
Abstract
Abnormal metabolism is a fundamental hallmark of cancer and represents a therapeutic opportunity, yet its regulation by oncogenes remains poorly understood. Here, we uncover that JMJD1C, a jumonji C (JmjC)-containing H3K9 demethylase, is a critical regulator of aberrant metabolic processes in homeobox A9 (HOXA9)-dependent acute myeloid leukemia (AML). JMJD1C overexpression increases in vivo cell proliferation and tumorigenicity through demethylase-independent upregulation of a glycolytic and oxidative program, which sustains leukemic cell bioenergetics and contributes to an aggressive AML phenotype in vivo. Targeting JMJD1C-mediated metabolism via pharmacologic inhibition of glycolysis and oxidative phosphorylation led to ATP depletion, induced necrosis/apoptosis and decreased tumor growth in vivo in leukemias co-expressing JMJD1C and HOXA9. The anti-metabolic therapy effectively diminished AML stem/progenitor cells and reduced tumor burden in a primary AML patient-derived xenograft. Our data establish a direct link between drug responses and endogenous expression of JMJD1C and HOXA9 in human AML cell line- and patient-derived xenografts. These findings demonstrate a previously unappreciated role for JMJD1C in counteracting adverse metabolic changes and retaining the metabolic integrity during tumorigenesis, which can be exploited therapeutically.
Similar content being viewed by others


Introduction
Malignant transformation rewires cell metabolism by directly regulating key metabolic enzymes/pathways that provide a selective growth advantage for tumor cells [1, 2]. Identifying oncogenes that drive cancer cell metabolism and defining their mechanisms of action are of particular importance, since they are not only essential for understanding cancer biology, but are also critical in developing effective inhibitors to target the disease.
Glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) are two major energy-producing pathways that promote tumor progression. Unlike normal cells, where glycolysis is induced by hypoxia, cancer cells metabolize glucose to lactate at a high rate to meet increased energy needs even in oxygen-rich (aerobic) conditions [3]. Glucose is the predominant energy fuel for most cancers and a cell’s metabolism can change based on the availability of this substrate [4]. When glucose is abundant within the tumor microenvironment, high rates of aerobic glycolysis, also known as the Warburg effect, convert glucose into pyruvate and subsequently produce considerable amounts of ATP required for cancer cell survival [4]. OXPHOS is an equally important metabolic transformation property and uses pyruvate, the final product of glycolysis, as the primary carbon source to generate ATP through the tricarboxylic acid (TCA) cycle in the mitochondria. A minor inhibition of ATP production can compromise cancer cell growth and depletion of ATP induces necrotic or apoptotic cell death [4, 5]. Evidence is emerging to support a link between cancer metabolism and clinical outcomes; aggressive tumors with coexistence of glycolytic and oxidative metabolism display increased metabolic plasticity that promotes tumorigenesis and metastasis [6, 7].
High rates of glycolysis and OXPHOS have also been detected in patients with AML contributing to cell proliferation and chemoresistance [8, 9]. We have previously reported a possible link between upregulation of mitochondrial OXPHOS enzymes and leukemogenesis in mixed lineage leukemia (MLL)-rearranged AML (e.g., MLL-AF9) [10], a drug-refractory subtype of leukemia, in which HOXA9/MEIS1 and JMJD1C are co-expressed and serve as important mediators of leukemia stem cells (LSCs) [11, 12]. HOXA9 and MEIS1 are homeodomain transcription factors that induce the expansion of normal hematopoietic stem cells (HSCs) and cooperate to transform HSCs into LSCs producing AML in mice [13, 14]. HOXA9 is overexpressed in greater than 50% of AML patients and predicts poor prognosis [15, 16]. JMJD1C functions as a HOXA9 cofactor and physically interacts with HOXA9 to modulate downstream genes essential for LSC functions [11]. Loss of JMJD1C impairs the maintenance of MLL-rearranged AML but has minimal effects on HSC self-renewal; [11] thus, targeting JMJD1C and its key downstream pathways provides a possible therapeutic opportunity. JMJD1C is a histone H3K9 demethylase, but so far, no enzymatic activity has been reported in HOXA9-dependent AML [11, 17]. This implicates additional/alternative mechanisms of action for JMJD1C in promoting tumor progression. Here, we uncover a previously unknown function for JMJD1C in cancer metabolism that supports leukemic cell proliferation and survival in HOXA9-dependent AML.
Methods
More information on methods can be found in the Supplemental Information.
Statistical analysis
Statistical significance of differences was determined by an unpaired two-tailed Student’s t test for comparison between two groups and log-rank test for Kaplan–Meier survival curves using GraphPad Prism 6.0 (La Jolla, CA, USA). Data are presented as mean ± SEM. *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001, NS = not significant, Student’s t test.
Results
JMJD1C exerts in vivo demethylase-independent tumor-promoting function through its metabolism-associated domain
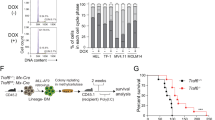
To directly investigate a pro-leukemogenic role for JMJD1C in HOXA9-dependent AML, we generated HOXA9/MEIS1 pre-LSCs, an early stage of LSC development [12, 13], by co-transducing HSC-enriched LSK cells with HOXA9-GFP and MEIS1-puro. We found that enforced expression of JMJD1C and JMJD1C-S, a short isoform lacking N-terminal zinc finger (ZF) domain, improved the serial replating ability of LSK-HOXA9/MEIS1 pre-LSCs and the S phase population (Supplemental Figure S1A), indicative of increased in vitro self-renewal and cell proliferation. Importantly, overexpression of JMJD1C and JMJD1C-S reduced the disease latency, respectively contributing to a more aggressive phenotype in HOXA9/MEIS1-driven AML (Fig. 1a) via enhancing in vivo cell proliferation in BL6 mice (Supplemental Figure S1B). Given the lack of the ZF domain in JMJD1C-S and its importance in the enzymatic activity of JmjC domain-containing proteins to specifically demethylate H3K9 [18], our data implicate a possible H3K9 demethylase-independent function for JMJD1C in HOXA9-dependent AML. This is in line with western blots exhibiting no noticeable differences in global H3K9me1/2/3 levels upon overexpression of JMJD1C/JMJD1C-S in LSK-HOXA9/MEIS1 leukemic cells (Supplemental Figure S2) and agrees with several recent studies that could not detect methylation alterations in JMJD1C-depleted HOXA9/MEIS1 and MLL-AF9 leukemic cells following extensive biochemical assays, including ChIP-seq and western blots [11, 17].

The metabolism-associated domain is required for JMJD1C to promote leukemogenesis in vivo in HOXA9-dependent AML. a Kaplan–Meier survival curve of BL6 mice transplanted with LSK-HOXA9/MEIS1 pre-LSCs overexpressing JMJD1C/JMJD1C-S. b Bioluminescence imaging and total flux (photons/sec; p/s) of MOLM-13 xenograft mice (n = 3)
To further assess the functional domains of JMJD1C in vivo, we generated two JMJD1C mutants that either eliminated the JmjC domain (∆JmjC; demethylase-associated) or deleted 121aa in the N-terminal region of the thyroid-hormone receptor (TR)-binding domain (∆N-TR; metabolism-associated). The impact of JMJD1C mutants on tumor burden was examined in an aggressive xenograft mouse model of human MLL-AF9 (MOLM-13) AML, which is a robust and effective assay system for in vivo functional studies given its extremely short latency (Supplemental Figure S3A). Confocal immunofluorescence confirmed overexpression and nuclear localization of JMJD1C mutants (Supplemental Figure S3B). Similar to the loss of the ZF domain, in vivo bioluminescence imaging revealed that deletion of the JmjC domain did not affect the ability of JMJD1C to promote leukemic cell engraftment and to produce a more aggressive leukemia in NOD/SCID/IL2R gamma-null (NSG) mice (Fig. 1b; Supplemental Figure S3C). Given that the JmjC domain is a prerequisite for the demethylase activity of JMJD1C, this result further supports a demethylase-independent role for JMJD1C in HOXA9-dependent AML. Conversely, partial deletion of the TR domain was sufficient to abrogate the proleukemogenic activity of JMJD1C in vivo (Fig. 1b; Supplemental Figure S3C), indicating possible involvement of the metabolism-associated domain in JMJD1C tumor-promoting function.
JMJD1C-regulated genes are involved in metabolic processes in HOXA9-dependent AML
To determine key molecular pathways induced by JMJD1C in HOXA9-driven leukemogenesis, we performed gene expression profiling of JMJD1C-overexpressing LSK-HOXA9/MEIS1 pre-LSCs. A total of 110 differentially expressed genes were identified with a significance cut-off of the false discovery rate ≤ 0.05 and fold change ≥2 (Fig. 2a). Subsequent Gene Ontology (GO) term analysis [19] revealed strong enrichment of metabolism-associated biological processes (Fig. 2a). Likewise, the gene set enrichment analysis [20] identified JMJD1C-induced enrichment of gene sets associated with energy metabolism including glycolysis, pyruvate metabolism and mitochondrial TCA cycle, as well as the mTOR pathway (Supplemental Figure S4A), which is required for the maintenance of mitochondrial oxidative activities [21]. These data implicate JMJD1C-mediated control of glycolytic and oxidative processes in the regulation of energy (ATP) production in HOXA9-dependent AML.

JMJD1C modulates bioenergetics through upregulation of genes/pathways involved in metabolic processes in HOXA9/MEIS1 AML. a Heat map of the top 110 genes differentially regulated by JMJD1C based on microarray data (n = 4); GO-term analysis displaying the top 14 most enriched GO terms of genes upregulated by JMJD1C (fold enrichment > 10 and P < 0.05). b Western blots of key metabolic enzymes upon JMJD1C overexpression in leukemic cells from primary LSK-HOXA9/MEIS1 AML. c Total ATP levels, OCR and ECAR in LSK-HOXA9/MEIS1 leukemic cells overexpressing JMJD1C (n = 3)
JMJD1C upregulates key glycolytic and oxidative enzymes associated with human cancers
JMJD1C (also known as TRIP8) was originally described as a TR-binding protein directly interacting with TRs, which could serve as DNA-binding transcription factors with a known regulatory function of energy metabolism by inducing OXPHOS gene transcripts and cross-talking to glycolysis [22,23,24,25]. We have previously shown a possible link between upregulation of OXPHOS enzymes (e.g., ND2) and MLL-AF9 leukemogenesis, and reported elevated expression of OXPHOS genes in a panel of primary human AML samples [10]. Here, we found that JMJD1C overexpression induced a substantial increase in mitochondrial complex 1 enzyme ND2 and complex III enzyme CYTB in LSK-HOXA9/MEIS1 leukemic cells (Fig. 2b), supporting a possible role for JMJD1C in the regulation of mitochondrial OXPHOS.
Our data also showed that JMJD1C upregulated several key glycolytic enzymes identified by microarray analysis, including ALDOC, PGK1, PKM2, and LDHA (Supplemental Figure S4B). Importantly, JMJD1C promoted phosphorylation of PKM2 at tyrosine Y105 (Fig. 2b), which facilitates the formation of the dimeric form of PKM2 [26]. The dimerization of Y105-p-PKM2 is a common event in human cancers and enhances glycolysis to support cancer progression [26], representing a valuable therapeutic target. Notably, JMJD1C knockout in primary MLL-AF9 leukemic cells did not cause changes in H3K9me2 levels on the loci of JMJD1C-modulated metabolic enzymes (Supplemental Figure S5A; GSE75577 [11]). This is consistent with our observation in human MLL-AF9 AML (MOLM-13) cells that deletion of the demethylase-associated JmjC domain did not affect the ability of JMJD1C to upregulate essential metabolic enzymes (i.e., Y105-p-PKM2 and CYTB); conversely, partial deletion of the metabolism-associated TR domain completely abolished the expression-promoting function of JMJD1C (Supplemental Figure S5B). These data underline H3K9 demethylase-independent regulation of metabolic pathways by JMJD1C in HOXA9-dependent AML.
JMJD1C enhances rates of glycolysis and OXPHOS for ATP generation
We next examined the direct effect of JMJD1C on cellular metabolism, and observed that JMJD1C overexpression significantly elevated ATP production compared to empty vector control (Fig. 2c). Since the total cellular ATP pool is predominantly produced by OXPHOS and glycolysis, we performed bioenergetic analysis of oxygen consumption rate (OCR) for OXPHOS and extracellular acidification rate (ECAR) for lactate production and glycolysis. Our data revealed increased levels of OCR and ECAR upon JMJD1C overexpression in LSK-HOXA9/MEIS1 leukemic cells (Fig. 2c). JMJD1C thus has the ability to increase ATP production through coordinated regulation of OXPHOS and glycolysis in HOXA9-dependent AML.
JMJD1C protects leukemic cells from high glucose-induced impairment of OXPHOS
Given the fact that glucose is the principal energy source for cancer cell proliferation and survival, and our observation that JMJD1C-regulated genes were enriched in the glucose metabolic process (Fig. 2a), we tested the effect of JMJD1C on glucose-induced alterations in metabolism. Our data showed that exposure to high glucose (10 mM) induced a metabolic shift resulting in increased ECAR but decreased OCR in LSK-HOXA9/MEIS1 leukemic cells, while enforced expression of JMJD1C rescued leukemic cells from high glucose-induced suppression of OXPHOS and consequently sustained high rates of lactate production and oxygen consumption when glucose availability was high (Supplemental Figure S6). Collectively, JMJD1C may confer metabolic flexibility in leukemic cells, allowing them to adapt to microenvironmental changes.
JMJD1C exerts an origin-specific function in modulating dimeric PKM2-dependent glycolysis
We have previously demonstrated that stem cell-derived LSK-HOXA9/MEIS1 and LSK-MLLAF9 pre-LSCs produce histopathologically and immunophenotypically similar diseases; however, MLL-AF9 AML is more aggressive than HOXA9/MEIS1 AML [13, 27]. While MLL-AF9 can transform both HSCs and granulocyte–monocyte progenitor (GMP) [12, 13, 28], the origin of pre-LSCs determines tumor aggressiveness [29, 30]. To evaluate whether the cellular origin affects JMJD1C function, we generated origin-specific pre-LSCs by transforming LSK or GMP cells with MLL-AF9-GFP followed by transduction with JMJD1C-targeting shRNAs or scrambled-control, and transplanted origin-specific MLL-AF9 pre-LSCs into BL6 mice to develop primary AML. GFP+ leukemic cells isolated from primary AML were subsequently transplanted into secondary recipients (Supplemental Figure S7A). JMJD1C depletion significantly extended survival of mice receiving LSK-MLLAF9 leukemic cells but had no effects on those receiving GMP-MLLAF9 cells (Fig. 3a). These data support an origin-specific role for JMJD1C in maintaining stem cell-derived MLL-AF9 AML, which is known to be highly aggressive with extensive chemoresistance as compared to GMP-derived MLL-AF9 AML [29, 31].

Inhibition of dimeric p-PKM2 by Shik suppresses glycolysis leading to a preferential use of OXPHOS over glycolysis for ATP production in an origin-specific manner. a Kaplan–Meier survival curve of BL6 mice receiving LSK-MLLAF9 or GMP-MLLAF9 leukemic cells from primary AML mice transplanted with origin-specific pre-LSCs. b ECAR and OCR following exposure of LSK-MLLAF9 LSCs to DMSO vs. Shik; total ATP levels in LSK-MLLAF9 LSCs treated with Shik for 48 h; ECAR levels in Shik-treated LSK-MLLAF9 LSCs following glucose stimulation (n = 3)
To identify an effective treatment strategy targeting JMJD1C-mediated metabolism, and given a known role of dimeric PKM2 as a key regulator of aerobic glycolysis in tumor progression [26], we investigated the effect of pharmacologic inhibition of dimeric PKM2 on metabolism in LSK-MLLAF9 LSCs [32]. GFP+ c-Kit-enriched LSK-MLLAF9 LSCs isolated from primary AML mice were treated with Shikonin (Shik), a natural compound from Chinese herbs that is a selective inhibitor of dimeric PKM2 with broad anticancer effects in multiple cancer types but exhibits no toxicity to normal cells [33, 34]. Shik treatment induced a significant reduction in ECAR accompanied with increased OCR following exposure to 100 nM Shik (Fig. 3b; Supplemental Figure S7B), a concentration sufficient to reduce the colony formation by ~50% (Supplemental Figure S7C). Likewise, a previous report reveals that genetic deletion of PKM2 causes a metabolic shift from glycolysis towards OXPHOS in MLL-AF9 leukemic cells [35]. As expected, inhibition of dimeric PKM2 by Shik led to no significant difference in total ATP production in LSK-MLLAF9 LSCs (Fig. 3b), largely due to increased OXPHOS partially compensating for the loss of glycolytic ATP production that in turn overcomes Shik-induced ATP depletion. Furthermore, Shik treatment prevented glucose-induced increase in lactate production in LSK-MLLAF9 LSCs (Fig. 3b). These findings underline an origin-specific function of JMJD1C in modulating dimeric PKM2-dependent glycolysis and implicate the importance of targeting both glycolysis and OXPHOS for ATP depletion in HOXA9-dependent AML.
Pharmacologic co-suppression of glycolysis and OXPHOS impairs leukemic stem cells
To efficiently reduce ATP production in order to impair LSCs, we performed combined inhibition of glycolysis and OXPHOS by Shik together with ABT-263, a BCL-2 inhibitor that has a known function to target OXPHOS selectively compromising LSCs in human AML but does not affect in vivo engraftment of normal HSCs in NSG mice at a concentration of 250 nM [8]. Compared to single-agent or control treatments, the combination of Shik and ABT-263 caused a significant decrease in colony formation, cell viability, ATP levels and bioenergetics while producing additive effects on necrosis/apoptosis in LSK-MLLAF9 LSCs (Fig. 4a–d; Supplemental Figure S8A). Furthermore, enforced expression of JMJD1C conferred significant protection of HOXA9/MEIS1 leukemic cells against metabolic impairment and prevented the decrease of ATP levels, cell viability and colony formation, caused by the combination treatment (Supplemental Figure S8B–D). These data are in line with JMJD1C knockout-induced decrease in ECAR and OCR in LSK-MLLAF9 LSCs (Supplemental Figure S8E–G), underscoring the dependence of stem cell-derived AML LSCs on both glycolytic and oxidative pathways. Our observations implicate the therapeutic value of targeting JMJD1C-driven metabolic pathways in AML treatment.

Co-suppression of glycolysis and OXPHOS by Shik and ABT-263 reduces ATP production impairing MLL-AF9 LSCs. a Colony number following 5 days treatment in methylcellulose (n = 3). Percentage of viable cells measured over a 96-h period for treated LSK-MLLAF9 LSCs (normalized against DMSO treated cells). Data were obtained from a representative experiment performed three times independently in replicates of four, mean ± SEM. b Total ATP levels in LSK-MLLAF9 LSCs treated for 48 h (n = 4). c ECAR and OCR following exposure of LSK-MLLAF9 LSCs to DMSO, 200 nM ABT-263 vs. a combination of 100 nM Shik and 200 nM ABT-263 for 24 h (n = 4). d Percentage of late apoptotic/necrotic cells (n = 3), following 72 h treatment
Co-inhibitory treatment targets human AML cells in vivo whereas single-agent treatment induces a metabolic shift between glycolytic and oxidative pathways
We next investigated the effect of Shik and/or ABT-263 treatment on human leukemia cell lines, including MLL-AF9 AML (THP-1 and MOLM-13), non-MLL AML (HL-60), CML (K562), ALL (Jurkat), and lymphoma (U937) cells. Our data along with analysis of a microarray dataset [36] revealed differential expression of JMJD1C and HOXA9 in these human leukemia cell lines (Supplemental Figure S9A–C). This provides a valuable assay system in understanding how endogenous expression of JMJD1C and HOXA9 determines drug responses of human leukemic cells (Table 1).
Our in vitro data showed that the inhibitory effect of combined treatment with Shik and ABT-263 was largely dependent upon co-expression of JMJD1C and HOXA9, as the combination selectively reduced cell viability and induced necrosis/apoptosis in human THP-1 and MOLM-13 cells that express high levels of JMJD1C and HOXA9, but had little effects on human HL-60, K562, U937 and Jurkat cells that lack either JMJD1C or HOXA9 (Supplemental Figure S10A, B). Consistently, our in vivo results from human cell line-derived xenograft (CDX) mouse models showed that 2-day ex vivo combination pretreatment of human leukemic cells reduced tumor burden in MOLM-13 xenografts but not in U937 and HL-60 xenografts, as determined by in vivo bioluminescence imaging (Supplemental Figures S11 and S12). The ex vivo pretreatment data were confirmed by direct in vivo treatment of MOLM-13 xenografts, which led to decreased proliferation of human hCD45+hCD33+ myeloid cells in treated NSG mice (Supplemental Figure S13A, B). These findings provide evidence supporting treatment specificity in targeting human AML cells co-expressing JMJD1C and HOXA9.
Of note, consistent with the modest single-agent activity observed in vitro, 2-day ex vivo single-agent pretreatment with Shik/ABT-263 exhibited no inhibitory effects on in vivo engraftment in MOLM-13 xenografts (Supplemental Figure S14A). This could be attributed to single-agent-induced metabolic shift in the balance between OXPHOS and glycolysis, as evidenced by the observation that ABT-263 reduced ND2 expression but elevated Y105-p-PKM2; conversely, Shik decreased Y105-p-PKM2 but increased ND2 (Supplemental Figure S14B). These data underline the necessity for co-inhibition of JMJD1C-mediated glycolytic and oxidative pathways as a therapeutic strategy in AML treatment.
Co-inhibitory treatment reduces leukemia burden in an AML patient-derived xenograft while sparing normal hCD34+ hematopoietic stem/progenitor cells in NSG mice
To directly investigate the clinical potential of combination treatment, we used an established patient-derived xenograft (PDX) mouse model AML-491 [37], in which primary human AML cells from a relapsed patient with aberrant cytogenetics were transplanted into NSG mice to generate serially transplantable PDX cells. PDX AML-491 stably expressing enhanced firefly luciferase (FLuc) and mCherry were transplanted into NSG mice for a 4-day combination treatment. In vivo bioluminescence imaging showed that the combination of Shik and ABT-263 substantially reduced leukemia burden and also greatly diminished the percentage of human CD34+ AML stem/progenitor cells in mouse BM indicating treatment-induced impairment of LSC compartment (Fig. 5a, b; also see Supplemental Figures S15 and S16 for two independent repeats). High levels of HOXA9 and JMJD1C were observed in human myeloid hCD33+ cells of PDX AML-491 compared to two independent remission samples from AML patients with normal karyotype (Fig. 5c). In contrast, combined treatment with Shik/ABT-263 showed no noticeable inhibitory effects on normal human CD34+ stem/progenitor cells in NSG mice (Supplemental Figure S17). Our data demonstrate a therapeutic window existing for the combination treatment targeting key metabolic pathway components specific to human leukemias, which largely depend on co-expression of JMJD1C and HOXA9.

In vivo treatment with Shik and ABT-263 reduces tumor burden in AML PDX mice. a Bioluminescence imaging and total flux for PDX mice treated with DMSO vs. combination of Shik and ABT-263 for four consecutive days. Note: the mice did not develop full AML during the course of the study. b Percentage of hCD34+ cells engrafted in the BM of PDX mice in response to in vivo treatment. c qPCR (n = 3) confirming high levels of HOXA9 and JMJD1C in hCD33+ myeloid cells of PDX mice compared to remission samples from two AML patients
Discussion
Increasing evidence shows that glycolysis and OXPHOS are not always mutually exclusive in a tumor; instead, they may coexist and contribute differently to ATP production in response to changes in the microenvironment such as glucose availability [1, 7, 38]. Here, we reveal a previously unknown role for JMJD1C in coupling glycolytic and oxidative metabolism to leukemic cell proliferation and subsequent acquisition of an aggressive malignant phenotype in AML. Disrupting JMJD1C-driven metabolic machinery provides a unique therapeutic opportunity for AML treatment.
AML is a devastating form of blood cancer. HOXA9 is overexpressed in more than 50% of AML patients and predicts poor prognosis [15, 16]. Notably, JMJD1C has been reported to act as an H3K9 demethylase and transcriptional activator in AML that lacks HOXA9 expression (e.g., AML1-ETO) [11, 39]; conversely, its demethylase activity could not be detected in AML that depends on HOXA9 (e.g., MLL-AF9) [11, 17]. Likewise, we observed a dispensable role of demethylase-associated domain in JMJD1C tumor-promoting action during HOXA9-dependent leukemogenesis. This implicates a novel function for JMJD1C in gene regulation where the physical interaction with HOXA9 [11] is likely to alter the protein conformation of JMJD1C, which favors the use of its metabolism-associated domain leading to subsequent promotion of cancer cell metabolism over the demethylase activity. JMJD1C may have a dual function exerting its pro-oncogenic properties through either a metabolism- or demethylase-dependent mechanism that is largely determined by endogenous expression of its binding partner HOXA9.
In addition to the nature of activated oncogenes, the availability of energy substrates (i.e., glucose) within the tumor microenvironment also determines the metabolic circuitry of cancer cells during tumor progression. Leukemic cells with high-glucose metabolism are associated with chemoresistance in human AML [9]. We found that high glucose promotes the Warburg effect, causing increased utilization of glycolysis and inhibition of OXPHOS in HOXA9/MEIS1 cells. However, enforced expression of JMJD1C protects leukemic cells from high glucose-induced impairment of OXPHOS resulting in their dependency on both glycolysis and OXPHOS for ATP production, a metabolic phenotype consistent with JMJD1C-enhanced disease aggressiveness in HOXA9-dependent AML. JMJD1C thus has a critical function in attenuating adverse effects on cancer metabolism to maintain sufficient ATP supply for tumorigenesis.
HSC-derived leukemias are more resistant to chemotherapy than GMP-derived leukemias [29, 30]. Our studies show that leukemia cell of origin not only influences the drug response, but also determines pathway activation and metabolic processes in AML. We have previously documented that GMP-derived MLL-AF9 AML largely depends on aberrant activation of G protein (Gaq)/β-catenin signaling to elevate OXPHOS for ATP synthesis via modulation of mitochondrial complex 1 genes [10]. Here, we demonstrate that HSC-derived MLL-AF9 AML relies on the JMJD1C pathway to increase ATP production through upregulation of glycolytic and mitochondrial complex enzymes. Intracellular ATP levels are an important determinant of chemoresistance in cancer [40]; increased ATP induced by JMJD1C may confer a drug resistant phenotype to HSC-derived MLL-AF9 LSCs, which generate an aggressive AML associated with poor clinical outcome [31]. The observation that treatment-induced phenotypic deficiency in human AML in vivo resembles the JMJD1C knockout in established HSC-derived MLL-AF9 AML [11] underscores the target specificity of the combination of ABT-263 and shikonin.
Our data have established a positive link between drug responses and endogenous expression of JMJD1C and HOXA9 in human AML CDX mouse models; this has been confirmed in a preclinical PDX model. These results provide important insights into the cellular response to drug application. ABT-263 has been used as a single agent in several clinical trials in patients with relapsed cancer [41, 42]. One limitation is that ABT-263 not only suppresses OXPHOS [8], but also inhibits BCL-XL that reduces platelet lifespan, causing dose-dependent thrombocytopenia in patients [41, 42]. On the other hand, ABT-199, a selective BCL-2 inhibitor sparing platelets [43], has been approved by the FDA for the treatment of certain CLL subtypes and could be an alternative treatment option for further studies.
In sum, we demonstrate for the first time a critical role of JMJD1C in regulating cancer metabolism. This study provides proof-of-concept evidence to support targeting JMJD1C-driven metabolic machinery as a promising anticancer therapeutic approach for HOXA9-dependent AML.
References
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–5.
Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–20.
Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
Vander Heiden MG, Chandel NS, Schumacker PT, Thompson CB. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell. 1999;3:159–67.
Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015;22:577–89.
Yu L, Lu M, Jia D, Ma J, Ben-Jacob E, Levine H, et al. Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res. 2017;77:1564–74.
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–41.
Chen Y, Ma X, Zhang M, Wang X, Wang C, Wang H, et al. Gadd45a regulates hematopoietic stem cell stress responses in mice. Blood. 2014;123:851–62.
Lynch JR, Yi H, Casolari DA, Voli F, Gonzales-Aloy E, Fung TK, et al. Gaq signaling is required for the maintenance of MLL-AF9 induced AML. Leukemia. 2016;30:1745–8.
Zhu N, Chen M, Eng R, DeJong J, Sinha AU, Rahnamay NF, et al. MLL-AF9-and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J Clin Investig. 2016;126:997–1011.
Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–22.
Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–3.
Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J, Lawrence HJ, et al. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood. 2002;99:121–9.
Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7.
Andreeff M, Ruvolo V, Gadgil S, Zeng C, Coombes K, Chen W, et al. HOX expression patterns identify a common signature for favorable AML. Leukemia. 2008;22:2041–7.
Sroczynska P, Cruickshank VA, Bukowski JP, Miyagi S, Bagger FO, Walfridsson J, et al. shRNA screening identifies JMJD1C as being required for leukemia maintenance. Blood. 2014;123:1870–82.
Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–95.
Cho RJ, Campbell MJ. Transcription, genomes, function. Trends Genet. 2000;16:409–15.
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32(Database issue)):D277–80.
Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–40.
Scheller K, Seibel P, Sekeris CE. Glucocorticoid and thyroid hormone receptors in mitochondria of animal cells. Int Rev Cytol. 2003;222:1–61.
Liu YY, Brent GA. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol Metab. 2010;21:166–73.
Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269–304.
Lee JW, Choi HS, Gyuris J, Brent R, Moore DD. Two classes of proteins dependent on either the presence or absence of thyroid hormone for interaction with the thyroid hormone receptor. Mol Endocrinol. 1995;9:243–54.
Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73.
Dietrich PA, Yang C, Leung HH, Lynch JR, Gonzales E, Liu B, et al. GPR84 sustains aberrant beta-catenin signaling in leukemic stem cells for maintenance of MLL leukemogenesis. Blood. 2014;124:3284–94.
Blanpain C. Tracing the cellular origin of cancer. Nat Cell Biol. 2013;15:126–34.
Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–60.
George J, Uyar A, Young K, Kuffler L, Waldron-Francis K, Marquez E, et al. Leukaemia cell of origin identified by chromatin landscape of bulk tumour cells. Nat Commun. 2016;7:12166.
Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. 2016;30:43–58.
Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–68.
Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011;30:4297–306.
Chao TK, Huang TS, Liao YP, Huang RL, Su PH, Shen HY, et al. Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS ONE 2017;12:e0182166.
Wang YH, Israelsen WJ, Lee D, Yu VW, Jeanson NT, Clish CB, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell . 2014;158:1309–23.
Banerji V, Frumm SM, Ross KN, Li LS, Schinzel AC, Hahn CK, et al. The intersection of genetic and chemical genomic screens identifies GSK-3alpha as a target in human acute myeloid leukemia. J Clin Invest. 2012;122:935–47.
Vick B, Rothenberg M, Sandhofer N, Carlet M, Finkenzeller C, Krupka C, et al. An advanced preclinical mouse model for acute myeloid leukemia using patients’ cells of various genetic subgroups and in vivo bioluminescence imaging. PLoS ONE 2015;10:e0120925.
Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–83.
Chen M, Zhu N, Liu X, Laurent B, Tang Z, Eng R, et al. JMJD1C is required for the survival of acute myeloid leukemia by functioning as a coactivator for key transcription factors. Genes Dev. 2015;29:2123–39.
Zhou Y, Tozzi F, Chen J, Fan F, Xia L, Wang J, et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012;72:304–14.
Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–9.
Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–96.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8.
Acknowledgments
We thank Bing Liu, Jin Yi Lim, Emma Ramsay, and the staff of the Mark Wainwright Analytical Centre for their assistance, and the CCI tumor bank for human AML samples. This work was supported by NHMRC APP1128824 and Cancer Council NSW RG15-11 (J.Y.W.).
Author contributions
J.R.L., B.S., P.C. and H.H.L.L. performed experiments; A.P. and P.H. partially contributed to bioenergetic analysis; B.V., I.J., K.S., T.T., T.L., M.H., M.D.N., A.J.W. and J.W. contributed to experimental reagents; J.Y.W. wrote the paper, managed the project, and supported the work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lynch, J.R., Salik, B., Connerty, P. et al. JMJD1C-mediated metabolic dysregulation contributes to HOXA9-dependent leukemogenesis. Leukemia 33, 1400–1410 (2019). https://doi.org/10.1038/s41375-018-0354-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0354-z
- Springer Nature Limited
This article is cited by
-
Epigenetic roles of KDM3B and KDM3C in tumorigenesis and their therapeutic implications
Cell Death & Disease (2024)
-
The emerging roles of histone demethylases in cancers
Cancer and Metastasis Reviews (2024)
-
Metabolic dependencies of acute myeloid leukemia stem cells
International Journal of Hematology (2024)
-
JMJD1C promotes smooth muscle cell proliferation by activating glycolysis in pulmonary arterial hypertension
Cell Death Discovery (2023)
-
Role of HOXA9 in solid tumors: mechanistic insights and therapeutic potential
Cancer Cell International (2022)