
Abstract
Cyclic-AMP (cAMP) exerts suppressive effects in the innate and adaptive immune system. The PD-1/PD-L1 immune checkpoint downregulates T-cell activity. Here, we examined if these two immunosuppressive nodes intersect. Using normal and malignant lymphocytes from humans, and the phosphodiesterase 4b (Pde4b) knockout mouse, we found that cAMP induces PD-L1 transcription and protein expression. Mechanistically, we discovered that the cAMP effectors PKA and CREB induce the transcription/secretion of IL-10, IL-8, and IL-6, which initiate an autocrine loop that activates the JAK/STAT pathway and ultimately increase PD-L1 expression in the cell surface. This signaling axis is disarmed at two specific nodes in subsets of diffuse large B-cell lymphoma, which may help explain the variable PD-L1 expression in these tumors. In vivo, we found that despite its immunosuppressive attributes, the PDE4 inhibitor roflumilast did not decrease the clinical activity of checkpoint inhibitors, an important clinical observation given the approved use of these agents in multiple diseases. In summary, we discovered that PD-L1 induction is a part of the repertoire of immunosuppressive actions mediated by cAMP, defined a cytokine-mediated autocrine loop that executes this action and, reassuringly, showed that PDE4 inhibition does not antagonize immune checkpoint blockade in an in vivo syngeneic lymphoma model.
Similar content being viewed by others


Introduction
Cyclic-AMP (cAMP) is a broadly acting second messenger that relays extra-cellular signals in a cell type and context specific manner. Through binding to effectors, notably PKA (protein kinase A), elevation of intracellular cAMP leads to rapid post-translational modifications in the cytosol, and architectural changes in discrete cellular membrane microdomains. In addition, primarily via the PKA-mediated phosphorylation of CREB (cAMP response element-binding protein), cAMP elicits specific gene transcription programs [1].
In cells of the innate and adaptive immune system, cAMP exerts suppressive effects [2, 3]. In B and T lymphocytes, sustained elevation of cAMP counters the B-cell receptor (BCR) and T-cell receptor (TCR) function, respectively, at least in part by suppressing the activity of downstream kinases [4,5,6,7,8]. In addition, cAMP provides tonic negative constraint on the synthesis and secretion of pro-inflammatory cytokines in monocytes and neutrophils, and limits the LPS-driven stimulation of innate immune response [2, 3]. Adenylate cyclases produce most of the cellular cAMP [1]. In turn, a superfamily of enzymes, the phosphodiesterases (PDE), hydrolyze cAMP into inert 5′AMP and terminates signaling. In cells of the immune system, the PDE4 family accounts for most of the cAMP hydrolysis [9]. This feature, alongside a catalytic structure amenable for specific small molecule-based inhibition, has led to the development and FDA-approval of PDE4 inhibitors as anti-inflammatory and immunosuppressive agents [2]. Further, in part due to cAMP’s role in suppressing the BCR, these agents are also being tested for the treatment of mature B-cell malignancies [10,11,12].
Antigen-specific T lymphocytes can recognize and eliminate aberrant cells, including cancer cells. Alongside this capability, a system of immune checkpoints has evolved to regulate the quantity and activity of T cells, thus establishing peripheral tolerance and limiting overt tissue damage. A prominent immune checkpoint system is executed by the PD-1 (programmed cell death protein 1)–PD-L1/2 (programmed cell death protein ligand 1/2) interaction [13]. PD-1, a co-inhibitory receptor expressed on the surface of antigen-stimulated T cells, interacts ligands PD-L1 and PD-L2. This engagement inhibits T-cell proliferation, survival, cytokine production, and other effector functions [13]. Cancer cells hijack this system by expressing PD-L1 on their surface, binding to cognate PD-1 in activated T cells, thus foiling anti-cancer immunity, which can be successfully reengaged with checkpoint inhibitors, primarily anti-PD-1 and anti-PD-L1 antibodies [14]. Considering the role of PD-L1 in halting anti-cancer T-cell activity, significant effort has been placed in identifying regulators of PD-L1 expression. Oncogenes and tumor suppressors, as well as pro-inflammatory cytokines, e.g., IFN-γ, have been shown to induce PD-L1 expression, aiding biomarker identification toward improved therapeutic usage [15].
A putative role for cAMP on PD-L1 expression has not yet been directly examined. An interplay between two broadly immunosuppressive nodes—the cAMP signaling pathway and the PD-1/PD-L1/2 immune checkpoint—is predictable and it would be consistent with cAMP’s role as a mediator of inputs that suppress T cells [4]. Testing this possibility is important as it may unveil a novel regulatory axis that modulates PD-L1 expression and immune checkpoint function. In addition, this knowledge will advance our understanding of the mechanism of action of FDA-approved PDE4 inhibitors, and may guide decisions on their use in approved [16,17,18,19,20,21,22] and in investigational [12] settings.
Methods
Detailed materials and methods are provided as supplemental data.
Cell lines and primary tumor samples
DLBCL cell lines were cultured as reported [23]; the DLBCL PDX models have been characterized earlier [24], and herein were analyzed ex vivo. De-identified primary human lymph nodes were obtained from the Department of Pathology [25], and their use was approved by the Institutional Review Board of the UTHSCSA. All patients signed a consent form.
Mice
The Pde4b knockout (KO) mouse was reported before [26]. To generate murine B-cell lymphoma models, A20 cells were inoculated subcutaneously in Balb/c mice [27, 28]. Mice were treated with roflumilast (5 mg/kg) or vehicle control (PBS/DMSO), administered daily intra-peritoneally (IP), and/or with anti-mouse PD-L1 (clone 10 F.9G2) or rat IgG2b isotype control, administered IP (200 μg per injection) at 2–3 days intervals. Studies were approved by the Institutional Animal Care and Use Committee of UTHSCSA.
Fluorescence-activated cell sorting (FACS) analysis
Used to detect PD-L1 in human and murine cells, and to characterize immune cell subpopulations in the mouse. Cells were acquired using a BD FACSCelesta (BD Bioscience) or with a Cytek Aurora (Cytek BioSciences). Data were analyzed with FlowJo software v10.6.2 (FlowJo LLC), as we described [29].
Immunoblotting analysis
Performed to detect PD-L1, pSTAT3 (Y705) pSTAT1 (Y701), pCREB (S133) in human and murine cells, as described [30].
RNA isolation, cDNA synthesis and q-RT-PCR
RNA was isolated and cDNA synthesized from primary human lymph nodes, murine splenocytes, DLBCL cell lines and PDX models, as we described [31]. Q-RT-PCRs were performed as reported [32], and gene expression calculated using the 2–∆∆Ct method [33]. Oligonucleotide sequences are listed in Supplemental Table 1.
Chemokine array profiling and cytokine quantification
DLBCL cells were treated with DMSO or forskolin (40 µM) for 8 h and supernatant used for the screening of 105 cytokines/chemokines (Human XL Cytokine Array Kit, R&D Systems), or for the direct detection of IL-10, IL-8 and IL-6.
Luciferase reporter assay
A PD-L1 promoter reporter construct (pGL3-PD-L1-3kb) was described before [34], and luciferase and beta-galactosidase quantified as we reported [35].
Statistical analysis
Performed with the GraphPad Prism 8 software. P value ≤0.05 was considered significant
Results
Cyclic-AMP induces PD-L1 expression in DLBCL cell lines
We used forskolin, an adenylyl-cyclase activator, to increase cAMP levels in DLBCL cell lines and well-characterized DLBCL PDX models analyzed ex vivo [24], and measured PD-L1 expression using FACS. In all ABC (activated B-cell)-like DLBCL models, we detected a significant increase in PD-L1 expression in the cell surface (mean increase 2.2-fold, range 1.5–4.1) in association with higher cAMP levels (Figs. 1A and S1A, B). Conversely, no PD-L1 expression, or increase, was detected in GCB (germinal center B-cell)-like DLBCL cell lines or PDXs, even though cAMP was readily elevated and PKA activated in these models (Fig. S1C–F). Using q-RT-PCR, we determined that cAMP induces PD-L1 expression at RNA level (Fig. 1B), and with western blot (WB) we found that an increase in PD-L1 expression could also be detected in whole cell lysates (Fig. 1C). To validate the concept that forskolin effects are mediated by cAMP, we used the synthetic, cell-permeable, 8-Bromo-cAMP molecule and recapitulated the induction of PD-L1 expression detected with forskolin (Fig. 1D). In addition, blocking PKA activity with its inhibitor H-89, abrogated cAMP effects, establishing PKA as the effector of cAMP-induced PD-L1 expression (Fig. 1E). We concluded that cAMP, in a PKA-dependent manner, induces PD-L1 transcription and protein expression in subsets of DLBCL.

A FACS analysis of cell-surface expression of PD-L1 in DLBCL cell line and PDX models in low vs. high cAMP status, DMSO or forskolin (Frsk 20-40 μM, 16 h). Mean fluorescence intensity (MFI) of three replicates is shown in the right panel (full data in Fig. S1B). B q-RT-PCR of PD-L1 in the same DLBCL cell models in ctrl (DMSO) vs. Frsk (40 μM, 8–16 h). C western blot (WB) analysis of PD-L1 expression in DLBCL models with low vs. high cAMP levels (− and + Frsk, 40 μM, 16 h). Differential baseline PD-L1 expression is shown in Fig. S1GD WB of PD-L1 in the SU-DHL2 cell line exposed to the synthetic cell-permeable 8-bromo-cAMP for 16 h. E WB of PD-L1 and phospho-CREB (S133) in the SU-DHL2 cell line exposed to DMSO, forskolin (20 μM) or forskolin + the PKA inhibitor H-89 (20 μM) for 4 h. In A and B, data shown are mean ± SD of triplicates. *p < 0.05, **p < 0.01, ***p < 0.001, two-sided Student’s t test. In C–E densitometric quantification of biological replicates is shown below the WBs; data shown are mean ± SD, p values are from two-sided Student’s t test.
Modulation of PD-L1 expression by the cAMP-PDE4 axis in vivo
Of the members of the PDE4 family, PDE4B is a prominent regulator of cAMP levels in lymphocytes [36, 37]. Thus, we used a Pde4b KO mouse model to test the effects of this enzyme on the expression of PD-L1 in murine B and T cells. We collected spleens from 8 to 16 weeks old, sex matched, Pde4b WT or KO mice (n = 10) and used FACS to examine surface PD-L1 expression in CD19+ and CD3+ subpopulations. The percentage of B and T cells expressing PD-L1 was significantly higher in Pde4b KO mice than in WT littermates (Fig. 2A); WB of total splenocytes confirmed higher expression of PD-L1 in Pde4b KO cells (Fig. 2B). To expand on the DLBCL models and mouse data, we examined the interplay between cAMP/PDE4 signals and PD-L1 expression in freshly obtained primary human specimens, including reactive lymph nodes and B-cell lymphomas. We found that ex vivo activation of cAMP signals with forskolin, in combination with the FDA-approved PDE4 inhibitor roflumilast, induced PD-L1 expression at RNA and protein levels (Fig. 2C); unfortunately, additional materials were not available for FACS analysis, but the correlation between WB and FACS data in DLBCL cell lines/PDX and Pde4b KO models (Figs. 1A, C and 2A, B) indicate that in these primary samples PD-L1 expression increased in the cell surface as well. We concluded that genetic and pharmacological modulation of the cAMP-PDE4B axis influences PD-L1 expression in untransformed murine B and T lymphocytes and in primary human reactive B cells and lymphomas.

A FACS analysis of cell-surface PD-L1 expression in splenic B (CD19+) and T (CD3+) cells from Pde4b+/+ (WT) and −/− (KO) mice (n = 10). Data shown are mean ± SD; p values are from a two-sided Student’s t test, *p < 0.05. B WB of PD-L1 in spleen cells of Pde4b KO mice or WT littermates C left panel—Q-RT-PCR-based quantification of PD-L1 in 10 primary human samples—3 reactive lymph nodes and 7 mature B-cell lymphomas (MCL, MZL, DLBCL, SLL, LPL, FL and CLL) cultured for 8 h in the presence of vehicle (DMSO) or forskolin (20 μM) + roflumilast (10 μM) (F + R); each sample data point represents the mean of quantifications performed in triplicate; data shown are mean ± SEM; p value was calculated with one-way ANOVA and Tukey’s post-test, **p < 0.01; right panel—WB of PD-L1 in two representative human primary samples from which enough material was available for protein isolation. Densitometric quantification is shown below the WB in (B and C).
Secreted factor(s) mediate cAMP effects on PD-L1 expression
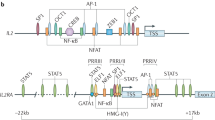
PKA phosphorylates and activates the transcription factor CREB (cAMP-response element-binding protein), which binds to cAMP-response elements (CRE) in promoters of its target genes, and induces transcription [1]. Thus, to explore the mechanism(s) by which cAMP induces PD-L1 transcription/expression, we first investigated if through PKA/CREB, cAMP could directly modulate PD-L1 transcription. Using the “regulatory function” of the UCSC browser and Ensembl, as we reported before [35], and the ConSite web interface for prediction of regulatory elements [38], we mapped five putative CRE sites to the well-characterized PD-L1 promoter [34] (Fig. S2). Next, using a luciferase reporter construct, we found that elevation of intracellular cAMP readily phosphorylated CREB, but it did not induce reporter activity; conversely, IFNγ, used here as a positive control for PD-L1 promoter activity, consistently increased luciferase levels (Fig. S2). We concluded that PD-L1 promoter is not responsive to cAMP and that the putative CRE sites are nonfunctional.
The JAK/STAT pathway is central to the regulation of PD-L1 expression [15, 39]. Therefore, we asked whether this signaling module could be involved in the cAMP/PDE4-mediated induction of PD-L1. In DLBCL cell lines and PDX models, elevation of cAMP levels resulted in pronounced STAT3 phosphorylation, and pSTAT3 was also higher in the spleen of Pde4b KO vs. WT mice (Fig. 3A,B). Confirming the participation of JAK signals in this process, we showed that a pan-JAK inhibitor blunted induction of PD-L1 (Fig. 3C). As the JAK/STAT signaling pathway is an important transducer of extra-cellular stimuli, we postulated that cAMP might promote the secretion of “factor(s)”, which in an autocrine fashion could activate JAK/STAT and induce PD-L1 expression. In support to this hypothesis, we found that the conditioned media from DLBCL cells with transient elevation of cAMP, but not from isogenic models with low cAMP, induced PD-L1 expression and STAT3 phosphorylation (Fig. 3D). We concluded that cAMP promotes the secretion of extra-cellular factors, which likely via an autocrine loop mediated by the JAK-STAT pathway, induce PD-L1 expression.

A WB analysis of pSTAT3 (Y705) and total STAT3 in DLBCL cell lines and PDX model following exposure to forskolin (Frsk, 40 μM, 4–16 h). The more modest increase in pSTAT3 in OCI-Ly3 reflects the constitutive activity of STAT signals in this cell line—densitometric quantification (pSTAT3/STAT3) of biological replicates is shown on the right, data shown are mean ± SD of replicates, p values are from two-sided Student’s t test. B WB of pSTAT3 (Y705) and total STAT3 in spleen cells of Pde4b KO mice or WT littermates; densitometric quantification (pSTAT3/STAT3) is shown below the WBs. C WB of PD-L1 and pSTAT3 (Y705) in DLBCL cell lines exposed to DMSO, forskolin (Frsk, 40 µM), JAKi (JAK inhibitor pyridone 6, 5 μM) or their combination for 16 h. STAT3 phosphorylation in the RIVA occurs early and it is labile, thus it is detected after 4 h exposure to forskolin (A), but not after 16 h (C); the OCI-Ly3 pSTAT3/STAT3 displays in (A and C) are from the same WB; densitometric quantification is shown below the WBs (PD-L1/β-actin; pSTAT3/STAT3), data shown are mean ± SD of biological replicates, p values are from one-way ANOVA. D WB of PD-L1 and pSTAT3 (Y705) in DLBCL cell lines cultured for 8 h in conditioned media (CM). The forskolin-free CM was obtained from DLBCL cell lines exposed to DMSO or forskolin (Frsk, 40 µM) for 1 h, followed by a drug wash-off and replenishment with fresh media. densitometric quantification of biological replicates (n = 5, 3 for DHL2, 2 for U2932) is shown on the right (PD-L1/β-actin; pSTAT3/STAT3); data are mean ± SD, p value is from a two-sided Student’s t test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Cyclic-AMP induces the expression and secretion of multiple cytokines that increase PD-L1 levels
Part of cAMP’s suppressive effects on the immune system is mediated by cytokines [2]. Thus, we investigated if cytokines could function as intermediaries in the cAMP driven, JAK/STAT mediated, induction of PD-L1. First, we utilized a protein array to profile the pattern of cytokine secretion in isogenic DLBCL cells in low vs. high cAMP status. In this screening strategy, focusing on cytokines that displayed significant basal signal intensity, reflective of their abundance, and which changed between the two tested conditions, we found that the secretion of IL-10, IL-8 and IL-6 were induced by cAMP (5.7-, 1.5-, and 1.6-fold increase, respectively, Fig. 4A). We validated this observation by directly quantifying the secretion of each of these cytokines in an extended panel of DLBCL models (Fig. 4B). We also determined that the induction of IL-10, IL-8, and IL-6 occurs at RNA level, and it is PKA activity dependent (Fig. S3A, B). In addition, we detected the cAMP-mediated induction of cytokines transcription in primary human lymphoid tissues, and in normal murine lymphocytes using either a combination of forskolin and roflumilast, or synthetic cell-permeable 8-Br-cAMP (Fig. 4C, D). Next, we showed that recombinant IL-10, IL-8, and IL-6 recapitulated the effects of cAMP on PD-L1 expression (Fig. 4E) and confirmed that cytokine-mediated signaling can activate the PD-L1 promoter (Fig. S3C). Then, to firmly link cAMP to the cytokines-mediated autocrine loop and subsequent PD-L1 induction, we used blocking antibodies directed at IL-10 and IL-6 and/or their receptors, as well as against the IL-8 receptor, CXCR2. Confirming the contributions of all cytokines examined as mediators of cAMP effect, blockade of each individual signal node, significantly blunted cAMP-mediated PD-L1 induction (Fig. 4F). Notably, in GCB-DLBCL cell models, although cAMP activated PKA/CREB (Fig. S1F), it did not induce cytokines expression/secretion to the levels found in ABC-DLBCL (which on average was three orders of magnitude higher in ABC-DLBCL than GCB-DLBCL models), nor it activated the JAK/STAT signals (Fig. S3D–E). Interestingly, although IFNγ engaged the JAK/STAT pathway in GCB-DLBCL models, it also did not upregulate PD-L1 (Fig. S3F). These data suggest that in GCB-type DLBCLs, lymphoma cell-intrinsic defects in multiple signaling nodes (i.e., cAMP/cytokine secretion axis, and JAK-STAT activation/PD-L1 promoter transcription module) contribute to the lack of PD-L1 expression. Lastly, we explored public datasets, including cohorts of epithelial tumors, such as melanoma, colon and lung cancer, and DLBCL, and found a significant correlation between PD-L1 levels and expression of IL-10, IL-8, and IL-6 and/or their receptors (Fig. S3F). We concluded that cAMP in a PKA-dependent manner drives the expression of multiple cytokines, which initiates an autocrine loop that activates the JAK/STAT pathway and induces PD-L1 expression.

A Cytokine array exposed to conditioned media from the SU-DHL2 cell line cultured with DMSO (Ctrl) or forskolin (cAMP, 40 μM) for 8 h. Increased levels of IL-6, IL-8, and IL-10 in high cAMP supernatant is indicated by red arrow; increased IL-1 abundance is also noted B Top—ELISA-based quantification of IL-10 in the supernatant of DLBCL cell lines exposed DMSO (Ctrl) or forskolin (Frsk, 40 μM) for 8 h. Bottom—ELISA-based quantification of IL-6 and IL-8 in the supernatant of DLBCL cell lines exposed DMSO (Ctrl) or forskolin (Frsk, 40 μM) for 8 h. C q-RT-PCR quantification of IL-10 and IL-6 in primary human reactive lymph nodes or mature B-cell tumors cultured for 8 h in presence of vehicle (DMSO) or roflumilast (10 μM) + forskolin (20 μM), R + F. D q-RT-PCR quantification of Il-10 and Il-6 in murine splenocytes cultured for 4 h in presence of vehicle (DMSO) or roflumilast (10 μM) forskolin (40 μM) (R + F), and 8-Br-cAMP (500 μM). E WB analysis of PD-L1 in DLBCL cell lines exposed to recombinant IL-10 (10 ng/ml), IL-6, or IL-8 (both at 40 ng/ml) for 4–16 h. F WB analysis of PD-L1 in SU-DHL2 cells exposed to forskolin (Frsk, 20 μM), in the presence of isotypes or anti-IL-10 + IL-10 receptor antibodies (5 μg/each) for 8 h (left panel), anti-IL-6 (10 μg) + IL-6R (5 μg) antibodies for 4 h (middle panel), or anti-CXCR2 antibody (10 µg) for 4 h (right panel). Densitometric quantification is shown below the WBs in (E and F); data shown are mean ± SD of biological replicates, p values are from one-way ANOVA. In B p values are from two-sided Student’s t test; in C and D each sample data point is the mean of quantifications performed in triplicate and p values are from one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
PDE4 blockade does not limit the activity of immune checkpoint inhibitors in a syngeneic murine model of B-cell lymphoma
Given their broad anti-inflammatory and immunosuppressive effects, PDE4 inhibitors are FDA-approved for the treatment of chronic obstructive pulmonary disease (COPD) (roflumilast) and psoriatic arthritis (apremilast) [19, 21]. These observations raised the possibility that the suppressive effects on cells on the innate and adaptive immune system, may cause pharmacological inactivation of PDE4 to abrogate the activity of immune checkpoint inhibitors in vivo. To examine this possibility, we developed a syngeneic A20 murine B-cell lymphoma model in Balb/c mice (n = 60, three cohorts of 20 mice). Importantly, differently from human DLBCL cell lines [6,7,8], the growth of A20 cells are not inhibited by roflumilast (Fig. S4A), thus allowing for a better understating of the role the tumor immune microenvironment in the therapeutic response. Following tumor engraftment, mice were randomized into four treatment arms (n = 15 mice/arm) to receive vehicle control (DMSO and isotype antibody), roflumilast (5 mg/kg/day IP), anti-PD-L1 (200 μg IP, 5 doses, alternating every 2–3 days), as described [40], or the combination of roflumilast and anti-PD-L1. Tumors grew unencumbered in the control and roflumilast-only groups but were similarly and significantly suppressed in the anti-PD-L1 or roflumilast/anti-PD-L1 arms (Fig. 5A), suggesting that the PDE4 inhibitor-mediated immunosuppression does not abrogate the beneficial anti-lymphoma effects of immune checkpoint inhibitors in this model. This difference in tumor growth translated in improved survival for the mice in the anti-PD-L1 or roflumilast/anti-PD-L1 arms (Fig. S4B). To better understand the pattern of response obtained in these assays, and to define how PDE4 inhibition may influence anti-cancer immune response, we used FACS to examine the tumor microenvironment. The growth inhibition detected in the anti-PD-L1 arm was, expectedly, closely associated with a significant increase in CD3+ cells in the tumor milieu (Fig. 5B), which has been previously shown to directly account for these effects [40]. In addition, as predicted given its role in suppressing TCR-signal and T-cell survival, the CD3+ T-cells infiltrate was significantly smaller in roflumilast-only treated mice (Fig. 5B). Remarkably, despite their similar clinical response, the tumors of mice in the combination arm (roflumilast + anti-PD-L1) had a significantly less pronounced T-cell infiltrate than the anti-PD-L1 only group (Fig. 5B). To investigate the potential reasons for this intriguing observation, we characterized the CD4/CD8 subpopulations in the tumor microenvironment. Curiously, PDE4 inhibition with roflumilast (as single agent or in combination with anti-PD-L1), increased the proportions of cytotoxic CD8+ T cells and decrease the CD4+ T-cell infiltrate and, consequently, decreased the CD4/CD8 ratio (Figs. 5C and S4B, C), a profile suggested to associate with improved outcome in cancer [41]. Surely, in isolation this qualitatively distinct tumor infiltrating lymphocyte (TIL) profile was not sufficient to suppress tumor growth, as lymphomas grew unimpeded in the roflumilast-only arm (Fig. 5A). However, this TIL profile may help explain how even with significantly fewer infiltrating T cells, the tumors in the combination arm responded similarly to those treated with anti-PD-L1 only (Fig. 5A). Lastly, even though the A20 cell model constitutively express high PD-L1 levels, in accordance with data from normal and malignant human and murine lymphocytes, we detected a modest but significant additional increase in the expression of PD-L1 in the surface of the murine A20 lymphomas in vivo (Fig. S4D). Still, this finding likely has little clinical impact since roflumilast led to a decrease in T-cell infiltration in the microenvironment. We concluded that despite its broad immunosuppressive attributes, the PDE4 inhibitor roflumilast did not decrease the clinical activity of checkpoint inhibitors in a mouse model of B-cell lymphoma. In this model, PDE4 inhibition led to the emergence of a favorable anti-tumor immune profiles including a relative increase in CD8+ cytotoxic T cells.

A Tumor volume in Balb/c mice engrafted subcutaneously with the A20 lymphoma cells and randomized in four treatment groups (n = 60 total, 15/treatment arm). Data are mean ± SEM, p value is from two-way ANOVA Tukey’s post-test. B FACS analysis of CD3 expression in tumors harvested at the end of the therapeutic trial (n = 47). Data shown are mean ± SEM of the percentage of CD3+ cell in each treatment arm, p value is from two-sided Student’s t test. C Ratio of the percentages of CD4 and CD8 cells determined by FACS in tumors harvested at the end of the therapeutic trial (n = 47). Data shown are mean ± SEM, p value is from two-sided Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
We discovered that the second messenger cAMP modulates PD-L1 expression and defined the mechanistic basis for this action. Cyclic-AMP is a well-established suppressor of innate and adaptive immune cells [2, 4]. To date, these effects have been mainly associated with cAMP’s ability to dampen the synthesis of pro-inflammatory cytokines by neutrophils and monocytes, and to “tone-down” BCR and TCR function [2,3,4, 12]. Our data suggest that induction of PD-L1 expression, with potential engagement of PD-1/PD-L1 pathway and inhibition of cytotoxic T-cell activity, is an additional mechanism by which cAMP executes immunosuppression.
We identified an elaborate autocrine loop as the mechanistic basis for the cAMP/PD-L1 interplay. In human and murine, normal and malignant, lymphocytes, cAMP induces the expression and secretion of the IL-10, IL-8, IL-6, which activated JAK/STAT signals, resulting in transcriptional activation of PD-L1. (Fig. 6) The interplay between IL-10 and cAMP/PDE4 has been recognized before [2, 42], and linking them to PD-L1 expression expands the portfolio of immune-suppressive activities that are cAMP-driven and IL-10 executed. Conversely, considering the mainly pro-inflammatory role of IL-6, its cAMP-mediated transcription/secretion is less intuitive. This apparent contradiction may be in part explained by the context-dependency of cAMP effects. For example, while cAMP dampens LPS-mediated IL-6 induction in cells of the innate immune system, it can also directly promote IL-6 transcription/secretion [43,44,45], as we detected in our models. IL-8 is a chemokine produced by monocytes, neutrophils, fibroblasts, and tumor cells, which plays an important role within the tumor microenvironment as a chemotactic factor [46]. This expression pattern explains the more restricted effects of cAMP on IL-8 transcription/secretion, which in our examination was limited to cancer cells. Analogous to the IL-6 paradigm, despite its primarily pro-inflammatory role [46], we found that IL-8 readily induces PD-L1 in DLBCL, in agreement with earlier reports in gastric cancer [47]. However, contrary to IL-6, the link between cAMP and IL-8 expression is likely to be indirect and may involve IL-1 (as preliminarily shown in Fig. 4A), a cAMP-induced cytokine that transcriptionally activates IL-8 [48, 49]. Interestingly, very recently, systemic and tumor-associated IL-8 was identified as modulator of the cellular profile of the tumor microenvironment, and associated with poor response to immune checkpoint blockade [50, 51]. It is possible that the cytokines involved as intermediaries in the cAMP-PD-L1 interplay will vary according to the tissue type. However, encouragingly, we also found a significant correlation between PD-L1 and IL-10, IL-8, IL-6 expression in melanoma, lung and colon cancer. Together, we proposed that, at least in a B-cell tumor model, there is a cytokine hierarchy at the intersection of cAMP production and PD-L1 expression, wherein the immunosuppressive IL-10 plays a dominant role, while IL-6 and IL-8 contribute, possibly as a part of feedback negative loop to limit their own pro-inflammatory effects.

Increased intracellular levels of cAMP via PKA-CREB transcriptionally activates the IL-10, IL-8, and IL-6 genes. Secretion of these cytokines engage their receptors in an autocrine fashion, leading to JAK activation and STAT phosphorylation. Phosphorylated STATs bind to and activate the PD-L1 promoter, induce PD-L1 transcription and its cell-surface expression. Physiologic stimuli and pharmacological agents that impinge on this pathway are also shown.
We developed part of our data in DLBCL cell lines and in the ex vivo analysis of PDX models. This aspect of the work provided an opportunity to add mechanistic understanding to the known association between PD-L1 expression and ABC-DLBCL (non-GCB DLBCL), which contrasts with a low/null PD-L1 levels in GCB-DLBCL [52, 53]. While cAMP levels and PKA activity were equally inducible in ABC-like and GCB-like DLBCL cell lines, as we reviewed earlier [12], in the latter the expression/secretion of cytokines was not elicited. This blockade prevented the establishment of the autocrine loop identified in ABC-DLBCLs and, as a result, cAMP did not activate JAK/STAT or induced PD-L1 expression in GCB DLBCLs. Notably, bypassing the cAMP-signaling network with the potent PD-L1 regulator IFNγ also did not induce PD-L1 expression in the GCB models, although in this instance STAT was readily engaged. Thus, in respect to induction of PD-L1 expression in GCB-DLBCL, at least two regulatory nodes appear to be deficient: (a) cytokine expression and engagement of the autocrine loop and, (b) the activation of the PD-L1 promoter by phosphorylated STATs. These findings point to a putative architectural/functional silencing of cytokines and PD-L1 promoters in the GCB-DLBCL, which may reflect the B-cell developmental stage from which these tumors derive, i.e., their cell of origin. These observations are relevant because they challenge a current paradigm, in which it is implied that since GCB-DLBCLs often display a “non-inflamed” immune environment, the lack of PD-L1 expression in the lymphoma cell would be secondary to the absence of local stimuli [52]. Indeed, as our data were generated in cell lines and thus without the confounding immune microenvironment, we could establish that lymphoma cell-intrinsic defects are at least partially responsible for the low/null PD-L1 expression in subsets of DLBCL. Nonetheless, it is important to highlight that irrespective of the molecular subtype, DLBCL display very low overall response to checkpoint inhibitors [52, 54]. In addition, the interplay between cAMP/PDE4 and PD-L1 expression that we describe herein, is unlikely to play a dominant role in this lack of response.
PDE4 inhibitors, which are FDA-approved for COPD and psoriatic arthritis, elevate cAMP levels in cells on the innate and adaptive immune system, and display broad immunosuppressive properties, which we showed may at least part be due to PD-L1 expression. Although increase in PD-L1 levels in tumor cells has been considered a desired biomarker for checkpoint inhibitors activity [13,14,15, 52], this concept needs to be contextualized and, herein, our postulate was that the output of PDE4 inhibition, with the potential to suppress cytotoxic secretion and the immune microenvironment, could in fact associate poor response to immune checkpoint blockade. Remarkably, roflumilast did not decrease the anti-lymphoma activity of an anti-PD-L1 antibody, but this “neutral” outcome was achieved in unexpected ways. The efficacy of anti-PD-L1 used as single agent was, predictably, associated with a substantial T-cell infiltrate in the tumor microenvironment. Conversely, although the anti-lymphoma efficacy of the roflumilast/anti-PD-L1 combination was not significantly inferior to anti-PD-L1 alone, the T-cell infiltrate was quantitatively modest and instead displayed a subtle, and potentially consequential, qualitative changes, including an increase in CD8 cytotoxic T cells and decrease in CD4 T-cells. Certainly, the changes in the TIL profile mediated by PDE4 inhibition are not sufficient to promote anti-tumor immunity, as roflumilast was ineffective as a single agent. However, they may explain how even in the absence of a prominent T-cell infiltrate, the roflumilast/anti-PD-L1 combination still maintained anti-cancer activity. In the future, it would be important to expand these observations and better characterize the subpopulation of TILs, as well as define the role of cAMP signals on macrophages, natural killer and myeloid-derived suppressor cells, as they all play a part on the response to checkpoint blockade. In extrapolating our finding to the clinical realm, two observations may be pertinent: (1) COPD patients, who are prescribed roflumilast “on label”, are at a significantly higher risk for lung cancer. Yet, data from the three pivotal roflumilast trials, which include more than 9000 patients [16,17,18], did not show an increase in cancer diagnosis, even though cAMP is immunosuppressive and could compromise natural anti-cancer immunity. This observation may reflect the qualitative changes in the tumor infiltrate that we described, which could offset the overall immunosuppression. (2) Strategies to establish a de novo inflamed tumor microenvironment, which could improve therapeutic responses to checkpoint inhibitors, are highly sought. In these models, PD-L1 expression in tumor cells is a regulatory counter response to T-cell activity and is mediated in large part by IFNγ. Although somewhat paradoxically, given its anti-inflammatory role, we speculate that PDE4 inhibitors may have a role to play in these approaches, as it could: a) sustain PD-L1 expression independent of IFNγ signals via traditionally suppressive cytokines, i.e., IL-10, (b) it could limit undesirable systemic inflammatory responses and, (c) it could establish a favorable local anti-tumor immune profile, as we found in the A20 model.
In summary, the data reported here expand the understanding of the mechanistic basis of cAMP-mediated immunosuppression, by defining the existence of a hitherto unappreciated interplay with the PD-1/PD-L1 immune checkpoint axis (Fig. 6). These observations may help guide the clinical usage of FDA-approved PDE4 inhibitors.
References
Sassone-Corsi P. The cyclic AMP pathway. Cold Spring Harb Perspect Biol. 2012;4:a011148.
Raker VK, Becker C, Steinbrink K. The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Front Immunol. 2016;7:123.
Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol. 2008;39:127–32.
Mosenden R, Tasken K. Cyclic AMP-mediated immune regulation-overview of mechanisms of action in T cells. Cell Signal. 2011;23:1009–16.
Cooney JD, Lin AP, Jiang D, Wang L, Suhasini AN, Myers J, et al. Synergistic targeting of the regulatory and catalytic subunits of PI3Kdelta in mature B-cell malignancies. Clin Cancer Res. 2018;24:1103–13.
Kim SW, Rai D, Aguiar RC. Gene set enrichment analysis unveils the mechanism for the phosphodiesterase 4B control of glucocorticoid response in B-cell lymphoma. Clin Cancer Res. 2011;17:6723–32.
Kim SW, Rai D, McKeller MR, Aguiar RC. Rational combined targeting of phosphodiesterase 4B and SYK in DLBCL. Blood. 2009;113:6153–60.
Smith PG, Wang F, Wilkinson KN, Savage KJ, Klein U, Neuberg DS, et al. The phosphodiesterase PDE4B limits cAMP-associated PI3K/AKT-dependent apoptosis in diffuse large B-cell lymphoma. Blood. 2005;105:308–16.
Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem. 2003;278:5493–6.
Kelly K, Mejia A, Suhasini AN, Lin AP, Kuhn J, Karnad AB, et al. Safety and pharmacodynamics of the PDE4 inhibitor roflumilast in advanced B-cell malignancies. Clin Cancer Res. 2017;23:1186–92.
Butler MJ, Aguiar RCT. Biology informs treatment choices in diffuse large B cell lymphoma. Trends Cancer. 2017;3:871–82.
Cooney JD, Aguiar RC. Phosphodiesterase 4 inhibitors have wide-ranging activity in B-cell malignancies. Blood. 2016;128:2886–90.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Prestipino A, Zeiser R. Clinical implications of tumor-intrinsic mechanisms regulating PD-L1. Science Transl Med. 2019;11:eaav4810.
Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. 2019;76:359–70.
Rabe KF, Bateman ED, O’Donnell D, Witte S, Bredenbroker D, Bethke TD. Roflumilast-an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2005;366:563–71.
Calverley PMA, Rabe KF, Goehring U-M, Kristiansen S, Fabbri LM, Martinez FJ, et al. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374:685–94.
Fabbri LM, Calverley PM, Izquierdo-Alonso JL, Bundschuh DS, Brose M, Martinez FJ, et al. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet. 2009;374:695–703.
Wedzicha JA, Calverley PM, Rabe KF. Roflumilast: a review of its use in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:81–90.
Papp K, Cather JC, Rosoph L, Sofen H, Langley RG, Matheson RT, et al. Efficacy of apremilast in the treatment of moderate to severe psoriasis: a randomised controlled trial. Lancet. 2012;380:738–46.
Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83:1583–90.
Schett G. Apremilast in psoriatic arthritis. Clin Exp Rheumatol. 2015;33 Suppl 93:S98–100.
Bouamar H, Abbas S, Lin AP, Wang L, Jiang D, Holder KN, et al. A capture-sequencing strategy identifies IRF8, EBF1, and APRIL as novel IGH fusion partners in B-cell lymphoma. Blood. 2013;122:726–33.
Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;30:183.
Lin AP, Abbas S, Kim SW, Ortega M, Bouamar H, Escobedo Y, et al. D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nat Commun. 2015;6:7768.
Suhasini AN, Wang L, Holder KN, Lin AP, Bhatnagar H, Kim SW, et al. A phosphodiesterase 4B-dependent interplay between tumor cells and the microenvironment regulates angiogenesis in B-cell lymphoma. Leukemia. 2016;30:617–26.
Rai D, Kim SW, McKeller MR, Dahia PL, Aguiar RC. Targeting of SMAD5 links microRNA-155 to the TGF-beta pathway and lymphomagenesis. Proc Natl Acad Sci USA. 2010;107:3111–6.
Bouamar H, Jiang D, Wang L, Lin AP, Ortega M, Aguiar RC. MicroRNA 155 control of p53 activity is context dependent and mediated by Aicda and Socs1. Mol Cell Biol. 2015;35:1329–40.
Ortega M, Bhatnagar H, Lin AP, Wang L, Aster JC, Sill H, et al. A microRNA-mediated regulatory loop modulates NOTCH and MYC oncogenic signals in B- and T-cell malignancies. Leukemia. 2015;29:968–76.
Jiang D, Aguiar RC. MicroRNA-155 controls RB phosphorylation in normal and malignant B lymphocytes via the noncanonical TGF-beta1/SMAD5 signaling module. Blood. 2014;123:86–93.
Elkashef SM, Lin AP, Myers J, Sill H, Jiang D, Dahia PLM, et al. IDH mutation, competitive inhibition of FTO, and RNA methylation. Cancer Cell. 2017;31:619–20.
Li C, Kim SW, Rai D, Bolla AR, Adhvaryu S, Kinney MC, et al. Copy number abnormalities, MYC activity, and the genetic fingerprint of normal B cells mechanistically define the microRNA profile of diffuse large B-cell lymphoma. Blood. 2009;113:6681–90.
Jung I, Aguiar RC. MicroRNA-155 expression and outcome in diffuse large B-cell lymphoma. Br J Haematol. 2009;144:138–40.
Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47:1083–1099.e1086.
Qiu Z, Lin AP, Jiang S, Elkashef SM, Myers J, Srikantan S, et al. MYC regulation of D2HGDH and L2HGDH influences the epigenome and epitranscriptome. Cell Chem Biol. 2020;27:538–50.
Manning CD, Burman M, Christensen SB, Cieslinski LB, Essayan DM, Grous M, et al. Suppression of human inflammatory cell function by subtype-selective PDE4 inhibitors correlates with inhibition of PDE4A and PDE4B. Br J Pharmacol. 1999;128:1393–8.
Gantner F, Gotz C, Gekeler V, Schudt C, Wendel A, Hatzelmann A. Phosphodiesterase profile of human B lymphocytes from normal and atopic donors and the effects of PDE inhibition on B cell proliferation. Br J Pharmacol. 1998;123:1031–8.
Sandelin A, Wasserman WW, Lenhard B. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 2004;32:W249–252.
Prestipino A, Emhardt AJ, Aumann K, O’Sullivan D, Gorantla SP, Duquesne S, et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Science Transl Med. 2018;10:eaam7729.
Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci USA. 2015;112(Mar):E966–72.
Idos GE, Kwok J, Bonthala N, Kysh L, Gruber SB, Qu C. The prognostic implications of tumor infiltrating lymphocytes in colorectal cancer: a systematic review and meta-analysis. Sci Rep. 2020;10:3360.
Liopeta K, Boubali S, Virgilio L, Thyphronitis G, Mavrothalassitis G, Dimitracopoulos G, et al. cAMP regulates IL-10 production by normal human T lymphocytes at multiple levels: a potential role for MEF2. Mol Immunol. 2009;46:345–54.
Zhang Y, Lin JX, Vilcek J. Synthesis of interleukin 6 (interferon-beta 2/B cell stimulatory factor 2) in human fibroblasts is triggered by an increase in intracellular cyclic AMP. J Biol Chem. 1988;263:6177–82.
Szabo-Fresnais N, Blondeau JP, Pomerance M. Activation of the cAMP pathway synergistically increases IL-1-induced IL-6 gene expression in FRTL-5 thyroid cells: involvement of AP-1 transcription factors. Mol Cell Endocrinol. 2008;284:28–37.
Koga K, Takaesu G, Yoshida R, Nakaya M, Kobayashi T, Kinjyo I, et al. Cyclic adenosine monophosphate suppresses the transcription of proinflammatory cytokines via the phosphorylated c-Fos protein. Immunity. 2009;30:372–83.
Atta ur R, Harvey K, Siddiqui RA. Interleukin-8: an autocrine inflammatory mediator. Curr Pharm Des. 1999;5(Apr):241–53.
Sun L, Wang Q, Chen B, Zhao Y, Shen B, Wang H, et al. Gastric cancer mesenchymal stem cells derived IL-8 induces PD-L1 expression in gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell death Dis. 2018;9:928.
Sung SS, Walters JA. Increased cyclic AMP levels enhance IL-1 alpha and IL-1 beta mRNA expression and protein production in human myelomonocytic cell lines and monocytes. J Clin Investig. 1991;88:1915–23.
Kaplanski G, Farnarier C, Kaplanski S, Porat R, Shapiro L, Bongrand P, et al. Interleukin-1 induces interleukin-8 secretion from endothelial cells by a juxtacrine mechanism. Blood. 1994;84:4242–8.
Schalper KA, Carleton M, Zhou M, Chen T, Feng Y, Huang SP, et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat Med. 2020;26:688–92.
Yuen KC, Liu LF, Gupta V, Madireddi S, Keerthivasan S, Li C, et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat Med. 2020;26:693–8.
Kline J, Godfrey J, Ansell SM. The immune landscape and response to immune checkpoint blockade therapy in lymphoma. Blood. 2020;135:523–33.
Kiyasu J, Miyoshi H, Hirata A, Arakawa F, Ichikawa A, Niino D, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood. 2015;126:2193–201.
Ansell SM, Minnema MC, Johnson P, Timmerman JM, Armand P, Shipp MA, et al. Nivolumab for relapsed/refractory diffuse large B-cell lymphoma in patients ineligible for or having failed autologous transplantation: a Single-arm, phase II study. J Clin Oncol. 2019;37:481–9.
Acknowledgements
RCTA was supported by RP150277, RP170146 (Cancer Prevention and Research Institute of Texas), TRP 6524-17(Leukemia and Lymphoma Society), R01ES031522 (NIEHS/NIH), and I01BX001882 (Veterans Administration Merit Award). The FACS core facility is supported by P30 CA054174 (NCI/NIH).
Author information
Authors and Affiliations
Contributions
BS designed, performed, and interpreted assays; PE, JM, and A-PL performed and interpreted experiments; SJ and ZQ provided support for in vivo assays; KNH, procured and characterized primary tumors; RCTA conceived the project, designed, and interpreted assays, wrote manuscript, which was reviewed and approved by all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sasi, B., Ethiraj, P., Myers, J. et al. Regulation of PD-L1 expression is a novel facet of cyclic-AMP-mediated immunosuppression. Leukemia 35, 1990–2001 (2021). https://doi.org/10.1038/s41375-020-01105-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-020-01105-0
- Springer Nature Limited
This article is cited by
-
cAMP-PKA/EPAC signaling and cancer: the interplay in tumor microenvironment
Journal of Hematology & Oncology (2024)
-
Activation of melanocortin-1 receptor signaling in melanoma cells impairs T cell infiltration to dampen antitumor immunity
Nature Communications (2023)
-
The miR-199a-5p/PD-L1 axis regulates cell proliferation, migration and invasion in follicular thyroid carcinoma
BMC Cancer (2022)
-
MYC, mitochondrial metabolism and O-GlcNAcylation converge to modulate the activity and subcellular localization of DNA and RNA demethylases
Leukemia (2022)
-
Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics
Nature Reviews Clinical Oncology (2022)