
Abstract
The R98S mutation in ribosomal protein L10 (RPL10 R98S) affects 8% of pediatric T-cell acute lymphoblastic leukemia (T-ALL) cases, and was previously described to impair cellular proliferation. The current study reveals that RPL10 R98S cells accumulate reactive oxygen species which promotes mitochondrial dysfunction and reduced ATP levels, causing the proliferation defect. RPL10 R98S mutant leukemia cells can survive high oxidative stress levels via a specific increase of IRES-mediated translation of the anti-apoptotic factor B-cell lymphoma 2 (BCL-2), mediating BCL-2 protein overexpression. RPL10 R98S selective sensitivity to the clinically available Bcl-2 inhibitor Venetoclax (ABT-199) was supported by suppression of splenomegaly and the absence of human leukemia cells in the blood of T-ALL xenografted mice. These results shed new light on the oncogenic function of ribosomal mutations in cancer, provide a novel mechanism for BCL-2 upregulation in leukemia, and highlight BCL-2 inhibition as a novel therapeutic opportunity in RPL10 R98S defective T-ALL.
Similar content being viewed by others


Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a genetically complex and aggressive hematologic cancer caused by the accumulation of somatic mutations in developing T-cells [1]. Outcomes of T-ALL improved with the advent of high-dose, multi-agent chemotherapy regimens, resulting in long-term event free survival (EFS) rates above 80% in pediatric patients [2]. However, relapses due to therapy resistance or initial therapy failure remain a problem and are the most common causes of death. To further improve therapy, targeted treatments are necessary that specifically interfere with the cellular processes deregulated in genetic subgroups of pediatric T-ALL patients.
Ribosomes are the cellular components that execute protein translation of mRNAs. Mutations in 4 different ribosomal proteins (RPL5, RPL10, RPL22, and RPL11) have been described in T-ALL, with the highest incidence of genetic lesions occurring at residue R98 of ribosomal protein L10 (RPL10) [3,4,5]. The RPL10 R98S missense mutational hotspot was identified in 8% of pediatric T-ALL cases [4]. The R98 residue is located at the base of an essential flexible loop near the ribosomal catalytic core in the 60S subunit. This loop is required for proper 60S assembly and translational fidelity, and we have previously demonstrated that mutant RPL10 R98S yeast and mouse lymphoid cells display impaired ribosome formation and proliferation [4, 6]. Besides its function in regulating translation-associated functions, RPL10 has a known extra-ribosomal function of binding the transcription factor AP-1 (proto-oncogene c-Jun), thereby interfering with c-Jun transcriptional activation [7, 8]. In addition, RPL10 has been proposed to inhibit the activity of the Src family kinase Yes [9]. Moreover, a study investigating the RPL10 H213Q mutation associated with autism spectrum disorder indicates a function for RPL10 in cell metabolism [10]. In the context of T-ALL, we recently found RPL10 R98S mutations to be mutually exclusive with JAK/STAT mutations [11]. While existing literature on RPL10 points to several promising candidates for therapeutic intervention, no therapeutic strategy has been validated in a mouse model so far. Moreover, while the observed hypoproliferation phenotype associated with the RPL10 R98S mutation has been tied to ribosome assembly defects in yeast, no causal mechanisms for this proliferation phenotype have been elucidated in mammalian cells yet.
In the present study, we describe the RPL10 R98S ribosomal mutation associated phenotypes, and that hypoproliferation of RPL10 R98S cells is influenced by mitochondrial dysfunction. A leukemic cell survival benefit is facilitated by the R98S mutant ribosome specific IRES-mediated BCL-2 translation. Finally, we demonstrate that the RPL10 R98S specific elevated BCL-2 expression sensitizes RPL10 R98S leukemia to the clinical inhibitor ABT-199, and that ABT-199 suppresses leukemia progression in vivo.
Methods
Cell cultures
Ba/F3 and Jurkat cells (DSMZ) were cultured in RPMI-1640 (Life technologies) with 10–20% Fetal Bovine Serum (FBS) (Thermo Fisher Scientific). Mouse lymphoid-B-cell Ba/F3 RPL10 WT and RPL10 R98S mutant clones were generated as previously described [11]. CRISPR/Cas9 genome editing was used to introduce the RPL10 R98S point mutation in the Jurkat T-ALL cell line as described in Supplementary data file.
MTS assay
Cell proliferation was assessed using the CellTiter 96 Aqueous One Solution Cell Proliferation assay (MTS) (Promega). Cells were plated at a density of 0.25×106 in the presence or absence of Nac/H2O2 for 48 h.
Generation of a conditional Rpl10 R98S knock in mouse line (Rpl10cKI R98S)
Generation of this mouse model was performed by the company Polygene AG (Rümlang, Switzerland) and was previously described [11].
Serial re-plating assay
Rpl10cKI R98S mice were crossed with Mx1-Cre mice (Figure S1A). Lineage negative cells were isolated (EasySep Mouse Hematopoietic Progenitor Cell enrichment kit, Stemcell Technologies) from 6 to 8 weeks old male Mx1-Cre Rpl10cKI R98S mice and from Mx1-Cre control mice. Cells were plated at 2000 cells/ml in Methocult (M3534, Stemcell Technologies) containing 1250 units/ml of interferonβ for complete recombination (IFNβ) (R&D systems) (Figure S1B). More details are in Supplementary data.
T-ALL patient samples
This study was approved by the ethics committees at the institutes involved. After getting written informed consent, the mononuclear cell fraction of BM from pediatric T-ALL patients was obtained (Table S4).
Proteomics
Cells from 3 monoclonal Ba/F3 cultures expressing WT or R98S RPL10 were lysed in lysis buffer (Cell Signaling Technology) with addition of 5 mM Na3VO4 and protease inhibitors (Complete, Roche). In total 20 µg of protein were processed for quantitative proteomics, as previously described [11]. Data are available via ProteomeXchange with identifier PXD005995 [11].
Immunoblotting
In total 1×106 cells were lysed (cell lysis buffer, Cell signaling technology) and denatured in 1 × Laemmli sample buffer (Bio-rad) containing 2-mercaptoethanol (Sigma Aldrich). Proteins were separated on Criterion Tris-Glycine eXtended gels (Bio-rad), transferred to PVDF membranes using the Trans-Blot Turbo system (Bio-rad), incubated with primary antibodies targeting Catalase (Cell Signaling #14097 & Abcam #ab76024), xanthine oxidase (Abcam #ab109235), Bcl-2 (Cell Signaling #2870), and with secondary antibody Goat Anti-Rabbit or Goat Anti-Mouse IgG-HRP (Thermo Fischer). Proteins were visualized using chemiluminescence on an Azure C600 (Azure Biosystems). Quantification was performed using LI-COR Image Studio Lite software version 5.2. Vinculin (Sigma Aldrich), tubulin or beta actin (Sigma Aldrich) was used to normalize for protein input.
13C Tracer analysis
Labeling experiments were performed in dialyzed serum for 48 h. 13C6-glucose tracer was purchased from Sigma-Aldrich. The cell quenching procedures and the metabolite preparations were described before [12]. Metabolites were derivatized and measured as described before [13, 14]. Mass distribution vectors were extracted from the raw ion chromatograms using a custom Matlab M-file, which applies consistent integration bounds and baseline correction to each ion. Data are corrected for naturally occurring isotopes. More details are in Supplementary data.
Flow cytometry
Cells were stained for 30 min with 5 nM dihydroethidium (Life technologies) for the detection of reactive oxygen species, with 400 nM mitotracker (Cell signaling technology) to detect active mitochondria, or with 4 uL human or mouse CD45 antibody (ebioscience #17-9459-42, BD biosciences #557659). The cells were analyzed using a MACSQuant VYB (Miltenyi) flow cytometer and FlowJo software. Viable cell counts/mL were determined by FSC/SSC performed with and without administration of hydrogen peroxide (H2O2, 20 μM, Sigma Aldrich) or N-acetyl-cysteine (NAC, 1–5 mM, Sigma Aldrich). Annexin V-RPE (IQ products), Propidium iodide (PI, Sigma Aldrich) and active caspase 3 (BD biosciences) were used as apoptotic markers in the experiments with doxorubicin, maritoclax (Selleckchem) and/or ABT-199 (LC Laboratories).
Ribosomal fractionation and qRT-PCR
Cell lysates from three Ba/F3 RPL10 WT and three R98S clones were applied on a sucrose gradient and centrifuged in order to get a distribution of monosomes and polysomes as described before [11]. Seven fractions of equal volume were collected (Figure S7B). For technical reasons, the three fractions corresponding to the highest polysomes were subsequently combined and considered as 1 fraction. RNA was extracted from equal volumes of the 5 remaining fractions followed by cDNA synthesis. On each fraction, qRT-PCR was performed for Bcl-2 and Hprt in quadruplicate. Primer sequences: mouse Bcl-2 forward ATC GCC CTG TGG ATG ACT GAG, reverse CAG CCA GGA GAA ATC AAA CAG AGG, and mouse Hprt forward CAT TAT GCC GAG GAT TTG G, reverse GCA AGT CTT TCA GTC CTG T.
Xenografts in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice
NSG animal experiments were approved by the local ethics committee (P179/2015). Human T-ALL patient samples were engrafted by tail vein injections of 106 cells. Blood samples were obtained every 2 weeks from the facial vein. Animals reaching > 90% human CD45 expressing cells in their peripheral blood were sacrificed. Terminal cardiac bleeding was performed under pentobarbital (150 mg/kg) anesthesia to obtain blood plasma.
BCL-2 IRES reporter assay
The BCL-2 IRES plasmids were a kind gift from Dr. Adi Kimchi (Weizmann institute of science) [15]. IRES activities were tested in CRISPR-Cas9 engineered RPL10 WT and RPL10 R98S Jurkat cells using dual luciferase reporter assays (Promega) as detailed in Supplementary section.
Statistics
Statistical analyses were performed using IBM SPSS 23 (IBM Analytics) software. A two-tailed student’s t-test was used to compare RPL10 WT and RPL10 R98S mutational groups for all in vitro and in vivo measurements based upon Levene’s Test for Equality of Variances. When data of multiple independent mouse experiments were combined, we made use of the Mann–Whitney U test due to unequal distribution of the data as mentioned in the text where applicable. The statistical tests used are indicated for each experiment that has been performed.
Results
Proteomic profiling of RPL10 R98S cells identifies an increase in peroxisomal β-oxidation
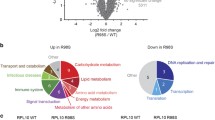
We previously described that RPL10 R98S impairs proliferation of lymphoid Ba/F3 cells under exponential growth conditions [4]. Moreover, in a recent study we described a quantitative proteomics mass spectrometry analysis of these RPL10 WT and R98S Ba/F3 clones [11]. Yet, the underlying mechanism of hypoproliferation as a cellular phenotype of the RPL10 R98S mutation remains unknown. To gain insights into the causes of hypoproliferation, we explored KEGG pathway analysis of the proteomics dataset. The peroxisome and associated metabolic pathways were upregulated in the Rpl10 R98S Ba/F3 cells (Figure 1a, b, Table S1/2, FDR < 0.1). For two of these genes, elevated mRNA expression levels were detected (Figure S1). Peroxisomes are membrane-enclosed cellular organelles in which β-oxidation of very long-chain fatty acids occurs. During peroxisomal β-oxidation high levels of hydrogen peroxide (H2O2) are produced, which form a selective substrate for the anti-oxidant catalase (Fig. 1b), a marker for peroxisome activity [16]. In accordance with higher protein expression of peroxisome enzymes, catalase protein expression was significantly elevated in Rpl10 R98S Ba/F3 cells (Figure S3A, student’s t-test, p = 0.045), in lineage negative (lin-) bone marrow (BM) cells extracted from MX1-Cre Rpl10 R98S mutant knock-in mice (Rpl10cKI R98S) (Figure S2A/B and S3B, student’s t-test, p = 0.003), and in RPL10 R98S mutant T-ALL xenografted patient samples (Figure S3C, student’s t-test, p = 0.020), but not in isogenic Jurkat T-ALL cells RPL10 R98S CRISPR-Cas9 cells (Figure S3D, student’s t-test, p = 0.309). Elevated catalase expression was previously described as a leukemic cell phenotype in patients, and might therefore not be affected by RPL10 R98S in our Jurkat leukemic cell model [17, 18]. These data support that induced peroxisomal enzyme expression in RPL10 R98S mutant leukemia cells may lead to higher peroxisomal β-oxidation activity.

RPL10 R98S enhances peroxisomal activity and is associated with oxidative stress. a Heatmap representing significantly (p < 0.01) upregulated peroxisome proteins in RPL10 WT and R98S Ba/F3 clones. Values are normalized spectral counts as determined using the Scaffold analysis software. b Overview of the peroxisome dysregulation in RPL10 R98S mutant cells. c Flow cytometry analysis of ROS levels showing the percentage DHE expressing cells from the total population in different RPL10 R98S cell models: (left) Ba/F3 lymphoid cells, (middle) Jurkat T-ALL cells, (right) lin- BM CFC assay cells. Box-plots show the data median and error bars define data distribution
RPL10 R98S cells accumulate ROS causing impaired cell proliferation
While the peroxisomal β-oxidation associated H2O2 production can be scavenged by catalase, system overloading may lead to the accumulation of ROS (Fig. 1b) [16]. Rpl10 R98S mutant Ba/F3 cultures contained 25% more cells with high ROS levels (Figure 1c, S4A, student’s t-test, p = 0.018). Similarly, 20% more cells with high ROS levels were observed in RPL10 R98S as compared to RPL10 WT Jurkat cultures (Figure 1c, S4B, student’s t-test, p = 0.028), and 40% more cells with high ROS occurred in Rpl10 R98S BM as compared to WT BM (Figure 1c, S4C, student’s t-test, p = 0.022). To examine whether changes in mitochondrial activity contributed to higher ROS percentages in R98S cells, we explored 13C6-Glucose tracing into metabolites of the TCA cycle. Glucose tracer analysis [19] demonstrated a decrease in the labeled carbons in M + 4 to M + 6 (mass distribution vector) in citrate from 13C6-Glucose and a concomitant increase in M + 2 labeling in Rpl10 R98S Ba/F3 and RPL10 R98S Jurkat cells in comparison to WT cells (Fig. 2a, student’s t-test, all p < 0.05). This labeling pattern was consistently observed in other TCA cycle metabolites and suggests decreased oxidative phosphorylation in the TCA cycle (Figure S4D/E). In addition, Rpl10 R98S Ba/F3 cells presented a 40% reduction in basal ATP levels (Figure S4F, student’s t-test, p < 0.001). Lin- BM Rpl10 R98S colony forming cell (CFC) cultures displayed a reduced percentage of cells with active mitochondria (Fig. 2b, student’s t-test, p = 0.001). Altogether, these observations support that RPL10 R98S is associated with impaired mitochondrial function, and that elevated ROS levels are thus likely to originate from the enhanced peroxisomal activity in RPL10 R98S cells. Oxidative stress driven by cellular H2O2 ROS accumulation has been shown to lead to mitochondrial dysfunction, such as reduced mitochondrial respiration [21,22,23,24]. As oxidative stress has previously been shown to perturb the proliferation of leukemia cells [20], we explored neutralizing treatment with the antioxidant N-acetylcysteine (Nac). Nac treatment completely abolished the proliferation defect in Rpl10 R98S Ba/F3 cells (Fig. 2c). Collectively, these data demonstrate RPL10 R98S-specific enhancement of peroxisomal β-oxidation, which leads to oxidative stress through ROS accumulation, resulting in mitochondrial dysfunction and the associated proliferation defect.

Mitochondrial dysfunction and the associated proliferation defect in RPL10 R98S cells can be restored by neutralizing oxidative stress. a Metabolic tracing analysis (13C6-Glucose) measuring the mass distribution vector presented as % citrate derived from labeled glucose. Left: comparison of three independent Ba/F3 RPL10 WT clones versus three RPL10 R98S clones. Right: technical triplicates for RPL10 WT Jurkat and RPL10 R98S mutant Jurkat cells. Data are represented as mean ± SD. b Mitotracker flow cytometry analysis of Rpl10 WT and R98S CFC assay cells. Three independent CFC plates per BM donor were analyzed. Box-plots show the median and error bars define data distribution. c MTS cell proliferation assay analysis of Ba/F3 RPL10 WT and R98S clones either untreated or treated with 5 mM Nac (optical densitometry). Data are represented as mean ± SD. Statistical analysis *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001. # clone ID and donor mouse ID
RPL10 R98S mutant cells possess an increased cell survival that is associated with enhanced Bcl-2 expression
Under normal physiological conditions, cells encountering such high levels of oxidative stress would undergo programmed cell death. WT cells treated with H2O2 show reduced cell viability due to enhanced oxidative stress, while RPL10 R98S cells remained unaffected even upon further increase of oxidative stress levels (Figure S5A). To investigate if RPL10 R98S cells have adapted a general survival mechanism, we explored their cell survival capacity. Ba/F3 Rpl10 R98S mutant cells displayed a clear survival benefit as compared to RPL10 WT cells in overgrowth condition (cell culture having reached the growth plateau, Fig. 3a, student’s t-test, p < 0.001), and independent of differences in residual nutrients (Fig. 3b, student’s t-test, p < 0.001). RPL10 R98S mutation in T-ALL Jurkat cells caused hypoproliferation, similar to our observation in Ba/F3 R98S cells (Fig. 3c, student’s t-test, p < 0.001) [4]. Also RPL10 R98S Jurkat cells presented an enhanced cell survival capacity in overgrowth condition, supported by reduced levels of propidium iodide (PI) stained apoptotic cells (Figure 3d, S5B, student’s t-test, p < 0.001). In addition, R98S Jurkat clones were more resistant to apoptotic stimuli such as doxorubicin chemotherapy (Figure S5C). In line with previous data, lin- Rpl10 R98S bone marrow cells displayed a reduced expansion potential during the first replate (Figure S5D, Mann–Whitney U test, p = 0.043) in CFC assays. However, RPL10 R98S cells presented an enhanced replating capacity at the 3rd and 4th re-plate, while Rpl10 WT replating capacity was lost (Fig. 3e, student’s t-test, for all p < 0.05), supporting an enhanced survival phenotype.

The RPL10 R98S mutation promotes a survival benefit. a Viable cell counts (day 5) of Ba/F3 RPL10 WT clones versus R98S clones (overgrowth condition, triplicate measurement). b Ba/F3 RPL10 WT and R98S clones were grown at 2×106 for two days in exhausted media derived from overgrown Ba/F3 parental cells (viable cell counts). c Proliferation curve of RPL10 WT and RPL10 R98S Jurkat clones during 72 h of culturing (cell counts in quadruplicate/24 h). d Percentage of viable cells at day 5 of culture (overgrowth condition, triplicate measurement). e CFC assay using Rpl10 WT or Rpl10 R98S mouse lin- BM (CFC in duplicate for each mouse). Data are represented as mean ± SD. Statistical analysis *p-value < 0.05, ***p-value < 0.001. # clone ID and donor mouse ID
Proteome profiling showed an induction of anti-apoptotic Bcl-2 expression while other apoptosis regulators were unchanged or even decreased (Figure S5E/F). Immunoblot analysis confirmed increased BCL-2 expression in Rpl10 R98S Ba/F3 (Fig. 4a, b, student’s t-test, p < 0.001), RPL10 R98S CRISPR/Cas9 Jurkat cells (Fig. 4a, b, student’s t-test, p = 0.002), lin- BM Rpl10 R98S cells (Fig. 4a, b, student’s t-test, p < 0.001), and in RPL10 R98S mutant patient T-ALL xenografted samples (Fig. 4a, b, student’s t-test, p < 0.001). This BCL-2 upregulation was specific to the ribosomal RPL10 R98S mutation, as mutations in RPL5 and RPL22 did not enhance BCL-2 expression in Jurkat T-ALL cells (Figure S6).

RPL10 R98S mutant cells present enhanced BCL-2 protein expression. a Immunoblot analysis of BCL-2 in several RPL10 WT and RPL10 R98S mutant cell models. b Quantification of immunoblot analysis. Box-plots represent the median and error bars define data distribution for RPL10 WT (gray) and RPL10 R98S mutant (blue) samples. Statistical analysis **p-value < 0.01, ***p-value < 0.001. # clone ID and donor mouse ID
RPL10 R98S cells display increased IRES-mediated Bcl-2 translation
BCL-2 is an important factor in the defense against oxidative stress-induced apoptosis in leukemia cells [25]. While the Bcl-2 mRNA levels were unchanged between RPL10 WT and R98S Ba/F3 clones (Figure S7A), we observed a 1.6-fold enhanced ribosome binding to Bcl-2 mRNA (Fig. 5a, student’s t-test, p < 0.001). In addition, qRT-PCR analysis on fractions collected from monosomes versus polysomes showed a shift in Bcl-2 mRNA distribution towards the heavy polysome fraction in RPL10 R98S cells, supporting enhanced Bcl-2 translation efficiency by the R98S mutant ribosome (Figure S7B). BCL-2 protein translation can be cap-dependent or IRES-mediated. BCL-2 has previously been shown to be regulated by stress-induced IRES-mediated translation [26]. Alterations in IRES-dependent BCL-2 translation was examined using specific dual luciferase reporter assays (Figure S7C). This revealed that RPL10 R98S Jurkat cells possess a 5-fold increase in BCL-2 IRES translational activity as compared to WT cells (Fig. 5b, student’s t-test, p < 0.001). Upon cap-dependent translation inhibition, RPL10 R98S Jurkat cells even showed a 9-fold increase in BCL-2 IRES translation (Fig. 5b, student’s t-test, p < 0.001). Involvement of oxidative stress in the regulation of BCL-2 translation was explored by Nac treatment. In line with previous data, Jurkat leukemic RPL10 WT cells displayed reduced total and IRES-driven BCL-2 translation upon Nac treatment (Fig. 5c, student’s t-test, p < 0.001 for IRES and Cap-dependent translation, p = 0.011 for IRES-driven translation) [26]. However, the IRES-mediated BCL-2 reporter activity of RPL10 R98S mutant Jurkat leukemia cells remained unchanged after Nac treatment (Fig. 5c, student’s t-test, p = 0.054 for IRES and Cap-dependent translation, p = 0.549 for IRES-driven translation). These results emphasize that RPL10 R98S-mutant ribosomes have an intrinsically higher preference for IRES-dependent BCL-2 translation, which is likely to enhance their leukemic cell survival.

RPL10 R98S cells display elevated BCL-2 IRES-mediated translation. a Overall ribosome bound Bcl-2 mRNA levels relative to Hprt mRNA binding. Box-plots represent the median and error bars define data distribution for the overall Bcl-2/Hprt ratio in sucrose gradient fractions. b/c BCL-2 IRES reporter assay read outs of RPL10 WT and R98S clones. Data are represented as mean ± SD. b On the left, the overall IRES and Cap-dependent BCL-2 translation and on the right IRES-driven BCL-2 translation. WT and R98S numbers represent the different Jurkat clone ID’s. c In the presence or absence of Nac anti-oxidant treatment. RPL10 WT (gray) and RPL10 R98S (blue). Statistical analysis *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001
RPL10 R98S specific therapeutic targeting by BCL-2 inhibition
To investigate whether the elevated IRES-mediated BCL-2 translation in RPL10 R98S leukemic cells provides a potential new selective therapeutic opportunity, we tested the efficacy of the FDA approved clinical BCL-2 inhibitor ABT-199 (venetoclax, RG7601, GDC-0199). Lin- BM Rpl10 R98S cells presented a ~50% reduction in CFC capacity in the presence of ABT-199, while Rpl10 WT BM cells were not inhibited (Fig. 6a, p < 0.001). Jurkat RPL10 R98S cells were slightly more apoptotic upon ABT-199 treatment as compared to RPL10 WT clones (Figure S8A). In xenografted T-ALL samples, ABT-199 treatment induced apoptosis of RPL10 R98S T-ALL cells, while no cellular responses were detected in RPL10 WT T-ALL cells (Figure S8B). R98S cells were not generally sensitized to targeting of anti-apoptotic proteins, as RPL10 R98S lin- BM and Jurkat cells were less sensitive to MCL-1 inhibitor maritoclax as compared to RPL10 WT cells (Figure S9A/B/C/D). Moreover, RPL10 R98S cells could be re-sensitized to doxorubicin by the addition of ABT-199 (Figure S10). Combined, these findings highlight that BCL-2 driven enhanced cellular resistance of RPL10 R98S cells can be efficiently targeted by ABT-199 treatment.

RPL10 R98S cells show selective sensitivity to BCL-2 inhibition in vitro and in vivo. a CFC assay of Rpl10 WT or Rpl10 R98S knock in mouse lin- BM cells in the absence or presence of the Bcl-2 inhibitor venetoclax (ABT-199, 1 μM, in triplicate for two individual RPL10 WT and R98S donor mice). The box-plots present the median viable cell count and error bars define data distribution. b Experimental in vivo study design for xenografting T-ALL patient samples in NSG mice. c On the left, flow cytometry analysis of human CD45 cells in the blood of the animals and on the right, the associated spleen weights of mice injected with RPL10 WT X11 and X12 and RPL10 R98S X13 and X15 T-ALL that progressed to the leukemia disease end stage, comparing DMSO and ABT-199 treated animals to healthy control animals. Data are presented as individual data points. Statistical analysis *p-value < 0.05, ***p-value < 0.001
To further evaluate the effectiveness in vivo, RPL10 WT and R98S pediatric T-ALL samples were injected into NSG mice (Fig. 6b), receiving DMSO or ABT-199 (50 mg/kg) therapy once a week when human CD45% in the peripheral blood (PB) reached > 2%. The RPL10 WT X11 and X12 xenografted mice showed minimal sensitivity to ABT-199 (n = 5 DMSO versus n = 5 ABT-199), and the percentages of human CD45 T-ALL cells in the PB of the mice were identical between DMSO and ABT-199 treated groups (Fig. 6c, p = 0.149 and p = 0.428). The spleen weights were slightly reduced in the ABT-199 treated X11, but not in X12 RPL10 WT xenografted mice (Fig. 6c, p = 0.040 and p = 0.603). The X12 animals all presented >95% human CD45 positive engraftment in BM (Figure S11A). In contrast, the RPL10 R98S X13 and X15 xenografted mice treated with ABT-199 did not show any signs of human CD45 leukemia progression in their PB, while progressive disease was observed in DMSO treated animals (Fig. 6c and S11B/D, X13 n = 5 DMSO and n = 4 ABT-199 and X15 n = 5 versus n = 5). Splenomegaly caused by human leukemia progression in DMSO treated animals was almost completely suppressed in ABT-199 treated animals (Fig. 6c, X13 and X15 p < 0.001), and the spleens of ABT-199 treated animals were only slightly larger than the spleens from healthy NSG mice (Fig. 6c). Flow cytometry analysis revealed a 30–50% suppression of human CD45 leukemia cell engraftment in the BM and the presence of 30–40% mouse CD45 cells in ABT-199 treated animals, whereas DMSO treated animals presented >95% human leukemia infiltration in the BM with a complete lack of mouse CD45 expressing cells (Figure S11C/E, X13 hCD45 and mCD45 both p < 0.001, X15 hCD45 p = 0.009 and mCD45 p = 0.004). This was underscored by white femurs (lack of erythropoiesis) in the DMSO treatment animals, while the femurs of ABT-199 treated animals remained red in color (Figure S11C). Altogether, these findings support that the RPL10 R98S mutation induces IRES-driven BCL-2 translation that sensitizes RPL10 R98S pediatric T-ALL for BCL-2 targeted therapies such as ABT-199/Venetoclax.
Discussion
In this study, we describe that RPL10 R98S leukemic cells harbor a proliferative disadvantage due to enhanced cellular oxidative stress from the accumulation of ROS (Fig. 7). Impairment of mitochondria is a likely consequence of the oxidative stress, resulting in a robust decrease in ATP levels and TCA cycle metabolites. It remains to be determined by which mechanism RPL10 R98S cells upregulate peroxisomal activity causing hypoproliferation. The proliferation impairment observed in different ribosomal protein defective models has been linked to ribosome assembly defects [4, 27, 28]. Such defects can be caused by oxidative stress as well [29]. To our knowledge, this is the first demonstration that a ribosomal mutation causes hypoproliferation due to extensive oxidative stress and mitochondrial dysfunction. This defect can be rescued by anti-oxidant treatment or possibly by acquired secondary mutations.

Schematic model representing the leukemic RPL10 R98S mechanism. Two phenotypes can be observed; (1) enhanced peroxisomal function generating ROS, which causes a proliferation defect, and (2) a mutant ribosomal preference for BCL-2 IRES-mediated translation. Enhanced BCL-2 can be efficiently targeted by ABT-199/venetoclax treatment
While normal cells initiate programmed cell death upon H2O2 exposure, RPL10 R98S cells protect themselves via enhanced IRES-dependent BCL-2 translation. The resulting constitutive BCL-2 expression facilitates and enhances leukemic RPL10 R98S cell survival (Fig. 7). The exact role of BCL-2 in the observed phenotypes may be addressed in more detail by silencing and/or overexpression of BCL-2 in RPL10 WT and R98S cells, and we cannot exclude that other apoptosis regulators for which no protein expression data were available (such as BAD, BMF, HRK, NOXA or PUMA) may also play a role. Nevertheless, we show that RPL10 R98S cells are highly sensitive to BCL-2 inhibitors, which have limited reactivity to normal healthy cells and are in clinical usage for lymphoid malignancies [30,31,32,33,34]. Several research groups observed high expression of BCL-2 in early T-cell precursor (ETP)-ALL and/or associations with JAK/STAT mutant cases, rendering such samples sensitive to ABT-199 [35,36,37,38,39,40,41,42]. However, the RPL10 R98S mutation mainly occurs in mature non-ETP-ALL [43]. The T-ALL sample X11 in our study harbored an activating JAK3 mutation (M511I). This sample did not show detectable BCL-2 protein expression, and was not targeted by ABT-199 in vivo (Fig. 6c). We previously showed that RPL10 R98S enhances JAK/STAT signaling [11]. Our current observations support that RPL10 R98S regulates BCL-2 expression independently, preceding the effects of RPL10 R98S on JAK/STAT pathway activation that only occurred during later stages. The CRISPR-Cas9 engineered RPL10 R98S Jurkat cells show increased BCL-2 expression levels, while increased JAK/STAT pathway activation was not present. Moreover, oxidative stress has been shown to modulate JAK/STAT pathway regulation [44] and proteasome complexes [45]. We could observe that JAK1 protein expression was slightly reduced upon Nac treatment in RPL10 R98S Ba/F3 cells but not in RPL10 WT cells (Figure S12), providing another mechanism for enhanced JAK/STAT pathway activation in these cells.
The peroxisomal enzyme xanthine oxidase (XO) is involved in purine degradation, with uric acid as a waste product [16, 46]. Peroxisome-active RPL10 R98S leukemic cells may have contributed to the significantly higher uric acid levels in the blood at diagnosis of RPL10 R98S mutant T-ALL patients as compared to RPL10 WT T-ALL patients (RPL10 WT n = 38 and RPL10 R98S n = 15, Figure S13A, Mann–Whitney-U test, p = 0.041). In serum samples of RPL10 R98S T-ALL xenografted mice, we observed a trend for increased uric acid levels upon leukemia development, which was inhibited by ABT-199 treatment (Figure S12B). These observations suggest that uric acid levels at diagnosis might be indicative for RPL10 R98S mutant T-ALL cases.
So far, only few examples of altered IRES-driven translation in disease have been described. Changes in ribosomal RNA (rRNA) modifications can deregulate translation rates of IRES-containing genes in cancer: IGF1R, MYC, FGF1, FGF2 and VEGFA [47]. Moreover, hypo-pseudouridylation of rRNA in the congenital disease dyskeratosis congenita reduces IRES-translation of XIAP, TP53, CDKN1B, and BCL2L1 and increases VEGF translation [48,49,50]. Inactivation of Diamond-Blackfan anemia genes RPS19 and RPL11 impaired IRES-mediated translation of mRNAs involved in erythroid development [51]. The upregulation of IRES-dependent translation in RPL10 R98S cells seems to be specific for the BCL-2 IRES, as other known cellular IRES-containing genes showed no consistent increase in translation/expression in RPL10 R98S cells as revealed by mass spectrometry (Table S3) [52]. The unique preference of IRES-mediated translation specific to the RPL10 R98S mutation in leukemia provides a novel mechanism for BCL-2 upregulation.
Change history
08 March 2019
Following the publication of this article, the authors noted that Dr Laura Fancello was not listed among the authors. The corrected author list is given in the Correction. Additionally, the following was not included in the author contribution statement: ‘L.F. analyzed RNA sequencing data’. The authors wish to apologise for any inconvenience caused.
References
Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. 2017;129:1113–23.
Cooper SL, Brown PA. Treatment of pediatric acute lymphoblastic leukemia. Pediatr Clin North Am. 2015;62:61–73.
Rao S, Lee SY, Gutierrez A, Perrigoue J, Thapa RJ, Tu Z, et al. Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood. 2012;120:3764–73.
De Keersmaecker K, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat Genet. 2013;45:186–90.
Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–71.
Sulima SO, Patchett S, Advani VM, De Keersmaecker K, Johnson AW, Dinman JD. Bypass of the pre-60S ribosomal quality control as a pathway to oncogenesis. Proc Natl Acad Sci USA. 2014;111:5640–5.
Inada H, Mukai J, Matsushima S, Tanaka T. QM is a novel zinc-binding transcription regulatory protein: its binding to c-Jun is regulated by zinc ions and phosphorylation by protein kinase C. Biochem Biophys Res Commun. 1997;230:331–4.
Monteclaro FS, Vogt PK. A Jun-binding protein related to a putative tumor suppressor. Proc Natl Acad Sci USA. 1993;90:6726–30.
Oh HS, Kwon H, Sun SK, Yang CH. QM, a putative tumor suppressor, regulates proto-oncogene c-Yes. J Biol Chem. 2002;277:36489–98.
Chiocchetti AG, Haslinger D, Boesch M, Karl T, Wiemann S, Freitag CM, et al. Protein signatures of oxidative stress response in a patient specific cell line model for autism. Mol Autism. 2014;5:10.
Girardi T, Vereecke S, Sulima SO, Khan Y, Fancello L, Briggs JW, et al. The T-cell leukemia associated ribosomal RPL10 R98S mutation enhances JAK-STAT signaling. Leukemia. 2017;32:809–19.
Elia I, Broekaert D, Christen S, Boon R, Radaelli E, Orth MF, et al. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat Commun. 2017;8:15267.
Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, et al. Breast cancer-derived lung metastases show increased pyruvate carboxylase-dependent anaplerosis. Cell Rep. 2016;17:837–48.
Lorendeau D, Rinaldi G, Boon R, Spincemaille P, Metzger K, Jäger C, et al. Dual loss of succinate dehydrogenase (SDH) and complex I activity is necessary to recapitulate the metabolic phenotype of SDH mutant tumors. Metab Eng. 2016;43:187–97.
Marash L, Liberman N, Henis-Korenblit S, Sivan G, Reem E, Elroy-Stein O, et al. DAP5 promotes cap-independent translation of Bcl-2 and CDK1 to facilitate cell survival during mitosis. Mol Cell. 2008;30:447–59.
Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim Biophys Acta - Mol Basis Dis. 2012;1822:1363–73.
López-Pedrera C, Villalba JM, Siendones E, Barbarroja N, Gómez-Díaz C, Rodríguez-Ariza A, et al. Proteomic analysis of acute myeloid leukemia: identification of potential early biomarkers and therapeutic targets. Proteomics. 2006;6:S293–9.
Zelen I, Djurdjevic P, Popovic S, Stojanovic M, Jakovljevic V, Radivojevic S, et al. Antioxidant enzymes activities and plasma levels of oxidative stress markers in B-chronic lymphocytic leukemia patients. J BUON. 2010;15:330–6.
Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H, Clish CB, et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr Opin Biotechnol. 2015;34:189–201.
Kulkarni AP, Mittal SPK, Devasagayam TP, Pal JK. Oxidative stress perturbs cell proliferation in human K562 cells by modulating protein synthesis and cell cycle. Free Radic Res. 2009;43:1090–100.
Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013;8:2003–14.
Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem. 2001;276:23357–61.
Battisti V, Maders LDK, Bagatini MD, Santos KF, Spanevello RM, Maldonado PA, et al. Measurement of oxidative stress and antioxidant status in acute lymphoblastic leukemia patients. Clin Biochem. 2008;41:511–8.
Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008;270:1–9.
Lee JE, Sohn J, Lee JH, Lee KC, Son CS, Tockgo YC. Regulation of bcl-2 family in hydrogen peroxide-induced apoptosis in human leukemia HL-60 cells. Exp Mol Med. 2000;32:42–6.
Sherrill KW, Byrd MP, Van Eden ME, Lloyd RE. BCL-2 translation is mediated via internal ribosome entry during cell stress. J Biol Chem. 2004;279:29066–74.
Kirn-Safran CB, Oristian DS, Focht RJ, Parker SG, Vivian JL, Carson DD. Global growth deficiencies in mice lacking the ribosomal protein HIP/RPL29. Dev Dyn. 2007;236:447–60.
Teng T, Mercer CA, Hexley P, Thomas G, Fumagalli S. Loss of tumor suppressor RPL5/RPL11 does not induce cell cycle arrest but impedes proliferation due to reduced ribosome content and translation capacity. Mol Cell Biol. 2013;33:4660–71.
Shenton D, Smirnova JB, Selley JN, Carroll K, Hubbard SJ, Pavitt GD, et al. Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J Biol Chem. 2006;281:29011–21.
Cang S, Iragavarapu C, Savooji J, Song Y, Liu D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol. 2015;8:129.
Gerecitano JF, Roberts AW, Seymour JF, Wierda WG, Kahl BS, Pagel JM, et al. A phase 1 study of venetoclax (ABT-199/GDC-0199) monotherapy in patients with relapsed/refractory non-hodgkin lymphoma. Blood. 2015;126:254.
Seymour JF, Gerecitano JF, Kahl BS, Pagel JM, Wierda WG, Anderson M-A, et al. The single-agent Bcl-2 inhibitor ABT-199 (GDC-0199) in patients with relapsed/refractory (R/R) non-hodgkin lymphoma (NHL): responses observed in all mantle cell lymphoma (MCL) patients. Blood. 2013;122:1789.
Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–22.
Stilgenbauer S, Eichhorst B, Schetelig J, Coutre S, Seymour JF, Munir T, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768–78.
Ni Chonghaile T, Roderick JE, Glenfield C, Ryan J, Sallan SE, Silverman LB, et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014;4:1074–87.
Maude SL, Dolai S, Delgado-martin C, Vincent T, Robbins A, Selvanathan A, et al. Efficacy of JAK / STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125:1759–68.
Peirs S, Matthijssens F, Goossens S, Van I, de W, Ruggero K, de Bock CE, et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood. 2014;124:3738–47.
Frismantas V, Dobay MP, Rinaldi A, Tchinda J, Dunn SH, Kunz J, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood. 2017;129:26–38.
Peirs S, Frismantas V, Matthijssens F, Van Loocke W, Pieters T, Vandamme N, et al. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia. 2017;31:2037–47.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63.
Li G, Miskimen KL, Wang Z, Xie XY, Brenzovich J, Ryan JJ, et al. STAT5 requires the N-domain for suppression of miR15/16, induction of bcl-2, and survival signaling in myeloproliferative disease. Blood. 2010;115:1416–24.
Waibel M, Solomon VS, Knight DA, Ralli RA, Kim SK, Banks KM, et al. Combined targeting of JAK2 and Bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Rep. 2013;5:1047–59.
Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–8.
Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–52.
Aiken CT, Kaake RM, Wang X, Huang L, Stumpf MP, Mishto M, et al. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteom. 2011;10:R110.006924.
Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V. Regulation of uric acid metabolism and excretion. Int J Cardiol. 2016;213:8–14.
Marcel V, Ghayad SE, Belin S, Therizols G, Morel AP, Solano-Gonzàlez E, et al. P53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell. 2013;24:318–30.
Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego ERD. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006;312:902–6.
Montanaro L, Calienni M, Bertoni S, Rocchi L, Sansone P, Storci G, et al. Novel dyskerin-mediated mechanism of p53 inactivation through defective mRNA translation. Cancer Res. 2010;70:4767–77.
Rocchi L, Pacilli A, Sethi R, Penzo M, Schneider RJ, Treré D, et al. Dyskerin depletion increases VEGF mRNA internal ribosome entry site-mediated translation. Nucleic Acids Res. 2013;41:8308–18.
Horos R, IJspeert H, Pospisilova D, Sendtner R, Andrieu-Soler C, Taskesen E, et al. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood. 2012;119:262–72.
Mokrejs M. IRESite: the database of experimentally verified IRES structures (www.iresite.org). Nucleic Acids Res. 2006;34:D125–30.
Acknowledgements
We thank the patients who donated samples and the physician assistants, nurse practitioners, and clinicians who acquired the samples. We thank Prof. Adi Kimchi (Weizmann institute of science, Israel) for providing dual-luciferase reporter plasmids.
Author contributions
K.R.K. designed research, performed research, collected data, analyzed data and wrote the paper. S.O.S. conducted the BCL2 IRES-reporter assays and wrote and edited the manuscript. B.V. and S.V. generated the CRISPR-Cas9 Jurkat clones. T.G. and JOdB generated Ba/F3 clones containing human RPL10 WT or R98S, G.R. and S.F. performed and interpreted the 13C6-Glucose tracing experiment. J.V. facilitated the mice studies, and P.S. performed ATP analysis. A.U., A.V.M., C.J.H., J.P.P.M., and J.C. provided the pediatric T-ALL patient data and samples. P.V. and D.C. measured uric acid levels in serum samples. K.D.K. designed research, supervised the study and wrote the paper.
Funding
K.R.K. was supported by the Lady Tata Memorial Trust International Award for research in Leukemia. S.O.S. is recipient of an EMBO long-term postdoctoral fellowship and the EHA José Carreras Junior Research Grant. T.G. was supported by a fellowship “Emmanuel van der Schueren” from Kom op tegen Kanker. B.V. and S.V. are S.B. PhD fellow at FWO (Nos. 1S07118N and 1S49817N). GR is supported by consecutive PhD fellowships from the Emmanuel van der Schueren—Kom op tegen Kanker foundation and FWO. S.M.F. acknowledges funding support from the Concern Foundation (Conquer Cancer Now) and KU-Leuven Methusalem Co-funding. P.V. and D.C. have senior clinical investigator fellowships of the FWO Vlaanderen. This research was funded by an ERC starting grant (No. 334946), FWO funding (G084013N and 1509814N) and a Stichting Tegen Kanker grant (Grant No. 2012-176 and 2016-775) to K.D.K. and by the leukemia research grant 2017 from the “Me To You” foundation to K.R.K.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Kampen, K.R., Sulima, S.O., Verbelen, B. et al. The ribosomal RPL10 R98S mutation drives IRES-dependent BCL-2 translation in T-ALL. Leukemia 33, 319–332 (2019). https://doi.org/10.1038/s41375-018-0176-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0176-z
- Springer Nature Limited
This article is cited by
-
Ribosome profiling: a powerful tool in oncological research
Biomarker Research (2024)
-
FUS regulates a subset of snoRNA expression and modulates the level of rRNA modifications
Scientific Reports (2023)
-
Molecular profiling of a real-world breast cancer cohort with genetically inferred ancestries reveals actionable tumor biology differences between European ancestry and African ancestry patient populations
Breast Cancer Research (2023)
-
The plasticity of mRNA translation during cancer progression and therapy resistance
Nature Reviews Cancer (2021)
-
BCL-2 family deregulation in colorectal cancer: potential for BH3 mimetics in therapy
Apoptosis (2020)