediated inhibition of LSC. The mechanisms underlying stage specific differences in p53 regulation are not known and require further investigation.
Efforts for clinical development of autophagy inhibitors include testing of HCQ and derivatives targeting lysosomes, and of inhibitors specifically targeting key autophagy mediators including ULK1 [47] and VPS34 [48]. In clinical trials, HCQ failed to achieve consistent autophagy inhibition at maximum tolerated doses. Deletion or pharmacological inhibition of ULK1 was shown to selectively target CML LSCs when combined with TKI treatment. The current studies demonstrate the potential for autophagy inhibition to activate and impair FLT3-ITD AML LSC function. However, in this setting, oncogenic tyrosine kinase signaling was necessary for LSC activation and depletion, and TKI inhibition antagonized the effects of autophagy inhibition. On the other hand, autophagy inhibition enhanced TKI-mediated targeting of AML progenitors. These observations highlighted the complex and differing effects of autophagy inhibition and its ability to sensitize or antagonize effects of specific anti-leukemia treatments, at different stages of leukemia cell maturation and between different leukemias. Importantly, these studies provide a foundation for further investigation of strategies to deplete LSC regenerative capacity in AML and other malignancies.
Data availability
RNA sequencing data is deposited in NCBI’s Gene Expression Omnibus (GEO) and is accessible through GEO Series accession number: GSE169750.
References
Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129:1577–85.
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014;28:1586–95.
Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med. 2019;381:1728–40.
Levis M, Perl AE. Gilteritinib: potent targeting of FLT3 mutations in AML. Blood Adv. 2020;4:1178–91.
Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–3.
Cortes JE, Khaled S, Martinelli G, Perl AE, Ganguly S, Russell N, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20:984–97.
Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64.
Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–62.
Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95.
Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494:323–7.
Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543:205–10.
Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–76.
Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208:455–67.
White E. Autophagy and p53. Cold Spring Harb Perspect Med. 2016;6:a026120.
Rothe K, Porter V, Jiang X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int J Mol Sci. 2019;20:461.
Rudat S, Pfaus A, Cheng YY, Holtmann J, Ellegast JM, Buhler C, et al. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia. 2018;32:2189–202.
Folkerts H, Hilgendorf S, Wierenga ATJ, Jaques J, Mulder AB, Coffer PJ, et al. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017;8:e2927.
Chen X, Clark J, Wunderlich M, Fan C, Davis A, Chen S, et al. Autophagy is dispensable for Kmt2a/Mll-Mllt3/Af9 AML maintenance and anti-leukemic effect of chloroquine. Autophagy. 2017;13:955–66.
Liu Q, Chen L, Atkinson JM, Claxton DF, Wang HG. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. 2016;7:e2361.
Larrue C, Saland E, Boutzen H, Vergez F, David M, Joffre C, et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood. 2016;127:882–92.
Stankov MV, El Khatib M, Kumar Thakur B, Heitmann K, Panayotova-Dimitrova D, Schoening J, et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia. 2014;28:577–88.
Baquero P, Dawson A, Mukhopadhyay A, Kuntz EM, Mitchell R, Olivares O, et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia. 2019;33:981–94.
Helgason GV, Karvela M, Holyoake TL. Kill one bird with two stones: potential efficacy of BCR-ABL and autophagy inhibition in CML. Blood. 2011;118:2035–43.
Karvela M, Baquero P, Kuntz EM, Mukhopadhyay A, Mitchell R, Allan EK, et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy. 2016;12:936–48.
Sumitomo Y, Koya J, Nakazaki K, Kataoka K, Tsuruta-Kishino T, Morita K, et al. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood. 2016;128:1614–24.
Porter AH, Leveque-El Mouttie L, Vu T, Bruedigam C, Sutton J, Jacquelin S, et al. Acute myeloid leukemia stem cell function is preserved in the absence of autophagy. Haematologica. 2017;102:e344–7.
McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci USA. 2012;109:8253–8.
Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–51.
Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature. 2017;551:384–8.
Abraham A, Qiu S, Chacko BK, Li H, Paterson A, He J, et al. SIRT1 regulates metabolism and leukemogenic potential in CML stem cells. J Clin Investig. 2019;129:2685–701.
Cook AM, Li L, Ho Y, Lin A, Li L, Stein A, et al. Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood. 2014;123:2826–37.
Kaizuka T, Morishita H, Hama Y, Tsukamoto S, Matsui T, Toyota Y, et al. An Autophagic Flux Probe that Releases an Internal Control. Mol Cell. 2016;64:835–49.
Heydt Q, Larrue C, Saland E, Bertoli S, Sarry JE, Besson A, et al. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene. 2018;37:787–97.
Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27:502–15.
Li L, Wang ZV, Hill JA, Lin F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol. 2014;25:305–15.
Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–7.
van Galen P, Hovestadt V, Wadsworth Ii MH, Hughes TK, Griffin GK, Battaglia S, et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell. 2019;176:1265–81.e1224.
Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596–606.
Hsu P, Qu CK. Metabolic plasticity and hematopoietic stem cell biology. Curr Opin Hematol. 2013;20:289–94.
Pei S, Minhajuddin M, Adane B, Khan N, Stevens BM, Mack SC, et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell. 2018;23:86–100.
Nguyen TD, Shaid S, Vakhrusheva O, Koschade SE, Klann K, Tholken M, et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood. 2019;133:168–79.
Xie Y, Li J, Kang R, Tang D. Interplay Between Lipid Metabolism and Autophagy. Front Cell Dev Biol. 2020;8:431.
Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Investig. 2009;119:1109–23.
Helgason GV, Mukhopadhyay A, Karvela M, Salomoni P, Calabretta B, Holyoake TL. Autophagy in chronic myeloid leukaemia: stem cell survival and implication in therapy. Curr Cancer Drug Targets. 2013;13:724–34.
Koschade SE, Klann K, Shaid S, Vick B, Stratmann JA, Tholken M, et al. Translatome proteomics identifies autophagy as a resistance mechanism to on-target FLT3 inhibitors in acute myeloid leukemia. Leukemia. 2022;36:2396–2407.
Martin KR, Celano SL, Solitro AR, Gunaydin H, Scott M, O’Hagan RC, et al. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience. 2018;8:74–84.
Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10:1013–9.
Acknowledgements
This work was supported by NIH grant R01 CA172447 and R01 CA248794 to RB. We thank Drs Victor Darley-Usmar and Balu Chacko at the Comparative Mitochondrial Health Assessment Core for the cellular bioenergetics assays. We thank Vidya Sagar Hanumanthu for providing help with FACS sorting at the UAB Comprehensive Flow Cytometry Core; the UAB Animal resources Center for maintaining mice colonies; Maya Robinson and Amanda Mullens for obtaining and processing human samples; Mason Harris for assistance with animal studies. Immunofluorescence imaging was performed at the High-Resolution Imaging Facility at UAB.
Author information
Authors and Affiliations
Contributions
Conceptualization: SQ, RB; Methodology: SQ, HK, CY, AP, NA, and RB; Investigation: SQ, HK, NA, HL, JH, JY, MX, DC, RL, RW, and RB; Formal analysis: SQ, RB; Writing–Original Draft: SQ, RB; Writing–Review & Editing: RB, SQ, AP; Funding Acquisition: RB; Supervision: RB.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Qiu, S., Kumar, H., Yan, C. et al. Autophagy inhibition impairs leukemia stem cell function in FLT3-ITD AML but has antagonistic interactions with tyrosine kinase inhibition. Leukemia 36, 2621–2633 (2022). https://doi.org/10.1038/s41375-022-01719-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-022-01719-6
- Springer Nature Limited
This article is cited by
-
Metabolic dependencies of acute myeloid leukemia stem cells
International Journal of Hematology (2024)
-
Update on Small Molecule Targeted Therapies for Acute Myeloid Leukemia
Current Treatment Options in Oncology (2023)