
Abstract
We previously reported the benefit of lomustine addition to conventional chemotherapy in older acute myeloid leukemias with nonadverse chromosomal aberrations in the LAM-SA 2007 randomized clinical trial (NCT00590837). A molecular analysis of 52 genes performed in 330 patients included in this trial, 163 patients being treated with lomustine in combination with idarubicin and cytarabine and 167 without lomustine, identified 1088 mutations with an average of 3.3 mutations per patient. NPM1, FLT3, and DNMT3A were the most frequently mutated genes. A putative therapeutic target was identified in 178 patients (54%). Among five molecular classifications analyzed, the ELN2017 risk classification has the stronger association with the clinical evolution. Patients not treated with lomustine have an expected survival prognosis in agreement with this classification regarding the overall and event-free survivals. In strong contrast, lomustine erased the ELN2017 classification prognosis. The benefit of lomustine in nonadverse chromosomal aberrations was restricted to patients with RUNX1, ASXL1, TP53, and FLT3-ITDhigh/NPM1WT mutations in contrast to the intermediate and favorable ELN2017 patients. This post-hoc analysis identified a subgroup of fit elderly AML patients with intermediate cytogenetics and molecular markers who may benefit from lomustine addition to intensive chemotherapy.
Similar content being viewed by others


Introduction
The incidence of acute myeloid leukemia (AML) increases with age, the majority of patients being diagnosed after 60 years. In these older patients, the prognosis is poor, with a median overall survival (OS) usually shorter than 1 year [1, 2]. In patients deemed fit for intensive chemotherapy, no significant improvement in outcome has been achieved except with CPX-351, a dual-drug liposomal encapsulation of cytarabine and daunorubicin recently approved for secondary AML, whereas the combination of the BCL2 inhibitor venetoclax with azacitidine improved outcome of patients unfit for chemotherapy [3]. AML have been classified initially according to cytogenetics profiles into three risk categories: favorable, intermediate, and poor [4]. Favorable cytogenetics abnormalities (core binding factor leukemias and t(15;17) translocation) have long been established as a key predictor for improved clinical outcome as opposed to patients with complex or monosomal karyotypes. The intermediate cytogenetics risk group, representing ~60% of the patients, forms a heterogeneous group [4]. Molecular analysis, especially in this latter group, is highly complementary to cytogenetics. Mutations in epigenetic modifiers, such as ASXL1, DNMT3A, or TET2, are early oncogenic events, frequently found in clonal hematopoiesis [5,6,7]. Moreover, RUNX1, ASXL1, and TP53 mutations are poor prognostic markers in patients with intermediate cytogenetics risk and have been recently included in the European LeukemiaNet (ELN) 2017 prognostic classification for AML, which combines cytogenetics and molecular biology [8].
We previously reported the benefit of the addition of lomustine (also known as chlorethyl-cyclohexyl-nitrosourea) to conventional chemotherapy (idarubicin and cytarabine) for older patients with de novo AML and nonadverse cytogenetics in a randomized clinical trial that enrolled 459 patients [9]. Lomustine is an alkylating agent with a significant antileukemic activity linked to DNA damage and/or impairment of cell replication [10,11,12]. In the LAM-SA 2007 trial, its addition significantly improved the complete response (CR) or CR with incomplete recovery rate (84.7% vs. 74.9%, p = 0.01) and reduced the cumulative incidence (CI) of relapse (41.2% vs. 60.9%, p = 0.003) resulting in improved 2-year event-free (41% vs. 26%, p = 0.01) and OS (56% vs. 48%, p = 0.02).
Here, we report the molecular analysis of this prospective, randomized cohort of older AML patients selected for intensive chemotherapy. The specific profile of the mutations was investigated regarding their functional pathways. Finally, the impact of lomustine in this molecular landscape was investigated through five molecular classifications of AML defined mainly from younger patients [8, 13,14,15,16]. A strong benefit to the addition of lomustine was identified in a subset of patients in the ELN2017 adverse risk group [8] with intermediate cytogenetics, bearing RUNX1, ASXL1, TP53, and FLT3-ITDhigh/NPM1WT (RATFIN) mutations.
Materials and methods
Patients
The LAM-SA 2007 trial was registered at clinicaltrials.gov as NCT00590837. It involved 32 clinical centers of the French Innovative Leukemia Organization (FILO) study group that enrolled 459 patients from February 2008 to December 2011. All patients were older than 60 years and had been diagnosed with de novo AML. Patients had to be considered fit without adverse cytogenetics (defined after the analysis of 20 mitosis at least if no abnormal clone was identified) [17], promyelocytic leukemia nor isolated granulocytic sarcoma. The trial was conducted according to the Declaration of Helsinki and approved by the ethical committee of Bordeaux University Hospital and the Agence Française de Sécurité Sanitaire des Produits de Santé. All patients provided written informed consent at enrollment. Before initiation of the induction chemotherapy course, patients were registered and randomized to receive or not lomustine during induction and postinduction treatment phases [9]. The clinical analysis was performed with 424 patients as 35 were excluded, including 14 from a single center by decision of the data and safety monitoring board as a result of noncompliance with the chemotherapy regimen. Ten patients were reclassified to adverse cytogenetics, eight had myelodysplasia, two a Sorror score of 3, and one withdrew its informed consent [9]. Molecular analyses were performed centrally on samples stored at the FILOthèque, FILO tumor cell bank (DC 2009-944). DNA material was available for 330 patients (78%) with no difference of prognosis according to the DNA availability (Fig. S1).
Molecular analysis
The presence of FLT3-ITD was tested as described [18]. Electrophoregrams peaks were quantified using GeneMarker 2.2 (SoftGenetics, State College, PA, USA). CEBPA screening was performed by classical Sanger sequencing according to Pabst et al. [19]. Six recurrent and frequent mutations (ASXL1 exon 12, DNMT3A exon 23, FLT3 exon 20, IDH1 exon 4, IDH2 exon 4 and NPM1 exon 12) were sequenced using next generation sequencing and a multiplex PCR amplicon based library with the following primers: qI_Halo_ASXL1_R634_F2 (CCACCACGGAGTCCTCCT), qI_Halo_ASXL1_R634_R2 (GCCTCACCACCATCACCA), qI_DNMT3A_X23_F1 (CTGGCCAGCACTCACCCT), qI_DNMT3A_X23_R1 (TGTTTAACTTTGTGTCGCTACCTCA), qI_FLT3_X20_F3 (GTTTACCATGATAACGACACAACAC), qI_FLT3_X20_R3 (GATTGCACTCCAGGATAATACACA), qI_IDH1_X4_F1 (GGCTTGTGAGTGGATGGGTAA), qI_IDH1_X4_R2 (GCATTTCTCAATTTCATACCTTGCTTA), qI_IDH2_X4_F1 (GAAAGATGGCGGCTGCAGT), qI_IDH2_X4_R2 (CACCCTGGCCTACCTGGTC), qI_NPM1_X12_F1 (GAAGTGTTGTGGTTCCTTAAC) and qI_NPM1_X12_R1 (TGGACAACACATTCTTGGCA). The library was sequenced using a MiSeq sequencer (Illumina, San Diego, CA, USA) and Miseq Reagent kit V2 (paired-end sequencing 2 × 150 cycles). Alignment was performed using BWA aligner and variant calling was performed using FreeBayes and Mutect2 variant callers.
An extended DNA resequencing was performed using a Illumina NextSeq500 and Haloplex HS (Agilent, Santa Clara, CA, USA) targeted on the complete coding regions of 52 genes: ASXL1, ASXL2, ATM, BCOR, BCORL1, CBL, CCND2, CEBPA, CSF3R, CUX1, DDX41, DHX15, DNMT3A, EP300, ETV6, EZH2, FLT3, GATA1, GATA2, IDH1, IDH2, JAK2, KDM5A, KDM6A, KIT, KMT2D, KRAS, MGA, MPL, MYC, NF1, NPM1, NRAS, PHF6, PIGA, PPM1D, PRPF8, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, ZBTB7A, and ZRSR2. Data were processed through two algorithms from GATK (https://software.broadinstitute.org/gatk), HaplotypeCaller (scaling accurate genetic variant discovery to tens of thousands of samples, Poplin et al. [20]) and Mutect2 [21]. The mean depth was 2,190. Identified variants were curated manually and named according to the rules of the Human Genome Variation Society (hgvs.org). Molecular data have been stored in the European Nucleotide Archive (https://www.ebi.ac.uk/ena/).
Statistics
The clinical database was frozen in June 2015 and follow-up was updated in May 2018 [9]. Statistical analyses were conducted according to intention to treat. Categorical data were presented as percentages and compared using Fisher exact tests. Continuous data were presented as mean and standard deviation and compared using Mann and Whitney tests.
The endpoints considered were OS, event-free survival (EFS), CR, and relapse. For mutation impact, OS and EFS were studied using log rank tests for equality of survivor functions and graphically represented using Kaplan–Meier curves. For model evaluation, OS and EFS were studied using Cox models, whereas CR and relapse were studied using Fine and Gray models considering death as a competing event. The prognostic value of each score was assessed through their inclusion as categorical covariates in these models.
The impact of disease severity on lomustine benefit was assessed by considering the interaction between lomustine treatment and each studied score with the allogeneic stem-cell transplantation being introduced as a time-dependent covariate. The global effects of covariates introduced in the model were assessed through likelihood ratio tests and the effects of each modality of covariates were assessed through Wald tests. The proportional hazard assumption was checked through the use of the Schoenfeld residuals analysis. The differential impact of lomustine depending on disease severity was graphically represented using Kaplan–Meier graphs.
All tests were considered as two-sided considering a type I error set to 0.05. All analyses were performed using Stata 13.1 software (StataCorp, College Station, TX, USA).
Results
Molecular landscape in older AML patients with nonadverse cytogenetics
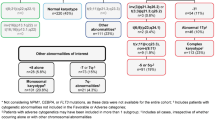
A molecular analysis of 52 genes was performed in 330 patients included in the LAM-SA 2007 clinical trial (78% of the cohort). Cytogenetic risk was intermediate for 281 patients (203 normal and 78 other intermediate karyotypes) and favorable for 22 [15 inv(16)/t(16;16) and 7 t(8;21)] (Fig. 1). Mutations were identified in 319 patients with an average of 3.3 mutations per patient (0–12 mutations per patient; Figs. 2 and 3 and Table S1). Eleven patients have no mutation detected, including 3 with inv(16) and 1 with a KMT2A rearrangement.

Patients included in the LAM-SA 2007 clinical trial and analyzed in the present study (K: karyotype).

Number of mutations (gray bars) and patients with mutations (black bars) per gene.

“Other genes” regroups genes not classified in the eight previous groups (ATM, BCOR, BCORL1, CCND2, CUX1, DDX41, DHX15, MGA, PPM1D, PRPF8, ZBTB7A). NK normal karyotype, IR_Other intermediate karyotype other than normal.
NPM1, FLT3, and DNMT3A were the most frequently mutated genes (Figs. 2 and 3 and Table S1). NPM1 was mutated in 113 patients (34%, 86 type A, 7 B, and 9 D), FLT3 in 98 (30%, 76 with ITD [1–4 per patient] and 27 with other mutations including 20 of the tyrosine kinase domain), and DNMT3A in 88 (27%, 43 located at the Arg882 hot spot amino acid). Mutations of IDH2 were present in 60 patients (18%, Arg140 codon in 48 patients and Arg172 codon in 12 patients) and of IDH1 in 36 (11%). Additional therapeutic targets were found in 15 patients (TP53 in 8, 2%; JAK2 in 4, 1%; KIT in 3, 1%). Overall, a putative therapeutic target for tyrosine kinase inhibitors, IDH inhibitors or TP53 activators was identified in 178 patients (54%).
Mutation associations in older AML patients are not random
The mutations detected were analyzed using functional categories (Table S2) [16]. Mutations of DNA methylation genes were the most frequent, occurring in 178 patients (54%; Figs. 3 and 4a). DNMT3A mutations were strongly associated to those of NPM1, IDH1, and IDH2 and strictly separate from EZH2 mutations (Figs. 3 and 4a). IDH1 and IDH2 mutations were strictly separate from TET2 mutations. One patient presented both IDH1 and IDH2 mutations (#308), the former being subclonal to the latter (variant allelic frequencies of 1% and 32%, respectively). In addition, IDH2 and TET2 mutations were strictly separate from DDX41 mutations. Finally for this category, univariate analysis for patients in both study arms showed that IDH1 mutations were associated with a significantly worse prognosis for OS (p = 0.021) and EFS (p = 0.019; Fig. S2).

a DNA methylation genes (DNMT3A, IDH1, IDH2, and TET2). b Signaling genes (CBL, CSF3R, FLT3, JAK2, KIT, KRAS, NRAS, NF1, PTPN11). c NPM1. d Transcription factors genes (CBFB–MYH11 and RUNX1–RUNX1T1 fusion genes; CEBPA, EP300, ETV6, GATA1, GATA2, MYC, RUNX1, SETBP1). e Splicing genes (SF3B1, SRSF2, U2AF1, ZRSR2). f Chromatin genes (KMT2A fusion gene; ASXL1, ASXL2, EZH2, KDM5A, KDM6A, KMT2D). g Cohesin genes (RAD21, SMC1A, SMC3, STAG2). h Antioncogenes (PHF6, TP53, WT1). i Other genes (ATM, BCOR, BCORL1, CCND2, CUX1, DDX41, DHX15, MGA, PPM1D, PRPF8, ZBTB7A).
Mutations of genes encoding tyrosine kinases and proteins of the RAS signaling pathway were identified in 51% of the patients, more frequently FLT3 in 98 patients (30%) and RAS genes in 75 (23%). RAS mutations were secondary events as demonstrated by their very low VAF (Fig. S3). FLT3 mutations were significantly associated with NPM1, WT1, and SMC3 mutations (Figs. 3 and 4b) and leukocytosis (p < 0.0001, Fig. S4). FLT3 mutations were strictly separate from DDX41 mutations, NRAS strongly separate from STAG2 mutations and KRAS from U2AF1 mutations (Figs. 3 and 4b). FLT3-ITD mutations were not linked to prognosis (OS, p = 0.49; EFS, p = 0.82; Fig. S5).
NPM1 mutations, present in 34% of the patients, were associated with a significantly better prognosis for OS (p = 0.027) and EFS (p = 0.020; Fig. S6). These mutations were significantly associated with FLT3, DNMT3A, TET2, IDH2, and PTPN11 mutations (Figs. 3 and 4c) and leukocytosis (p < 0.0001, Fig. S4) but strictly separate from DDX41 and RUNX1 mutations and CBFB–MYH11 fusion gene.
Transcription factors abnormalities were identified in 104 patients (32%), including 82 patients with intermediate cytogenetics (Figs. 3 and 4d). CBFB–MYH11 fusions were strongly associated with RAS and CSF3R mutations. RUNX1 mutations were significantly associated with EZH2, SF3B1, and BCORL1 mutations (in contrast to be significantly separate from BCOR mutations). CEBPA mutations were significantly associated with TET2 and STAG2 mutations and separate from WT1 mutations, although these differences were not significant when the number of CEBPA mutations per patient (19 mono-allelic vs. 10 bi-allelic) was specifically analyzed due to a lack of power (Fig. S7).
Mutations of genes involved in splicing were identified in 82 patients (25%, Figs. 3 and 4e), especially SRSF2 mutations (15%) that were significantly associated with ASXL1, TET2, STAG2, and CBL and separate from FLT3 mutations.
Chromatin regulators were mutated in 77 patients (23%, Figs. 3 and 4f), especially ASXL1 (15%). ASXL1 mutations were strongly associated with STAG2, SRSF2, EZH2, and ZRSR2 mutations and separate from FLT3, NPM1, and DNMT3A mutations. In univariate analysis, ASXL1 mutations were associated with a very poor prognosis in our series, especially for EFS (p = 0.0002; OS: p = 0.0069; Fig. S8).
Cohesin mutations were identified in 61 patients (18%, Figs. 3 and 4g), particularly STAG2 (12%). These mutations were significantly associated with leukopenia at diagnosis (p < 0.0001, Fig. S4) and mutations of ASXL1, SRSF2, CEBPA, EZH2, ETV6, and GATA2 and separate from DNMT3A and NRAS mutations.
Mutations of tumor-suppressor genes were more infrequent, identified in 28 patients (8%, Figs. 3 and 4h), the most frequent being WT1. Its mutations were strongly associated with those of FLT3 and separate from RAD21 and CEBPA mutations. Few patients had TP53 mutations (2%; Fig. 3), coherent with the exclusion of patient with adverse cytogenetics in this clinical trial.
The remaining genes, not classified in one of the eight categories detailed above, were found mutated in 67 patients (20%, Figs. 3 and 4i), the two most frequently mutated being BCOR (8%) and DDX41 (7%). BCOR mutations were strongly associated to BCORL1 mutations and separate from NPM1, U2AF1, and RUNX1 mutations. Patients with DDX41 mutations had a unique mutation pattern, without mutations of FLT3, NPM1, IDH2, nor TET2 and were strongly associated with leukopenia at diagnosis (p < 0.0001, Fig. S2). They might have had a better prognosis but the small number of cases led to a lack of statistical power (Fig. S9). This group of patients deserves a specific investigation.
Prognostic significance of molecular classifications in older AML patients
Different molecular classifications for AML have been proposed, either prognostic (Patel et al. [13], Papaemmanuil et al. [15], Bullinger et al. [16], and ELN2017 [8]) or ontogenic (Lindsley et al. [14]). Their impact in the LAM-SA 2007 clinical trial was evaluated (Fig. S10). The 3-month CI of CR was 85% (95% CI: 81–89%). Papaemmanuil et al.’s and Lindsley et al.’s classifications were significantly associated with variations of CR CI (respectively, p < 0.0001 and p = 0.0257). The 3- and 5-year CI of relapse (CIR) were respectively 73% (95% CI: 68–77%) and 79% (95% CI: 74–83%). The five classifications were significantly associated with the CIR (ELN2017 [p < 0.0001], Bullinger et al. [p < 0.0001], Papaemmanuil et al. [p < 0.0001], Patel et al. [p = 0.0047], and Lindsley et al. [p = 0.0152]). The 3- and 5-year EFS were 23% (95% CI: 19–27%) and 17% (95% CI: 13–21%), respectively. The five classifications were also significantly associated with EFS (ELN2017 [p = 0.0001], Patel et al. [p = 0.0006], Lindsley et al. [p = 0.0046], Bullinger et al. [p = 0.0096], and Papaemmanuil et al. [p = 0.0139]). The 3- and 5-year OS were 39% (95% CI: 35–44%) and 24% (95% CI: 20–29%), respectively. The ELN2017 classification was strongly associated to OS (p = 0.001) as well as to, to a lower extent, those of Patel et al. (p = 0.014) and Lindsley et al. (p = 0.050). Overall, ELN2017 was the best molecular classification to summarize the clinical evolution of the patients included in the LAM-SA 2017 clinical trial.
Impact of lomustine in the genomic landscape of AML
We evaluated the impact of these five classifications according to the treatment, 163 patients being assigned to arm A (with lomustine) and 167 to arm B (without lomustine). Regarding CR, a significant interaction between the Papaemmanuil classification and lomustine treatment was highlighted (p < 0.001), lomustine being significantly associated with a lower CR rate in the CBFB–MYH11 subgroup (p = 0.002) and a better CR rate in the IDH2 R172K subgroup (p < 0.001). Regarding CIR, a significant interaction between lomustine treatment and the ELN2017 classification was also highlighted (p = 0.027). Lomustine is significantly associated with a lower relapse rate in the subset of the ELN2017 adverse group with a nonadverse karyotype, i.e., with RATFIN mutations (p = 0.001) but not in the ELN2017 favorable nor intermediate groups (p = 0.879 and 0.861, respectively). A significant interaction with lomustine and the risk of relapse was also highlighted using the Papaemmanuil classification (p = 0.003) with a lower relapse rate in the subgroup of AML with mutated chromatin and/or RNA-splicing genes (p = 0.039) and a higher relapse rate in patients with RUNX1–RUNX1T1 fusion gene (p < 0.001). A significant interaction with lomustine was also highlighted using the Bullinger classification (p = 0.005) with a lower relapse rate in the chromatin-spliceosome group and TP53 mutations (p = 0.030 and p < 0.001, respectively). No interaction was observed between lomustine treatment and the classifications of Patel et al., Lindsley et al., Papaemmanuil et al., or Bullinger et al. for EFS (p = 0.867, 0.370, 0.232, and 0.127, respectively) nor OS (p = 0.896, 0.758, 0.261, and 0.127, respectively). However, there was a significant interaction between the ELN2017 classification and lomustine (p = 0.036 for EFS; p = 0.048 for OS; Fig. 5a, b), indicating that lomustine was significantly associated with a better EFS (p < 0.001) and OS (p = 0.023) in RATFIN mutations (Fig. 5c) but not intermediate (p = 0.162 for EFS; p = 0.599 for OS; Fig. 5d) nor favorable (p = 0.763 for EFS; p = 0.199 for OS, Fig. 5e) subgroups. The advantage of the lomustine addition was stronger in patients with TP53 and FLT3-ITDhigh/NPM1WT mutations (Fig. S11).

a ELN2017 risk classification of patients not treated with lomustine. b ELN2017 risk classification of patients treated with lomustine. c RATFIN mutated patients treated or not with lomustine. d ELN2017 intermediate risk classification of patients treated or not with lomustine. e ELN2017 favorable risk classification of patients treated or not with lomustine.
Discussion
This study corresponds to a post-hoc analysis of the LAM-SA 2017 phase 3 randomized trial and describes the genomic landscape of older AML patients with nonadverse cytogenetics risk selected for intensive chemotherapy. Whereas we confirmed previous studies regarding the distribution and gene-gene interactions of the most frequent mutations including FLT3-ITD, NPM1, DNMT3A, RUNX1, or ASXL1 [22, 23], we also described a rare subgroup of patients with DDX41 mutations, which is characterized by few co-mutations, leukopenia, and a probable better outcome in agreement with the recent publication of Sebert et al. [24].
The FILO study group has been using lomustine for decades with consistent favorable results regarding CR achievement after one course of induction and survival endpoints [9, 25]. However, adding a third cytotoxic agent to an anthracycline-cytarabine induction may increase general toxicity especially hematologic toxicity and infections. Although not significant, the early death rate was slightly increased in the lomustine arm in the LAM-SA 2017 trial [9]. Thus, defining patients who benefit most from lomustine is of considerable importance.
We assessed the prognostic impact of five recent molecular classifications [8, 13,14,15,16]. Overall, these classifications have been relatively effective in predicting EFS and risk of relapse in older AML patients although only ELN2017, Patel et al.’s, and Lindsley et al.’s classifications were associated with OS. Moreover, with regard to the main clinical endpoints, no consistent pattern of interaction between the impact of lomustine and most molecular classifications was observed except for the ELN2017 classification [8] suggesting that lomustine could benefit mostly to patients with RATFIN mutations (ELN2017 adverse risk with nonadverse risk cytogenetics) [8]. We acknowledge that this result could correspond to a type I error (i.e., a false positive result) linked to the multiplication of analyses or a true interaction between the ELN2017 score and lomustine treatment; therefore, these results should be confirmed by a prospective randomized trial in this specific subgroup of patients with RATFIN mutations.
Lomustine is an alkylating agent of the nitrosourea type that alkylates and cross-links DNA thereby inhibiting DNA and RNA synthesis [26]. DNA damage repair is mainly mediated by the O6-methylguanine DNA methyltransferase the expression of which is very low in AML compared to other cancers [26]. Moreover, lomustine activity is cell cycle-phase nonspecific, a property not shared by anthracyclines and cytarabine, that may be important to target noncycling cells of the AML clones. Of note, lomustine is lipophilic and crosses the blood–brain barrier. The addition of lomustine may explain the absence of prognosis of FLT3-ITD in our series, in contrast to the well-described worse prognosis of patients with FLT3-ITD [27]. TP53 allows the repair of interstrand cross-links through the upregulation of the DNA repair factors XPC and DDB2 [28]. In a model of glioma, the DNA double strand breaks generated by chloroethylating nitrosourea were not repaired when TP53 was mutated contrary to cells with normal TP53 [28]. Nevertheless, as RUNX1 and ASXL1 represent the most frequent high-risk mutations in this RATFIN group, the impact of lomustine could affect these mutations through mechanisms that remain to be elucidated.
As lomustine is used during front-line treatment, the ELN2017 status must be rapidly defined for an optimal use of this drug. This may be challenging especially for ASXL1 and RUNX1 genes. However, we have previously shown that waiting a short period of time before induction chemotherapy is safe in AML patients and allow a molecular testing before choosing the most appropriate induction regimen [29]. A recent study has also shown an interaction between genetic profiles and gemtuzumab ozogamycin efficacy indicating that molecular stratification is useful for a rational use of targeted therapies but also of cytotoxic agents in AML patients [30].
References
Juliusson G, Abrahamsson J, Lazarevic V, Antunovic P, Derolf A, Garelius H, et al. Prevalence and characteristics of survivors from acute myeloid leukemia in Sweden. Leukemia. 2017;31:728–31.
Oran B, Weisdorf DJ. Survival for older patients with acute myeloid leukemia: a population-based study. Haematologica. 2012;97:1916–24.
DiNardo CD, Wei AH. How I treat acute myeloid leukemia in the era of new drugs. Blood. 2020;135:85–96.
Grimwade D, Walker H, Harrison G, Oliver F, Chatters S, Harrison CJ, et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001;98:1312–20.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87.
Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47.
Pigneux A, Bene MC, Salmi LR, Dumas PY, Delaunay J, Bonmati C, et al. Improved survival by adding lomustine to conventional chemotherapy for elderly patients with aml without unfavorable cytogenetics: results of the LAM-SA 2007 FILO trial. J Clin Oncol. 2018;36:3203–10.
Gerson SL. Regeneration of O6-alkylguanine-DNA alkyltransferase in human lymphocytes after nitrosourea exposure. Cancer Res. 1988;48:5368–73.
Weiss RB, Tormey DC, Holland F, Weinberg VE, Lesnick G, Perloff M, et al. A randomized trial of postoperative five-versus three-drug chemotherapy after mastectomy: a Cancer and Leukemia Group B (CALGB) study. Recent Results Cancer Res. 1982;80:170–6.
Marsh JC. The effects of cancer chemotherapeutic agents on normal hematopoietic precursor cells: a review. Cancer Res. 1976;36:1853–82.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89.
Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–76.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35:934–46.
Dastugue N, Payen C, Lafage-Pochitaloff M, Bernard P, Leroux D, Huguet-Rigal F, et al. Prognostic significance of karyotype in de novo adult acute myeloid leukemia. The BGMT group. Leukemia. 1995;9:1491–8.
LaRochelle O, Bertoli S, Vergez F, Sarry JE, Mansat-De Mas V, Dobbelstein S, et al. Do AML patients with DNMT3A exon 23 mutations benefit from idarubicin as compared to daunorubicin? A single center experience. Oncotarget. 2011;2:850–61.
Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–70.
Poplin R, Ruano-Rubio V, DePristo M A, Fennell T J, Carneiro M O, Van der Auwera G A, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 201178; https://doi.org/10.1101/201178.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Eisfeld AK, Kohlschmidt J, Mrozek K, Blachly JS, Walker CJ, Nicolet D, et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years or older who respond favorably to standard chemotherapy: an analysis of alliance studies. Leukemia. 2018;32:1338–48.
Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Gorlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128:686–98.
Sebert M, Passet M, Raimbault A, Rahme R, Raffoux E, Sicre de Fontbrune F, et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood. 2019;134:1441–4.
Pigneux A, Harousseau JL, Witz F, Sauvezie M, Bene MC, Luquet I, et al. Addition of lomustine to idarubicin and cytarabine improves the outcome of elderly patients with de novo acute myeloid leukemia: a report from the GOELAMS. J Clin Oncol. 2010;28:3028–34.
Nikolova T, Roos WP, Kramer OH, Strik HM, Kaina B. Chloroethylating nitrosoureas in cancer therapy: DNA damage, repair and cell death signaling. Biochim Biophys Acta Rev Cancer. 2017;1868:29–39.
Whitman SP, Maharry K, Radmacher MD, Becker H, Mrozek K, Margeson D, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010;116:3622–6.
Batista LF, Roos WP, Christmann M, Menck CF, Kaina B. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res. 2007;67:11886–95.
Bertoli S, Berard E, Huguet F, Huynh A, Tavitian S, Vergez F, et al. Time from diagnosis to intensive chemotherapy initiation does not adversely impact the outcome of patients with acute myeloid leukemia. Blood. 2013;121:2618–26.
Fournier E, Duployez N, Ducourneau B, Raffoux E, Turlure P, Caillot D, et al. Mutational profile and benefit of gemtuzumab ozogamicin in acute myeloid leukemia. Blood. 2020;135:542–6.
Acknowledgements
We thank physicians from participating centers as well as central and local data managers for collecting and monitoring patients’ data. We thank Filothèque members for the conservation of patients samples; Catherine Lacombe, Rachid Boulghana, Laure Cabaret, Ségolène Diry, Krishshanthi Sinnadurai, and Shari Dini Mohamed.
French Innovative Leukemia Organization
J.-P. Marolleau26, A. Aleme26, F. Orsini-Piocelle27, N. Cadoux27, N. Ifrah28, M. Hunault28, C. Marie28, A. Al Jijakli29, G. Lepeu30, H. Zerazhi30, M. Beyrne30, A. Banos31, S. Labarrere31, E. Deconinck32, M. Peria32, A. El Yamani33, O. Kadiri33, B. Choufi34, M. Brument34, A. Pigneux35, T. Leguay35, P.-Y. Dumas35, C. Berthou36, G. Guillerm36, G. Drugmanne36, O. Tournilhac37, G. Roy37, B. Audhuy38, S. Camara38, D. Caillot39, M. Grandjean39, J.-Y. Cahn40, C.-E. Bulabois40, B. Fief40, N. Vey41, C. Ladraa41, V. Dorvaux42, M. Hagopian42, N. Fegueux43, C. Fenoll43, V. Sabadash43, M. Ojeda44, C. Haby44, F. Witz45, C. Bonmati45, M. Lhuire45, J. Delaunay46, P. Peterlin46, L. Airiau46, L. Mannone47, I. Touitou47, E. Jourdan48, D. Umuhire48, M. Alexis49, O. Michel49, F. Dreyfus50, D. Bouscary50, A. Cheung50, L. Sanhes51, F. Touhami51, E. Ribas51, M. Puyade52, M.-P. Gallego-Hernanz52, N. Hugon52, C. Himberlin53, L. Maggi53, T. Lamy54, A. Testu54, E. Tavernier55, S. Marchand55, B. Lioure56, C. Kravanja56, L. Benboubker57, D. Nollet57, M. Attal58, C. Recher58, A. Sarry58, A. Lhermitte59, G. Yrica59, D. Schwartz59, N. Le Montagner59, C. Fenoll59, V. Sabadash59, D. Nollet59, L. Auvray59, R. Delepine59, A. Fayault59
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the French Innovative Leukemia Organization (FILO) are listed below Acknowledgements.
Supplementary information
Rights and permissions
About this article

Cite this article
Largeaud, L., Cornillet-Lefebvre, P., Hamel, JF. et al. Lomustine is beneficial to older AML with ELN2017 adverse risk profile and intermediate karyotype: a FILO study. Leukemia 35, 1291–1300 (2021). https://doi.org/10.1038/s41375-020-01031-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-020-01031-1
- Springer Nature Limited