
Abstract
Acute promyelocytic leukemia (APL) is characterized by t(15;17)(q22;q21), resulting in a PML-RARA fusion that is the master driver of APL. A few cases that cannot be identified with PML-RARA by using conventional methods (karyotype analysis, FISH, and RT-PCR) involve abnormal promyelocytes that are fully in accordance with APL in morphology, cytochemistry, and immunophenotype. To explore the mechanisms involved in pathogenesis and recurrence of morphologically diagnosed APL, we performed comprehensive variant analysis by next-generation sequencing in 111 pediatric patients morphologically diagnosed as APL. Structural variant (SV) analysis in 120 DNA samples from both diagnosis and relapse stage identified 95 samples with RARA rearrangement (including 94 with PML-RARA and one with NPM-RARA) and two samples with KMT2A rearrangement. In the eligible 13 RNA samples without any RARA rearrangement at diagnosis, one case each with CPSF6-RARG, NPM1-CCDC28A, and TBC1D15-RAB21 and two cases with a TBL1XR1-RARB fusion were discovered. These uncovered fusion genes strongly suggested their contributions to leukemogenesis as driver alternations and APL phenotype may arise by abnormalities of other members of the nuclear receptor superfamily involved in retinoid signaling (RARB or RARG) or even by mechanisms distinct from the formation of aberrant retinoid receptors. Single-nucleotide variant (SNV) analysis in 77 children (80 samples) with RARA rearrangement showed recurrent alternations of primary APL in FLT3, WT1, USP9X, NRAS, and ARID1A, with a strong potential for involvement in pathogenesis, and WT1 as the only recurrently mutated gene in relapsed APL. WT1, NPM1, NRAS, FLT3, and NSD1 were identified as recurrently mutated in 17 primary samples without RARA rearrangement and WT1, NPM1, TP53, and RARA as recurrently mutated in 9 relapsed samples. The survival of APL with RARA rearrangement is much better than without RARA rearrangement. Thus, patients morphologically diagnosed as APL that cannot be identified as having a RARA rearrangement are more reasonably classified as a subclass of AML other than APL, and individualized treatment should be considered according to the genetic abnormalities.
Similar content being viewed by others

Introduction
Acute promyelocytic leukemia (APL), classified as M3 in the French–American–British system [1], is a distinct subtype of acute myeloid leukemia (AML) characterized by the expansion and accumulation of leukemic cells blocked at the promyelocytic stage of myelopoiesis [2]. It accounts for less than 10% of childhood AML [3]. This disease is characterized by a balanced reciprocal translocation between chromosomes 15 and 17, leading to the oncogenic fusion protein PML-RARA [4], which is required for a definitive APL diagnosis [5].
The karyotype of t(15;17) can be detected with conventional cytogenetic analysis in about 90% of APL cases [6]. In APL patients without a typical t(15;17) translocation, cryptic PML-RARA and variant translocations involving RARA gene may be present [7]. A cryptic PML-RARA fusion gene can be detected with reverse transcription PCR (RT-PCR) or fluorescence in situ hybridization (FISH) methods in most situations. For some extremely complex translocations, in which PML-RARA is failed to be identified by FISH or RT-PCR, next-generation sequencing (NGS) offers advantages in distinguishing them [8]. Besides, NGS is able to help to discover new partners of RARA. Recently, GTF2I, IRF2BP2, OBFC2A, and TBLR1 have been uncovered as new partners of RARA gene [9,10,11,12,13]. Thus, NGS has advantages in detecting structural variants (SVs) of APL by identifying cryptic PML-RARA or uncovering new fusion partner genes of RARA.
A few cases of APL without RARA gene rearrangement have been identified with abnormal promyelocytes that were fully in accordance with APL in morphology, cytochemistry, and immunophenotype [6, 14,15,16,17]. The existence of APL patients lacking RARA rearrangements suggests that this disease may arise by alternative mechanisms distinct from the formation of the X-RARA fusion. In this study, NGS was used to detect SVs and single-nucleotide variants (SNVs) of a large cohort of pediatric patients with morphological features resembling APL. Patients with and without RARA rearrangement identified by karyotype analysis, FISH, or RT-PCR were both included in this cohort. The uncovered genetic alternations at both initial diagnosis and relapse could help reveal the mechanisms involved in oncogenesis and recurrence and offer an opening to exploring alternative mechanisms distinct from the formation of the X-RARA fusion that are involved in the APL phenotype.
Patients and methods
Patients and samples
Between January 2000 and December 2016, a total of 136 patients were morphologically diagnosed with APL in the Department of Hematology and Oncology, Shanghai Children’s Medical Center (SCMC) in China. Patients were considered eligible for inclusion in the study to detect SVs by targeted capture sequencing only if the following criteria were satisfied: (1) Morphological features were consistent with or evocative of FAB type M3; (2) immunophenotypic features were consistent with M3; (3) DNA samples from bone marrow (BM) cells at initial diagnosis or relapse were eligible; and (4) written informed consent. On this basis, DNA samples from 111 patients were included, containing 101 primary samples from 101 patients, 4 relapse samples from 3 patients, and 15 matched samples from 7 patients, making a total of 120 samples (study flowchart and sample details, Fig. S1, Fig. S2 and Table S1). After the bioinformatics analysis of the SVs, patients were divided into two groups, one included patients with RARA rearrangement (considered as “typical APL”) and another one included patients without RARA rearrangement (considered as “atypical APL”). Somatic mutations (SNV and small indels) were analyzed in the two groups by targeted sequencing. Only patients who had DNA samples at complete remission (CR) eligible for germline control were included in the SNV analysis. On this basis, 84 DNA samples at diagnosis from 84 patients, 4 samples at relapse from 3 patients, and 18 matched samples from 8 patients were included making a total of 106 samples. In summary, 108 samples from initial diagnosis, 14 samples from relapse making a total of 122 DNA samples involved in the whole DNA sequencing study.
Clinical sample collection and genomic DNA library preparation
BM samples from disease/remission stages were collected from the Department of Hematology and Oncology, SCMC. The research was approved by the Ethics Committee at SCMC, Shanghai Jiao Tong University School of Medicine. Genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany; cat. 51106) according to the manufacturer’s instructions. The DNA sample was quantified by Qubit dsDNA BR Assay kit (Life Technologies, Carlsbad, CA, USA; Cat. No. Q32850), and DNA integrity was assessed by agarose gel electrophoresis. DNA was sheared on the Covaris M220 Focused-ultrasonicator (Covaris Company, Woburn, MA, USA). Fragmented DNA was repaired, ligated with Illumina adapters, size selected, amplified, and assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA).
Targeted sequencing
For a targeted capture library of SV analysis, a customized panel of biotinylated oligoprobes (MyGenostics, Baltimore, MD, MyGenosticsGenCap Enrichment Technologies) was designed to capture the whole gene regions of RARA and to cover the likely break regions of partner genes of RARA, including PML, PLZF, NuMA, NPM, STAT5b, PRKAR1a, FIP1L1, and BCOR. For analysis of SNVs, a customized panel of genes covered hotspot mutational regions of 1270 hematological malignancies and solid tumors that were either previously implicated in hematological malignancies or identified in earlier leukemia sequencing studies. The capture experiment was conducted according to the manufacturer’s protocol as described [18]. The captured DNA library was finally amplified and sequenced on Illumina X Ten sequencer for paired reads at 150 bp.
Bioinformatics analysis and validation based on DNA targeted sequencing
The detection of SVs, SNVs, and small indels was performed using paired-end sequence data. Paired-end reads of 150 bp were aligned to the hg19 reference genome using the Burrows–Wheeler Aligner (BWA-0.7.10). Duplicates were removed using picard-tools-2.17.0. Resulting bam files were further processed according to Genome Analysis Toolkit best practices (https://www.broadinstitute.org/gatk/guide/bp_step.php?p=1), by performing Indel Realignment and Base Quality Recalibration. A minimum mapping quality of 15 was considered for variant calling. SVs were called by speedseq (Version: 0.1.0). Somatic mutations were called using Mutect2. CR samples that were negative for RARA rearrangement were used as germline control. Mutations supported by either <5 reads or those supported by reads in only one direction were excluded. Also, variants with variant allele frequencies <0.05 were disregarded. Processed somatic variants were later filtered for false positives using the varscan2 fpfilter command. Final confident variants were annotated using annovar. Single-nucleotide polymorphisms (SNPs) commonly found in the general population were filtered out using dbSNP135, 1000 Genomes, and our in-house SNP databases, while retaining the clinically associated variants. Genetic mutations reported in this study were validated by Sanger sequencing.
Morphologic review
After DNA targeted sequencing, 19 patients identified without any RARA rearrangement were subjected to morphologic review. Morphologic reviewers were ignorant of the cytogenetic and molecular data; in the second step, the reviewed data were considered in the context of the clinical features.
RNA sequencing and bioinformatics analysis
As a complement, we performed RNA sequencing in the available RNA samples at diagnosis of the 19 patients without any RARA rearrangement to identify the SVs. We also randomly selected the same number of RNA samples from patients with RARA rearrangement for sequencing. Total RNA was extracted with TRIzol according to standard procedures and further purified using an RNeasy Micro Kit (QIAGEN, GmBH, Germany) and RNase-Free DNase Set (QIAGEN, GmBH, Germany). RNA was quantified using the Qubit fluorometer (Thermo Fisher, Carlsbad, CA, USA), while the quality of the RNA samples was measured using a Bioanalyzer instrument and RNA nano chips (Agilent, Santa Clara, CA, USA). Total RNA was then subjected to oligo(dT) selection using the Dynabeads® mRNA Purification Kit (Thermo Fisher) as per the manufacturer’s instructions. The aliquoted mRNA was then reverse transcribed into cDNA with random hexamers using standard techniques (Invitrogen). After limited cycle PCR, the RNA-seq libraries were size selected (350–700 bp fragments) in 2% agarose gel followed by purification with the QIAquick gel extraction kit (Qiagen). The cDNA libraries were loaded on a cBot (Illumina, Inc., San Diego, CA) for clustering on a flow cell and then sequenced using a Illumina X Ten sequencer for paired reads at 150 bp. Reads were aligned with TopHat2(v 2.1.1)/BowTie2 to the reference human genome hg19/GRCh37. deFuse (v 0.6.2) and ChimeraScan (v 0.4.5) packages were used to detect chimeric transcripts from RNA-seq data. To validate the suspected fusion genes by Sanger sequencing, standard PCR was performed on cDNA which was synthesized from total RNA using RT-PCR.
Treatment regimen
In our study, APL patients with RARA rearrangement received ATRA treatment combined with anthracycline-based chemotherapy (CT), and ATO was added as an important drug since 2007 (Treatment schedule and details of APL shown in Table S2). The 18 atypical APL patients without RARA rearrangement received induction treatment of APL regimen except one diagnosed before 2007 treated without ATO in induction course. Nine of them continued the APL regimen of ATRA-ATO-CT treatment followed by consolidation treatment and maintenance treatment with continuous anthracycline-based CT and intermittent ATRA-ATO. The other 9 patients switched to AML (non-M3) treatment regimen started with the second induction course followed by 4 consolidation courses of HDAra-C, combined with etoposide and daunomycin. Intermittent ATRA were given every 2 courses in addition (Treatment flowchart shown in Fig. S3).
Statistical analysis
The sensitivity of NGS in identifying RARA gene rearrangement was compared with karyotype analysis, FISH, and RT-PCR using the Pearson chi-square test (McNemar test). Demographics were compared between the patients with and without RARA rearrangement, using Pearson chi-square test for categorical variables and t test or Mann–Whitney U-test for continuous variables. Overall survival (OS) was calculated from date of diagnosis to time of death from any cause or time of last contact. Event-free survival (EFS) was calculated from date of diagnosis to last follow-up or first event (failure to achieve CR, relapse, second malignancy, or death from any cause, whichever occurred first). Relapse-free survival (RFS) was calculated from date of CR to last follow-up or relapse. The data were updated on December 30, 2017, as the reference date. Probabilities of OS and EFS were estimated using the Kaplan–Meier method. The significance of differences among the OS and EFS curves was estimated by the log-rank test (Mantel–Cox). Computations were performed using SPSS software version 19.0 (SPSS Inc., Chicago, IL, USA). The cumulative incidence of relapse (CIR) was estimated by the Kalbfleisch–Prentice method done by the R statistical software version 3.4.4. In this study, P < 0.05 (two-tailed) was considered to indicate statistical significance.
Results
Identification of structural variants based on DNA targeted sequencing
Targeted capture sequencing was used to characterize the SVs in 120 APL samples with 108 newly diagnosed samples and 12 relapse samples (Table S1). The clinical information and sequencing results are shown in Table S3. We achieved a mean depth of 642× (range 192–1357×), and an average 96% of nucleotides were covered by at least 50 reads.
DNA sequencing results showed that 94 samples were positive for RARA rearrangement and 26 were negative. Conventional methods (karyotype analysis, FISH, and RT-PCR) had identified 81 positives and 21 negatives making a total of 112 results. The final confirmation of RARA rearrangement were made by combining the results of NGS, Sanger sequencing, and conventional methods (Fig. 1). 80 of 94 NGS(+) and 24 of 26 NGS(−) were in accordance with the results of conventional methods. In the conflicting results, 7 samples of NGS(+)/conventional methods(−) were validated by Sanger sequencing and revealed 6 samples with PML-RARA and one with NPM-RARA; one NGS(−)/conventional methods(+) sample was considered as a positive one because of all positive results by conventional methods. Eight samples lacking results from conventional methods included 7 NGS(+) samples that were all validated by Sanger sequencing as PML-RARA positive and one NGS-negative sample considered to be an invalid result. Finally, 95 positive samples and 24 negative samples yielded a total of 119 valid results in RARA rearrangement. The sensitivities of karyotype analysis, FISH, RT-PCR, and NGS in discerning RARA rearrangement were 75.93%, 91.30%, 88.64%, and 98.95%, respectively. NGS was more sensitive compared with FISH, although the difference did not reach a statistical level (P = 1.000). However, NGS was significantly more sensitive when compared with karyotype analysis and RT-PCR (PNGS vs karyotype = 0.002; PNGS vs RT-PCR = 0.012) (Fig. 1c, Table S4).

Results of RARA rearrangement with each detecting method. a Topography of RARA rearrangement detected by karyotype analysis, FISH, RT-PCR, and NGS is depicted. The final results are identified in combination with Sanger sequencing. A total of 120 samples are displayed as columns. b The bars depict the absolute number of samples classified by RARA rearrangement. c The sensitivity of each detecting method in identifying RARA rearrangement is shown, NGS is more sensitive than the other detecting methods, when compared with karyotype analysis and RT-PCR, the differences have reached statistical levels (PNGS vs karyotype = 0.002; PNGS vs RT-PCR = 0.012)
Characteristics of PML-RARA breakpoints
Chr15:74287058–74337349 and chr17:38465446–38513894 (Homo sapiens, hg19, GRCh37, February 2009) were genomic breakpoint capturing regions of PML and RARA, respectively. The target region size was 98.7 kbp, and GenCap probes covered 95.8% of the regions. All of the chromosome17 breakpoints clustered within intron2 of RARA. Almost all of the chromosome15 breakpoints clustered within three regions of the PML gene: intron6 (breakpoint cluster region 1, or bcr1 to form the long isoform), exon6 (bcr2 to form the variant isoform), and intron3 (bcr3 to form the short isoform), which accounts for 43.3%, 15.6%, and 34.4%, respectively. In addition, new breakpoints in the PML gene were discovered in six children (6.7%), including exon7 in three children and intron5, intron7, and exon3 in another three children, respectively (Fig. 2).

PML-RARA breakpoint clustering regions. 93 samples were identified with PML-RARA in 90 cases by NGS (three cases afforded paired samples that had the same breakpoints). The breakpoint clustering regions of the 90 cases are summarized. The X axis represents the genomic position of the PML gene, and the Y axis represents the genomic position of the RARA gene
Complex variant translocations of t(15;17)
In addition to the two-way translocation of chromosomes 15 and 17, there were complex three-way and four-way translocations in three patients including t(1;7;15;17)(q22,q11,q22,q21), t(8;15;17)(p23,q22,q21), and t(15;16;17)(q22,p13,q21) respectively.
Furthermore, a large deletion of chromosome17 was discovered in one sample with PML-RARA, which formed a RARA-ITGB4 fusion. Two samples without PML-RARA were involved in KMT2A gene rearrangement, including t(1;11)(q21;q23) KMT2A-MLLT11 and t(1;19)(q23;p13) KMT2A-ELL.
Identification of structural variants based on RNA sequencing
According to the final confirmed results with SVs, 95 samples from 92 patients had RARA rearrangement and 24 samples from 19 patients lacked RARA rearrangement. As a complement, we performed RNA sequencing to detect the SVs on 13 eligible RNA samples at diagnosis from patients without RARA rearrangement and 13 random RNA samples with RARA rearrangement at diagnosis. In the 13 samples without RARA rearrangement, we identified three patients with CPSF6-RARG, NPM1-CCDC28A, and TBC1D15-RAB21, fusion respectively, and two patients both with a TBL1XR1-RARB fusion (Fig. 3). In the patient with t(1;11)(q21;q23) identified by DNA sequencing, we found RPRD2-KMT2A in addition to KMT2A-MLLT11. These fusion genes in samples without RARA rearrangement strongly suggested their contributions to leukemogenesis as driver alternations. In the 13 RNA samples with PML-RARA rearrangement, we also identified two samples with KMT2A-SEPT6 and ARID1B-WASH4P, respectively.

Schematic representation of four fusion proteins identified by RNA sequencing. a Schematic representation of NPM1, CCDC28A, and NPM1-CCDC28A proteins. b Schematic representation of CPSF6, RARG, and CPSF6-RARG proteins. c Schematic representation of TBL1XR1, RARB, and TBL1XR1-RARB proteins. d Schematic representation of TBC1D15, RAB21, and TBC1D15-RAB21 proteins
Analysis of somatic mutations
For further analysis of somatic mutations, the eligible DNA samples at CR stage as germline control should be taken into consideration, finally 80 samples from 77 APL patients with RARA rearrangement and 26 samples from 18 atypical APL patients without RARA rearrangement were studied by targeted capture sequencing (Table S1). We achieved a mean depth of 414× (range 58–666×), and an average 97% of nucleotides were covered by at least 50 reads.
To identify significantly mutated genes, we excluded from further analysis several genes that frequently exhibit a high rate of variants because of large size and constitute potential false-positives in cancer sequencing studies (Table S5). Overall, a total of 278 non-silent point mutations and indels in 128 genes were validated in 88 samples, including 120 mutations in 63 genes observed in 63 typical APL samples and 158 mutations in 77 genes observed in 25 atypical APL samples (Fig. S4a, detailed mutations in samples are shown in Table S6 and S7). Gene mutations were failed to be identified in 17 typical APL samples and one atypical APL sample. A median of two somatic mutations was observed in each sample (Fig. S4b). In both primary and relapse typical APL samples, a median of one somatic mutation was observed; in primary and relapse atypical APL samples, medians of two and four somatic mutations were identified, respectively.
Analysis of somatic mutations in APL with RARA rearrangement
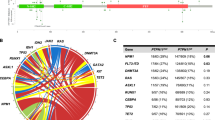
Overall, 111 non-silent point mutations and indels in 60 genes were validated in the 75 primary APL samples (details in Table S6). Recurrent mutations were observed in 14 genes, including FLT3, WT1, USP9X, NRAS, ARID1A, and KRAS (Fig. 4a, b). The FLT3 gene was the most frequently altered gene, accounting for 27% of the primary samples (20 out of 75 primary samples), including 5 samples (7%) with FLT3-ITD, 14 (19%) with missense mutations of FLT3, and one sample with a non-frameshift deletion (1%). The missense mutations of FLT3 predominantly occurred in the tyrosine kinase domains 1 and 2 and in the juxta-membrane domain, with D835 residue most frequently mutated (8/14 samples). In addition, mutations at N676 (2/14 samples), Q580 (1/14 samples), N841 (2/14 samples), and V491 (2/14 samples) were also revealed in FLT3.

Mutational profile between APL with and without RARA rearrangement. a Mutational topography of APL with and without RARA rearrangement is depicted. The mutated genes validated by Sanger sequencing are shown and color-coded for various types of mutation. Samples are displayed as columns and arranged to emphasize mutual exclusivity. Samples in which either no recurrent mutation was detected in our analysis pipeline or do not harbor fusion gene besides PML-RARA are not included in the matrix. Gene name in bold represents recurrently mutated genes, there are 14 genes and 8 genes recurrently mutated in APL with and without RARA rearrangement, respectively. b Bars represent the absolute number of recurrently gene mutations in APL samples with and without RARA rearrangement. c Comparison of mutational frequency of genes between APL with and without RARA rearrangement. “*” represents mutational rates of genes mutated at significantly different frequencies (Fisher’s exact test; P < 0.05). The same gene mutations shared in matched samples are not repeatedly included in the calculation of the mutation frequency
Other recurrently mutated genes included WT1 (13%), USP9X (9%), NRAS (7%), ARID1A (4%), KMT2E (4%), EED (4%), and KRAS (3%). Most WT1 mutations resulted in a truncated protein (one nonsense and six frameshift indels out of 10 mutations). In addition, two of the three missense substitutions were clustered in the C-terminus region encoding Zn-finger domains. All mutations of NRAS and KRAS were missense. All five NRAS mutations clustered at amino-acid residue G12, whereas KRAS mutations occurred at Q61 and A146. All three mutations in the ARID1A gene were nonsense mutations that resulted in loss of function.
Nine mutations in six genes were validated in the five relapse samples of four patients (one patient afforded both first and second relapse samples). WT1 was the only gene recurrently mutated in 4/5 samples (3/4 patients). The WT1 mutations at second relapse remained unchanged compared with the first relapse in the patient of APL-92 who relapsed twice. In the patient of APL-95, the WT1 mutation was newly acquired at relapse compared with initial diagnosis. All WT1 mutations resulted in loss of function (one nonsense and two frameshift indels).
Analysis of somatic mutations in atypical APL without RARA rearrangement
A total of 76 non-silent point mutations and indels in 64 genes were validated in the 15 primary atypical APL samples (details in Table S7). In the single sample in which mutations were not detected, the fusion gene NPM1-CCDC28A was identified, suggesting its contribution to leukemogenesis as driver alternations in atypical APL. Recurrent mutations were observed in five genes, including WT1 (17.6%), NPM1 (11.8%), NRAS (11.8%), FLT3 (11.8%), and NSD1 (11.8%) (Fig. 4).
A total of 82 non-silent point mutations and indels in 48 genes were validated in the nine relapse atypical APL samples. WT1, as the most frequently altered gene in primary samples, was also the most frequent lesion at relapse, at 44.4%. Other genes recurrently mutated in relapse samples included NPM1 (33.3%), TP53 (33.3%), and RARA (33.3%). Six patients of relapse afforded paired samples, in which TP53 and RARA mutations were specifically present at relapse, suggesting their relationship with relapse. One sample had WT1 mutations and two samples had NPM1 mutations that were shared between two stages from diagnosis to relapse, indicating re-emergence of a disease subclone following therapy. Another primary sample with a WT1 mutation acquired a new mutation in WT1 at the relapse stage. One sample with a NRAS mutation, two samples with FLT3 mutations, and another two samples with NSD1 mutations all lost these mutations at relapse (Fig. 4a).
Three mutations of RARA were detected in two relapse samples, all of which harbored mutations in the ligand-binding domain (LBD), with previously reported R272Q in one sample and G404fs in addition to S405P in the other. The RARA mutations were all detected post-ATRA treatment.
Mutational frequencies of recurrently mutated genes in the whole 80 APL sample set were compared with the 26 atypical APL samples (Fig. 4c). The same gene mutations shared in matched samples are not repeatedly included in the calculation of the mutation frequency. NPM1, TP53, NSD1, and ALK gene mutations were specifically observed in atypical APL, in which the difference in mutational frequency in NPM1 and TP53 reached statistical significance (P < 0.05). Mutations of RARA, WT1, and NRAS occurred at a higher frequency in atypical APL, in which the mutational frequency of RARA reached statistical significance (P < 0.05). Mutations in USP9X, EED, ADCY2, FAM166B, MYC, and SMARCB1 were not observed in atypical APL, and a decreased incidence of FLT3 alternations occurred in this cohort, although the difference did not reach statistical significance.
Clinical features of the pediatric APL patients
The presenting clinical and laboratory data for the 77 APL patients with RARA rearrangement (44 boys, 33 girls; median age 8.2 years, range 1.2–15.1 years) are listed in Table 1 (more details, Table S8). The 18 atypical APL patients without RARA rearrangement included 14 boys and 4 girls, with a median age of 6 years (range 4 months to 13.2 years) (more details, Table S9). The atypical APL samples were subjected to morphologic review, indicating morphological features in accordance with M3 in the FAB system (Fig. S5). The presenting clinical and laboratory details of these atypical APL patients were compared with 77 APL patients (Table 1). No differences related to sex, white blood cell count at diagnosis, or Hb level were observed between the two groups. The age at diagnosis of atypical APL was lower than that of APL (P = 0.004). Conversely, the former groups showed a significantly higher platelet and lactate dehydrogenase level than the latter (P = 0.01 and P = 0.001, respectively).
During the ATRA induction treatment, morphologically recognizable changes in most promyelocytes of patients with PML-RARA could be observed. The nucleus became larger, and fewer prominent primary granules along with the development of secondary granules were observed in the cytoplasm with condensation and segmentation of nuclei. An elevated population of metamyelocytes was evident, with indented or horseshoe-shaped nuclei and reduced nuclear/cytoplasmic ratio, although the nucleoplasm remained immature. We designated these promyelocytes with morphological changes as “ATRA-induced cells”. In atypical APL patients, the percentage of ATRA-induced cells was significantly lower than in samples with RARA rearrangement (P = 0.0001), although the typical morphological features of M3 were presented before treatment. The much lower percentage of “ATRA-induced cells” suggests that without the PML-RARA fusion gene, the atypical APL group was less sensitive or even unresponsive to ATRA. As a result, 6/18 (33.3%) patients still had more than 5% of immature leukemic cells after the first course of induction with ATRA-ATO-CT indicating a resistant disease. While only 2/77 patients (2.6%) in typical APL group had failure of induction. So the atypical APL had significantly higher rates of resistant diseases than typical APL (P < 0.001) (Table 1).
Prognosis of pediatric APL patients
In APL with RARA rearrangement, four patients presented recurrence, one relapsed during the maintenance treatment (14 months from diagnosis), and three relapsed after treatment (at 32, 33, and 33 months, respectively). One of the patients died of heart disease at the remission stage (28 months from diagnosis). With a median follow-up time of 42.6 months (range 12.6–153.4 months), the estimated 5- and 8-year OS was 96.1 ± 2.1%, whereas the EFS rates for the above periods were 90.9 ± 3.9% (Fig. 5a). The CIR was 7.3%.

Survival curves of APL. a Comparison of estimated probability of EFS and OS between APL with and without RARA rearrangement. b Comparison of estimated probability of EFS and OS between atypical APL treated with APL regimen (solid line) and AML regimen (dashed line)
Overall, 12 of 18 atypical APL patients achieved morphological CR after induction treatment, for an induction failure rate of 33.3%. The median duration for achieving CR was 37 days (27–163 days), and the whole cohort achieved CR. With a median follow-up time of 42.2 months (range, 10.6–170.1 months), the estimated 5-year and 8-year OS rates were 85.0 ± 10.2% and 63.8 ± 19.9%, respectively, whereas the EFS rates for the above periods were 57.4 ± 12.3% and 38.3 ± 17.7%, respectively (Fig. 5a). Relapse occurred in 7 patients (38.9%) who had achieved CR; 3 of them died after recurrence during the follow-up time, 3 of them given up treatment and lost to follow up, one of them continued the treatment and lost to follow up after second relapse. One child had a secondary malignancy 70 months after chemotherapy completed and lost to follow up. So a total of 5 patients lost to follow up after the event happened (more details, Table S9). The CIR of atypical APL was 42.6% which was much higher than the typical one (P = 0.0002) (Fig. S6). The survival rates of atypical APL were much worse than the atypical APL cases (POS < 0.001, PEFS < 0.001) (Fig. 5a).
Several fusion genes were discovered in atypical APL that had rarely or even never been previously reported. The patient with CPSF6-RARG, who did not experience morphological CR after induction treatment with the APL regimen (ATRA-ATO+DA), was switched to an AML (non-M3) regimen. CR still was difficult to achieve until the sixth course was finished (181 days from diagnosis), and 2 months after the chemotherapy completed (10 months from diagnosis), BM aspirate showed relapse. The two patients with TBL1XR1-RARB were both treated with the APL regimen. One of them maintained CR during the follow-up of 108 months from diagnosis; the other patient had a relapse in a neck lymph node 7 months following therapy end and experienced a second relapse in BM 15 months after re-induction. The two patients with NPM1-CCDC28A and TBC1D15-RAB21 each were treated with the AML regimen and maintained CR with a relapse-free time of 53 months and 55 months, respectively. Of the two patients with KMT2A rearrangement, one with t(11;19) KMT2A-ELL treated with the APL regimen remained CR with RFS of 168 months; the other one with KMT2A-MLLT11 and RPRD2-KMT2A treated with the AML regimen did not achieve CR until the fourth course was finished (131 days from diagnosis) and remained CR with a RFS of 30 months.
Nine of the 18 atypical APL patients were treated with APL regimen, and the other 9 patients switched to an AML (non-M3) regimen starting with the second induction course after the first induction course of APL regimen. We compared the survival rates and CIR of the two groups of patients with different treatment regimens (Fig. 5b and Fig. S7). Although patients treated with the APL regimen had a worse survival (estimated 5-year and 8-year OS rates were 83.3 ± 15.2% and 55.6 ± 24.8%, respectively; EFS rates for the above periods were 51.9 ± 17.6% and 25.9 ± 20.3%, respectively) and higher CIR (48.5%) compared to those treated with the AML (non-M3) regimen (estimated 5-year and 8-year OS rates both 88.9 ± 10.5%; EFS rates for these periods were both 63.5 ± 16.9%; CIR was 36.5%), the difference was not statistically significant (PEFS = 0.469; POS = 0.855; PCIR = 0.607).
Discussion
This study provides a comprehensive analysis of gene alternations in pediatric APL using NGS, which represents to our knowledge the largest mutational analysis in APL for children. Our targeted panel for SV detection was designed to capture the whole gene region of RARA and cover the likely break regions of its partner genes, including PML, PLZF, NuMA, NPM1, STAT5b, PRKAR1a, FIP1L1, and BCOR. SV results suggested a higher sensitivity of targeted sequencing based on our capture panel in discerning RARA rearrangement. In addition to RARA rearrangement, we identified two patients without RARA rearrangement, however, had a KMT2A rearrangement each, one with KMT2A-MLLT11 and another with KMT2A-ELL.
For patients in which a RARA rearrangement could not be identified by DNA sequencing and other methods, we performed RNA sequencing as a complement. In the 13 eligible RNA samples, we identified three patients with CPSF6-RARG, NPM1-CCDC28A, and TBC1D15-RAB21 fusion, respectively, and two patients both with TBL1XR1-RARB fusion, all fusion genes that rarely or never have been previously reported. These fusion genes identified in patients showing the M3 FAB subtype, however, without RARA rearrangement strongly suggested their contributions to leukemogenesis as driver alternations and that APL may arise by alternative mechanisms distinct from the formation of a PML-RARA fusion gene.
RARB and RARG are two members of the nuclear receptor superfamily that share high homology (90%) with RARA and are all involved in retinoid signaling. It has been reported that artificial PML-RARB and PML-RARG fusion both show oncogenic potential comparable to that of PML-RARA, with the ability to block myeloid differentiation and extend proliferative capacity, while conferring responsiveness to differentiation treatment with retinoid acid [19]. Two patients of AML displaying morphologic features resembling APL bearing CPSF6-RARG have been described recently [20]. Here, we reported a third case with CPSF6-RARG that we morphologically diagnosed as APL. Like the other two cases, the patient with CPSF6-RARG showed no response to induction with ATRA and ATO. Four cases of TBL1XR1-RARB have been discovered for the first time recently in RARA-negative APL, and showed oncogenic potency and partial response to retinoids [21]. Here, we also uncovered two cases of TBL1XR1-RARB. Both were treated with the APL regimen, and the patient carrying ATXN3 and MAP2K2 mutations experienced persistent CR, but the other patient with STAG2 mutations relapsed repeatedly. Thus, the prognosis of patients may be affected by TBL1XR1-RARB combined with other genetic abnormalities which needs further study with more cases.
NPM1-CCDC28A is a novel fusion we identified in leukemia. NPM1 is a RARA fusion partner and is presumed to play a role in ribosomal RNA processing or transport. In silico analyses support a contribution of CCDC28A to discrete stages of murine hematopoietic development and suggest selective enrichment of CCDC28A in the FAB M6 class of human AML [22]. A NUP98-CCDC28A fusion has been identified in T-cell acute lymphoblastic leukemia and AML, where it promoted the proliferative capacity and self-renewal potential of myeloid progenitors in in vivo studies [22]. NPM1-CCDC28A, as the only genetic alternation identified in the case, was surmised to play a crucial role in leukemogenesis and contribute to morphological features resembling APL.
TBC1D15-RAB21 fusion is reported for the first time in leukemia. The TBC1D15 gene, also known as GTPase-activating protein RAB7, encodes a member of the Ras-like proteins in the brain-GTPase-activating protein superfamily and functions as an oncoprotein controlling stem cell self-renewal through destabilization of the Numb–p53 complex. Rab21 is a member of the Rab subfamily, which has been identified as required for tumor-associated fibroblasts to promote the invasion of squamous carcinoma cells. However, no functional data on TBC1D15 and Rab21 have been reported in leukemia. The case with this fusion also harbored WT1, DNMT3A, and NSD1 mutations, so its supposed function in oncogenesis remains to be investigated.
The KMT2A gene (MLL1) on chromosome 11q23 is disrupted in a unique group of acute leukemias. Approximately 14% of pediatric AML harbors KMT2A translocations, which commonly are found in M4, M5, M0, and M1 subtypes [23]. AML bearing a KMT2A rearrangement with morphological features of M3 has rarely been reported. The two cases with KMT2A translocations mimicking APL that we identified both involved infants under age 12 months at diagnosis. The patient with t(11;19) KMT2A-ELL, which is found in 4.6% of MLL-rearranged pediatric AML and is categorized as intermediate risk [24, 25], had a favorable prognosis and maintained CR until the last follow-up, with a disease-free survival of 170 months. The other patient with t(1;11) KMT2A-MLLT11, which also is associated with a favorable prognosis [25], experienced a delay to CR until 131 days of treatment, but maintained CR until the last follow-up with a disease-free survival of 30 months.
Genetic abnormalities in APL are clinically valuable for selecting a treatment regimen and evaluating prognosis. Thus, high-throughput sequencing should be performed especially on APL patients whose RARA rearrangements are failed to be identified by conventional methods (i.e., karyotype analysis, FISH, RT-PCR). This is helpful to exclude the false-negative probability of conventional methods and to uncover new fusion genes as potential targets for treatment.
In addition to the common X-RARA, other abnormalities involved in retinoid signaling like RARB or RARG; or even other SVs distinct from the formation of aberrant retinoid receptors are able to perform as driver mutations, making APL a heterogeneous disease.
Preliminary whole genome/exome studies have revealed recurrently mutated genes such as FLT3, WT1, and NRAS may cooperate with PML-RARA in oncogenesis [26,27,28,29]. Previous NGS studies identifying genetic alternations were performed mostly in adult APL patients, and an analysis based on a large cohort involving children has not been reported. In our study, the topography of somatic mutations in APL with RARA rearrangement at diagnosis is defined by recurrent alterations of FLT3 (27%), WT1 (13%), USP9X (9%), NRAS (7%), ARID1A (4%), KMT2E (4%), EED (4%), and KRAS (3%). These recurrent gene mutations could cooperate with PML-RARA in the pathogenesis of APL. We also uncovered WT1 as the only gene recurrently mutated in the four relapse patients, and it is thought to be related to APL relapse. The profile of mutations was similar to the mutational landscape depicted by Madan et al. [26], which primarily involved adult patients.
Differences in the topography of somatic mutations could be observed between APL with and without RARA rearrangement. The recurrently mutated genes in the 17 primary samples of atypical APL included WT1 (17.6%), NPM1 (17.6%), NRAS (11.8%), FLT3 (11.8%), and NSD1 (11.8%); in the 9 relapsed samples, they included WT1 (44.4%), NPM1 (33.3%), TP53 (33.3%), and RARA (33.3%).
Recurrent TP53 and RARA mutations were specifically present in atypical relapse samples, which suggested a proposed relationship between the two mutated genes and relapse of atypical APL. Previous studies have identified that mutations clustered in the LBD of the RARA portion of the PML-RARA fusion protein lead to a diversity of alterations in binding to the ligand and to nuclear coregulators of transcription, triggering varying degrees of inhibition of retinoid-induced transcription [30, 31]. Without PML-RARA fusion, however, the two cases acquired RARA mutations, all located in the LBD region, after ATRA-based treatment. We speculate that molecular alterations in the LBD of RARA protein may also impair ATRA-binding capacity and lead to relapse. However, a functional analysis is necessary to confirm that these mutations play a role in the underlying mechanism of clinical ATRA resistance.
The 77 patients with RARA rearrangement had a particularly favorable outcome, with 8-year EFS and OS of 90.9 ± 3.9% and 96.1 ± 2.1%, respectively. Although FLT3 has been reported as a risk factor for survival rates in other studies [32, 33], neither FLT3-ITD nor overall FLT3 mutations seemed to affect the prognosis of APL in our study. Without RARA rearrangement, atypical APL may not be sensitive to ATRA and ATO treatment, so survival was much worse compared with the patients with RARA rearrangement. When evaluated the long-term outcomes of the patients, the OS I calculated may be better than that in the real situation because majority of relapse or resistant patients chose to give up the treatment due to big financial burden and increased the possibility of resistant disease, so they were lost to follow up after the event happened. For them lost to follow up was the end of observation instead of death in analysis of OS. Five out of 77 typical APL patients had events of relapse, resistant diseases or second malignancy in which 3 patients given up and lost to follow up accounting for 60%. While 5 out of 8 atypical APL patients who had events were lost to follow up accounting for 62.5%.
In atypical APL, there are six patients (33.3%) incapable of achieving morphological CR after the first course of induction based on ATRA treatment, showing ATRA-resistant. Clinical features and genetic abnormalities between ATRA-sensitive and ATRA-resistant patients are compared, however, no differences have reached the statistical level (Table S10). Larger amount of atypical APL cases are required to draw a conclusion.
To underscore the significance of the PML-RARA fusion, which may be cryptic or result from complex cytogenetic rearrangements other than t(15;17)(q24.1;q21.2), APL with this fusion has been renamed as APL with PML-RARA in the updated (2016) WHO classification [34]. The cases with morphological features resembling APL, however, that could not be identified with RARA rearrangement, are more reasonably classified as a subclass of AML. For example, the case with NPM1 mutations is classified as “AML with mutated NPM1” other than APL, and individualized treatment should be considered for them according to their genetic abnormalities.
In summary, to analyze APL-associated SVs, we performed NGS on samples from children who were morphologically diagnosed as APL. We identified several cases of RARA rearrangements and other new fusions involving RARB, RARG, and KMT2A that had not been recognized using conventional methods. The advantages NGS had in molecular diagnosis of APL suggested it could be applied combining with karyotype analysis, FISH, and RT-PCR for accurate diagnosis, especially when RARA rearrangement cannot be identified by the conventional methods. In addition to the common X-RARA, other abnormalities involved in retinoid signaling like RARB or RARG; or even other SVs distinct from the formation of aberrant retinoid receptors are able to perform as driver mutations, contributing to APL phenotype. We also revealed recurrent alternations of FLT3, WT1, USP9X, NRAS, and ARID1A in typical APL, which cooperated with the PML-RARA fusion gene in oncogenesis. Significant differences have been observed in mutational frequencies between APL with and without RARA rearrangement suggesting APL is a heterogeneous disease. We retrospectively analyzed treatment outcomes and demonstrated overall excellent outcomes of typical APL, which is much better than atypical APL. Thus, atypical APL is more reasonably classified into subclasses of AML other than APL, and individualized treatment should be considered for these patients according to the genetic abnormalities involved.
Data availability
The authors declare that all the data supporting the findings of this study are available within the paper and its Supplementary Information files.
References
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the acute leukaemias. French–American–British (FAB) co-operative group. Br J Haematol. 1976;33:451–8.
Thé HD, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A, et al. The PML-RAR fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991;66:675–84.
Gregory J, Feusner J. Acute promyelocytic leukemia in childhood. Curr Oncol Rep. 2009;11:439–45.
Redner RL. Variations on a theme: the alternate translocations in APL. Leukemia. 2002;16:1927–32.
O'Donnell MR, Tallman MS, Abboud CN, Altman JK, Appelbaum FR, Arber DA, et al. Acute myeloid leukemia, version 3.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017;15:926–57.
Grimwade D, Biondi A, Mozziconacci MJ, Hagemeijer A, Berger R, Neat M, et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Francais de Cytogenetique Hematologique, Groupe de Francais d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood. 2000;96:1297–308.
Adams J, Nassiri M. Acute promyelocytic leukemia: a review and discussion of variant translocations. Arch Pathol Lab Med. 2015;139:1308–13.
Welch JS, Westervelt P, Ding L, Larson DE, Klco JM, Kulkarni S, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011;305:1577–84.
Li J, Zhong HY, Zhang Y, Xiao L, Bai LH, Liu SF, et al. GTF2I-RARA is a novel fusion transcript in a t(7;17) variant of acute promyelocytic leukaemia with clinical resistance to retinoic acid. Br J Haematol. 2015;168:904–8.
Yin CC, Jain N, Mehrotra M, Zhagn J, Protopopov A, Zuo Z, et al. Identification of a novel fusion gene, IRF2BP2-RARA, in acute promyelocytic leukemia. J Natl Compr Canc Netw. 2015;13:19–22.
Shimomura Y, Mitsui H, Yamashita Y, Kamae T, Kanai A, Matsui H, et al. New variant of acute promyelocytic leukemia with IRF2BP2-RARA fusion. Cancer Sci. 2016;107:1165–8.
Won D, Shin SY, Park CJ, Jang S, Chi HS, Lee KH, et al. OBFC2A/RARA: a novel fusion gene in variant acute promyelocytic leukemia. Blood. 2013;121:1432–5.
Chen Y, Li S, Zhou C, Li C, Ru K, Rao Q, et al. TBLR1 fuses to retinoid acid receptor alpha in a variant t(3;17)(q26; q21) translocation of acute promyelocytic leukemia. Blood. 2014;124:936–45.
Duan Y, Nie J, Zhang Z, Zhou L, Zhu F, Zhang H, et al. A rare case with typical acute promyelocytic leukemia morphology associated with isolated isochromosome 17q without RARalpha rearrangement. Hematol Oncol Stem Cell Ther. 2013;6:42–5.
Hori T, Suzuki N, Hatakeyama N, Yamamoto M, Inazawa N, Miyachi H, et al. Infantile acute promyelocytic leukemia without an RARalpha rearrangement. Pediatr Int. 2011;53:1070–3.
Song X, Gong S, Chen J, Gao L, Yang J, Wang J. ATRA is effective to an acute promyelocytic leukemia patient without RARA gene rearrangement. Leuk Res. 2010;34:e190–3.
Wang HP, Xu H, Chen ZM, Tong XM, Qian WB, Jin J. t(X;17) as the sole karyotypic anomaly in a case of M(3r) subtype of acute promyelocytic leukemia without RARalpha rearrangement. Leuk Res. 2010;34:e55–7.
Jin Y, Wang X, Hu S, Tang J, Li B, Chai Y. Determination of ETV6-RUNX1 genomic breakpoint by next-generation sequencing. Cancer Med. 2016;5:337–51.
Marinelli A, Bossi D, Pelicci PG, Minucci S. A redundant oncogenic potential of the retinoic receptor (RAR) alpha, beta and gamma isoforms in acute promyelocytic leukemia. Leukemia. 2007;21:647–50.
Liu T, Wen L, Yuan H, Wang Y, Yao L, Xu Y, et al. Identification of novel recurrent CPSF6-RARG fusions in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood. 2018;131:1870–3.
Osumi T, Tsujimoto SI, Tamura M, Uchiyama M, Nakabayashi K, Okamura K, et al. Recurrent RARB translocations in acute promyelocytic leukemia lacking RARA translocation. Cancer Res. 2018;78:4452–8.
Petit A, Ragu C, Soler G, Ottolenghi C, Schluth C, Radford-Weiss I, et al. Functional analysis of the NUP98-CCDC28A fusion protein. Haematologica. 2012;97:379–87.
Chowdhury T, Brady HJ. Insights from clinical studies into the role of the MLL gene in infant and childhood leukemia. Blood Cells Mol Dis. 2008;40:192–9.
Coenen EA, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, et al. Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL-rearranged AML patients: results of an international study. Blood. 2011;117:7102–11.
Balgobind BV, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114:2489–96.
Madan V, Shyamsunder P, Han L, Mayakonda A, Nagata Y, Sundaresan J, et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia. 2016;30:1672–81.
Riva L, Ronchini C, Bodini M, Lo-Coco F, Lavorgna S, Ottone T, et al. Acute promyelocytic leukemias share cooperative mutations with other myeloid-leukemia subgroups. Blood Cancer J. 2013;3:e147.
Greif PA, Yaghmaie M, Konstandin NP, Ksienzyk B, Alimoghaddam K, Ghavamzadeh A, et al. Somatic mutations in acute promyelocytic leukemia (APL) identified by exome sequencing. Leukemia. 2011;25:1519–22.
Callens C, Chevret S, Cayuela JM, Cassinat B, Raffoux E, de Botton S, et al. Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia. 2005;19:1153–60.
Lou Y, Ma Y, Sun J, Ye X, Pan H, Wang Y, et al. Evaluating frequency of PML-RARA mutations and conferring resistance to arsenic trioxide-based therapy in relapsed acute promyelocytic leukemia patients. Ann Hematol. 2015;94:1829–37.
Cote S, Zhou D, Bianchini A, Nervi C, Gallagher RE, Miller WH Jr. Altered ligand binding and transcriptional regulation by mutations in the PML/RARalpha ligand-binding domain arising in retinoic acid-resistant patients with acute promyelocytic leukemia. Blood. 2000;96:3200–8.
Cicconi L, Divona M, Ciardi C, Ottone T, Ferrantini A, Lavorgna S, et al. PML-RARalpha kinetics and impact of FLT3-ITD mutations in newly diagnosed acute promyelocytic leukaemia treated with ATRA and ATO or ATRA and chemotherapy. Leukemia. 2016;30:1987–92.
Beitinjaneh A, Jang S, Roukoz H, Majhail NS. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations in acute promyelocytic leukemia: a systematic review. Leuk Res. 2010;34:831–6.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Acknowledgements
This study was supported by grants from National Natural Science Foundation of China (No. 81670136) and the National Key Research and Development Program of China (No. 2016YFC0902803).
Author information
Authors and Affiliations
Contributions
Ben-Shang Li and Jing-Yan Tang designed the research. Jie Zhao performed the experiments. Ben-Shang Li, Jian-Wei Liang, and Jie Zhao performed bioinformatics analyses of the sequencing data. Jie Zhao wrote the manuscript and analyzed the data. Jie Zhao, Hui-Liang Xue, Shu-Hong Shen, Jing Chen, Yan-Jing Tang, Li-Sha Yu, Huan-Huan Liang, and Long-Jun Gu collected the clinical information and samples.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhao, J., Liang, JW., Xue, HL. et al. The genetics and clinical characteristics of children morphologically diagnosed as acute promyelocytic leukemia. Leukemia 33, 1387–1399 (2019). https://doi.org/10.1038/s41375-018-0338-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0338-z
- Springer Nature Limited
This article is cited by
-
Current views on the genetic landscape and management of variant acute promyelocytic leukemia
Biomarker Research (2021)
-
Fusion gene map of acute leukemia revealed by transcriptome sequencing of a consecutive cohort of 1000 cases in a single center
Blood Cancer Journal (2021)
-
Gene mutations in acute promyelocytic leukemia early death in patients treated with arsenic trioxide alone
Clinical and Translational Oncology (2021)