
Abstract
Immune checkpoint inhibitors, as single-agent therapy, have shown modest clinical efficacy in the treatment of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS). As has been successfully shown in other less immunogenic hematologic malignancies, rationally designed combination approaches may be more effective than single-agent checkpoint inhibitors, and may be the approach to pursue in AML/MDS. Hypomethylating agents (HMAs) such as azacitidine, while enhancing anti-tumor immune response, concurrently dampen immune response by upregulating inhibitory immune checkpoint molecule expression. Immune checkpoint molecule upregulation may be an important mechanism of azacitidine resistance. These findings have resulted in multiple clinical trials combining HMAs with immune checkpoint blockade. Clinical trial data have shown encouraging response rates and durable responses without resorting to stem cell transplant. In this review, we discuss preclinical data supporting the use of these agents in combination, and focus on clinical and correlative data emerging from numerous clinical trials investigating HMA-immune checkpoint inhibitor combinations in AML/MDS.
Similar content being viewed by others


Introduction
One of the major factors driving anti-tumor immune responses is the activation of T cells through a complex and tightly regulated process. Activation of T cells is comprised of two important and necessary signals: First, is the presentation of antigen, essentially a peptide/major histocompatibility complex (MHC) complex, by an antigen-presenting cell (APC) to the T-cell receptor (TCR) present on T cells. Second, is the co-stimulatory signal, which is provided by the APCs through B7 molecules (B7-1 and B7-2) and their interaction in turn with CD28 present on the T cells. Only when both the signals are present via the APC–T cell interaction, parallel positive- and negative-signaling programs are initiated. On one hand it initiates intracellular signaling leading to cytokine production, cell-cycle progression, and upregulation of anti-apoptotic factors that cause T-cell proliferation and differention. On the other hand, it leads to the induction of inhibitory molecules including cytotoxic T-lymphocyte-associated-protein 4 (CTLA-4) and others on T cells, which ultimately triggers the termination of the activation response. There are several other co-stimulatory and inhibitory molecules, which regulate T-cell activation [1]. Some examples of other co-stimulatory molecules are 4-1BB and CD27 (expressed on T cells), CD80, and CD86 (expressed on APCs). Examples of other co-inhibitory molecules are PD-1, PD-L1, LAG3, TIM3, and VISTA. The most relevant and widely studied in clinical settings are CTLA-4, programmed cell-death protein PD-1 (expressed on activated and exhausted T cells) and and its ligands PD-L1 (expressed on many cell types including epithelial cells, immune cells, and endothelial cells) and PD-L2 (predominantly expressed on APCs) [1, 2].
CTLA-4 and PD-1/PD-L1 are immune checkpoint molecules that operate at different stages of T-cell activation and dampen T-cell anti-tumor response by different mechanisms of action. CTLA-4 is expressed on the T cells and is a homolog of CD28. It regulates T-cell activation during the initial stages of T-cell-mediated anti-tumor immune responses by binding with a higher avidity to both B7-1 and B7-2 molecules. This leads to attenuation of CD28 co-stimulatory signaling by directly competing with CD28 for binding to B7 molecules and initiating a negative-signaling network. The PD-1/PD-L1 signaling pathway predominantly modulates T-cell activity by inhibiting TCR signaling after T cells exit the circulation and migrate to tumor tissues, thus playing an important role in peripheral tolerance [3]. Signaling through PD-1 dampens T-cell activation following TCR/MHC engagement and CD28 activation [3, 4]. In normal physiologic states, PD-1 functions to limit T-cell effector responses to foreign antigens, infections and inflammation, preventing unchecked immune hyperactivation that would be detrimental to the host. In malignancies, the PD-1/PD-L1 pathway plays an important role in tumor immune evasion, thereby promoting tumor growth [5].
Clinical trials with antibodies targeting both the PD-1 and CTLA-4 pathways have demonstrated marked efficacy against a variety of solid tumors, and more recently in hematologic malignancies including classical Hodgkin’s lymphoma (cHL), non-Hodgkin’s lymphoma (NHL), and multiple myeloma (MM) [6, 7]. However, data suggest that the optimal benefits of immune checkpoint inhibitors in the broad majority of patients with NHL and MM were obtained not with single-agent checkpoint inhibitor therapy, but when combined with standard therapies to further improve the response rates, progression-free survival (PFS), and eventually overall survival.
Discussion
Immune check point therapies in hematological malignancies
cHL is particularly susceptible to PD-1 inhibition due to its unique pathophysiologic features, with immune infiltration, which is ineffective and malignant Reed-Sternberg cells that overexpress PD-L1. The Reed-Sternberg cells have chromosome 9p copy number alterations targeting PDL-1, PDL-2, which leads to increased PD-L1 and PD-L2 expression. In addition, 9p amplification also targets JAK2 leading to increased JAK-STAT signaling thereby further inducing transcription of PD-L1 and PD-L2 [8]. There is upregulation of PD-L1 and PD-L2 ligands by Epstein–Barr virus (EBV) infection in EBV positive cases of cHL [9]. A phase I trial involving 23 heavily pre-treated patients with cHL, who received the anti-PD-1 antibody nivolumab, reported high objective response rates of 87%, including 17% complete responses (CR) and 70% partial responses (PR) [10]. Comparable results were reported with another anti-PD-1 antibody pembrolizumab [11]. In preliminary results from a study in patients with refractory/relapsed cHL, the combination of brentuximab vedotin with nivolumab produced an overall response rate (ORR) of 100% in evaluable patients, with a CR rate of 62.5% [12]. In a recent report seven of eight evaluable patients with heavily pre-treated cHL achieved metabolic CR (87.5%) with anti-PD-1 antibody therapy (nivolumab or pembrolizumab); interestingly five patients had prior therapy with azacitidine and all achieved a CR suggesting an epigenetic priming effect to immune checkpoint inhibition [13]. Recently, other hematologic tumors with 9p copy number alterations and high-response rates to single-agent anti-PD-1/PD-L1 therapies have been identified including primary mediastinal B-cell lymphoma [14], primary central nervous system (CNS) and primary testicular lymphoma [15], and NK/T cell lymphomas [16].
In contrast to cHL, single-agent immune checkpoint blockade has demonstrated limited activity in NHL’s [17]. However, immune checkpoint antibodies have demonstrated marked synergism when combined with other agents in NHL’s, particularly follicular lymphoma. A phase II trial evaluating rituximab and pidilizumab in relapsed/refractory follicular lymphoma reported response rates of 66% (19/29 patients) including CR in 52% and PR in 14% [18]. Rituximab monotherapy in a similar population would be expected to produce a response rate of 35-40% with a CR rate of 10-15% [19]. Similarly, while the results with single-agent immune checkpoint blockade therapy in MM have been disappointing, with almost no objective single-agent responses in two phase I trials of nivolumab alone [20] and pembrolizumab alone [21], the combinations of pembrolizumab–lenalidomide–dexamethasone, and of pembrolizumab–pomalidomide–dexamethasone showed synergism in phase II trials, with objective response rates of 50–65% and median PFS of 16–18 months [22, 23]. This represents a doubling of the response rate and a 3–4 fold increase in PFS than has been reported with pomalidomide–dexamethasone therapy in patients with relapsed/refractory MM [24, 25]. In July 2017, the US FDA placed a clinical hold on three clinical trials [two phase III (KEYNOTE-185 and KEYNOTE-183) trials and one phase I (KEYNOTE-023) trial] using immunomodulatory agents lenalidomide or pomalidomide with or without pembrolizumab in patients with relapsed MM, after an independent data-monitoring committee discovered more patient deaths were observed in the pembrolizumab containing arm [26]. Following risks observed in the pembrolizumab clinical trials, a number of studies evaluating PD-1 inhibitor nivolumab and PD-L1 durvalumab in patients with relapsed MM and lymphoma were also placed on partial clinical hold by the US FDA in September 2017 [27, 28]. Patients enrolled in the trials on partial hold were allowed to continue on treatment if achieving clinical benefits. A randomized, multicenter, international study evaluating azacytidine, and durvalumab vs. azacytidine alone in patients with frontline high-risk myelodysplastic syndromes (MDS) and older AML (NCT02775903) and a randomized, multicenter trial of oral azacytidine (CC-486) alone or in combination with durvalumab in patients with MDS who had failed hypomethylating agent therapy (NCT02281084) were allowed by the FDA to continue enrollment. Recently, in December 2017, the FDA has removed the partial holds on two of the studies evaluating nivolumab with immunomodulatory agents in patients with relapsed MM.
T-cell repertoire and checkpoint expression in AML/MDS
T cells are present in both the bone marrow (BM) and the peripheral blood (PB) among patients with AML. The median T-cell population in the BM aspirates of patients with AML is 8–25% [29]. The T cells made up a significantly larger proportion of the CD45 + population (non-leukemic cells) in BM aspirates from patients with AML as compared with healthy donors. The T cells in patients with AML exhibit distinct phenotypic and genotypic differences from T cells of healthy donors. The proportion of CD8 T cells, the CD8/CD4 ratio, the T-regulatory cell population, and the CD4 + naive and memory cell populations increased in the CD45 + population in the BM of patients with AML compared with healthy donors [29, 30]. The T cells in the BM from patients with AML express activation markers (such as CD25, CD69, OX40) at a significantly higher rate than seen in healthy donors, indicating that the AML BM is an inflamed microenvironment [31, 32]. There is an increased overall expression of inhibitory surface molecules associated with T-cell exhaustion on CD4+ and CD8+ T cells of AML patients compared to controls [29, 33, 34]. Gene expression profiling studies on T cells from patients with AML have demonstrated aberrant T-cell activation patterns as compared to healthy donors [35]. The gene expression profile of T cells from AML was markedly different from that noted on T cells from healthy donors or T cells from patients with chronic lymphocytic leukemia (CLL) [35]. While T cells from patients with AML appeared to be more activated than healthy donor T cells, a comparison made with gene lists obtained in healthy T-cell activation suggested that gene expression changes in the AML-derived T cells were not purely attributable to upregulation of normal signaling that occurs in healthy T-cell activation [35]. Pathway analysis showed that the genes for T-cell activation and signaling that were upregulated in patients with AML were different from genes for T-cell activation previously noted to be upregulated in CLL, suggesting a potential need for differing strategies for evading T-cell-mediated immune surveillance in different malignancies [35, 36].
It has been demonstrated in murine models that, similar to many solid tumor malignancies, PD-1/PD-L1 pathways plays an important role in evading T-cell responses in leukemia [37]. Studies to evaluate the immune landscape in patients with AML have demonstrated overexpression of co-inhibitory immune checkpoints such as PD-1 and co-stimulatory checkpoints such as OX40 on T cells in BM aspirates from patients with AML compared with healthy donors [29]. Tan et al. [34] demonstrated that CD8+ T cells with an exhausted phenotype (i.e., coexpressing CD244 + or CD57+) were significantly increased in de novo AML group compared with healthy controls. Zhou et al. [38] identified T-cell immunoglobulin, and mucin domain-containing protein 3 (Tim3) co-expression on CD8 + T-cells was associated with a T-cell exhaustion phenotype that increased during disease progression, and that combined PD-1 and Tim3 blockade had an additive effect in reducing murine AML tumor burden and improving murine survival. Compared to healthy controls, patients with relapsed and newly diagnosed AML had a higher percentage of exhausted T-cell phenotype (PD-1 + TIM3 + and PD-1 + LAG3 + CD8 T cells) in the BM [39]. The PD-1 expression on the BM T cells was higher in multiply relapsed AML compared with first salvage AML, newly diagnosed AML or healthy donors (PD-1 on T-cells in multiply relapsed > first salvage > newly diagnosed > healthy donor), suggesting progressive T-cell exhaustion with more advanced AML [29]. Similar to PD-1 expression on T cells, blasts in the BM in AML expressed a higher percentage of PD-L1 compared to healthy controls [39]. Among newly diagnosed AML, the frequency of PD-L1-positive AML blast was highest in patients with AML with TP53 and adverse cytogenetics. Additionally, PD-L1 expression on leukemia/tumor cells was significantly higher in the relapse patients than in untreated patients [40]. a multivariate analysis for overall survival in patients with AML, PD-L1 promoter methylation (mPD-L1) added significant independent adverse prognosis to established prognostic factors such as high white blood cell count, high-risk karyotype, TP53 mutation, and FLT3 mutation [41].
For MDS a few studies have looked at checkpoint expression. PD-L1 and PD-L2, the ligands for PD-1, were overexpressed in higher-risk MDS. Treatment with decitabine increases the expression of these molecules in a concentration-dependent manner [42]. The expression of these ligands was also increased in MDS that had relapsed or was refractory to therapy with hypomethylating agents (HMAs) [42]. A further discussion of findings from these studies is included in the sections below.
Highlighting the immunosuppressive environment in AML and MDS
AML cells create an immunosuppressive environment and induce immune tolerance through several pathways, targeting of some of which has therapeutic implications in management [43]. Similarly to solid malignancies, AML cells have been demonstrated to have reduced T-cell and NK-cell function and cytotoxicity [35, 44], and an expanded population of T-regulatory cells [45]. There exists extensive cross-talk between AML cells and the surrounding hematopoietic microenvironment. Mesenchymal stromal cells (MSCs) play a vital role in the development and maintenance of the hematopoietic system [46]. They also have immune-modulating activity and can inhibit naive and memory T-cell responses to stimuli [47]. One of the mechanisms of T-cell response inhibition by human MSCs is the expression of IDO (indoleamine 2,3 dioxygenase), an interferon (IFN)-gamma induced enzyme that catalyzes the conversion of tryptophan to kynurine [48, 49]. Tryptophan depletion and kynurine production inhibits T-cell effector function [50]. In vitro studies have shown that forced IDO expression enhanced the inhibitory effect of MSCs on lymphocytes [51]. IDO-dependent immunosuppressive pathways may be an important mode of immune escape in AML via multiple mechanisms. IDO enzymatic activity was demonstrated to be increased in the blood of AML patients compared to controls [50]. T-cell regulatory cell frequencies were upregulated in AML expressing IDO [52]. Furthermore, AML cells were able to induce T-cell tolerance by converting CD4 + CD25- T cells into CD4 + CD25 + T(reg) cells through an IDO-dependent mechanism; supporting the observation that IDO may promote immune evasion through promotion of T regulatory cell function [52].
Proliferation assays have not revealed functional defects in the T cells of patients with AML with respect to proliferation or cytokine production, either at diagnosis or at relapse [53, 54]. Functional characterization studies have shown that PB T cells from patients with AML are able to proliferate and produce IFN-gamma when co-stimulated with anti-CD3 + anti-CD28 (additional engagement of co-stimulatory signals). In sharp contrast, simultaneously collected BM T cells from the same patient were unable to divide or produce IFN-gamma in the presence of anti-CD3 + anti-CD28 stimulation, but required addition of a PD-1 antibody to induce proliferation and IFN-gamma production. This demonstrated that immune exhaustion was more profound in the BM T cells and was reversible, at least in some cases, by blocking the inhibitory checkpoint pathways such as PD-1 [55]. These data suggest that decreased effector function may not be due to an inherent defect in T cells in AML, but rather due to increased inhibitory immune checkpoint expression on the T cells from long-term contact with AML cells or stromal elements in the leukemic BM [53, 56].
There is data to suggest that MSCs are functionally altered in MDS as well, and favor the survival of abnormal CD34 + hematopoietic stem cell progenitors (HSCs). Cross-talk between HSCs and MSCs has a reprogramming effect on MSCs, resulting in the upregulation of genes involved in inflammatory and cytokine signaling pathways and favoring engraftment of MDS CD34 + cells [57]. Also, MDS-derived MSCs have shown to express very high levels of Fas-L making them potential targets to apoptosis by Fas-expressing hematopoietic cells [58].
It has been demonstrated that CD33-expressing MDSCs (myeloid-derived suppressor cells), which have an effect on myeloid differentiation, are increased in MDS [59]. MDSCs mediate immunosuppression via reducing T-cell proliferation and functionality via CD33-S100A9 receptor-ligand pathway [60, 61]. In higher risk MDS, there appears to be an impairment of T-cell immune response with lower numbers of CD8 + and NK cells, and increased levels of T-regs [62,63,64]. The decline in the proportion of CD8 + T cells with increasing disease risk in MDS appears to be related to the expression of PD-1/PD-L1 [42]. These data suggest that similar to AML higher expression of PD-1/PD-L1 in BM in MDS may be associated with an inferior prognosis.
These data have led to the initiation of several clinical trials evaluating the potential therapeutic role of checkpoint inhibitors in AML and MDS. Preliminary data suggests that single-agent anti-PD-1 antibodies have limited activity in patients with AML and MDS [17, 65]. CTLA-4 inhibition may have single-agent activity in AML and MDS, as has been shown in post-stem cell transplant relapsed AML [66] and in MDS that is refractory/relapsed post-HMA agents [65]. Similar to follicular lymphoma and MM, rational combinations of immune checkpoint inhibitors with other standard anti-leukemic agents may be needed to improve the response rates, the durability of response, and the overall survival in patients with AML/MDS.
Hypomethylating agents and modulation of anti-tumor immune responses
HMAs, such as azacitidine and decitabine, have demonstrated diverse immune-modulating activities on tumor infiltrating lymphocytes and on leukemic cells [67] (Fig. 1). An important mechanism of tumor immune response evasion by cancer cells lies in their ability to alter the expression of tumor-associated antigens, resulting in deficient antigen presentation [68]. HMAs have favorable effects on anti-tumor immune response by upregulating a range of immunomodulatory pathway-related genes [69] including the expression of cancer testis antigens (CTAs) [69] [including melanoma-associated antigens (MAGE-1) and NY-ESO-1] [70, 71], increasing the expression of HLA class 1 on tumor cells allowing for improved tumor recognition [72], upregulating co-stimulatory molecules (CD28, CD40L) [72], and upregulating IFN-gamma pathway viral defense genes (IRF7, IFI27, IFI44, DDX41, STAT1, IFI6, and others). Goodyear et al. [73] demonstrated that treatment with azacitidine and valproate upregulated the expression of MAGE antigens on AML and myeloma cell lines and induced CD8 + cytotoxic T lymphocyte responses to MAGE antigens in patients previously lacking CTL responses. The achievement of CTL responses appeared to be associated major clinical responses. Apart from their effect on immune antigen expression, HMAs also impact T-cell population numbers. Costantini et al. [74] demonstrated that the percentage and absolute number of PB T-reg cells were significantly higher in patients with MDS than in healthy controls. The PB T-reg numbers in MDS decreased to normal after 9 months of azacytidine therapy. The PB T-regs were also higher in non-responders than responders to azacytidine. Interestingly, the PB Th1 and Th2 cells, but not Th-17 cells were reduced with azacytidine treatment. Among the CD8 + lymphocytes, there was a shift toward CD8 + /IFNγ + T subtype, which expresses a higher level of PD-1 [75].

Illustration of the effect of hypomethylating therapy on gene promoters. HMAs induce demethylation of methylated CpG islands of gene promoters regulating expression of several immune pathway-related genes including PD-L1, in tumor cells and PD-1 and CTLA-4 in T cells. Demethylation of these sites induces gene expression of PD-1 and CTLA-4 in T-cells, and PD-L1 in tumor cells. The expression of these co-inhibitory receptors leads to ‘‘exhausted’’ T cells with abrogation of anti-tumor response. At the same time HMA-induced demethylation has favorable effects on anti-tumor immunity including increased T-cell receptor and CD28 expression on T cells, increased expression of IFN-gamma viral defense genes, and enhanced tumor cell antigen and endoretroviral sequences (ERV) expression leading to activation of cellular antiviral response. Increased expression of PD-1/PD-L1 and CTLA-4 may be important mechanism of azacitidine resistance and potential targets for combinatorial approaches with immune check point inhibitors
Expression of PD-1 on T cells appears to be tightly coupled to the DNA methylation status of CpG sites in the PD-1 promoter [76]. DNA methylation status is inversely correlated with the expression of PD-1 protein. Treatment of activated CD8 T cells with azacitidine results in hypomethylation of the PD-1 promoter, which in turn leads to increased expression of PD-1 on T cells. This increased expression of PD-1 on T cells promotes exhaustion of tumor-specific T cells. Orskov et al. [77] showed treatment with azacitidine to be accompanied by DNA demethylation in the PD-1 promoter in 44% of patients with AML/MDS. Patients who did not respond to azacitidine therapy had a significantly higher baseline PD-1 promoter methylation compared to healthy controls, with consequently more demethylation in the promoter regions for PD-1 and PD-L1 on azacitidine therapy, resulting in an increased expression of PD-1/PD-L1 by flow-cytometry [77]. Demethylation of the PD-1 promoter correlated with a significantly worse ORR (8% vs. 60%, P = 0.014), and with trends towards a shorter OS (P = 0.11). Furthermore, PD-1 demethylation was reversible but occurred with each subsequent cycle of azacytidine. This process of PD-1 promoter hypomethylation is akin to the situation during chronic infections where the PD-1 promoter remains unmethylated, resulting in continuous PD-1 expression in the chronically activated T cells, leading to the eventual exhaustion of the T cells and termination of the inflammatory process [78].
Silencing of the immune-related genes is frequently mediated by DNA methylation, a process reversed by HMAs, leading to immune-related gene re-expression [67, 79]. Wrangle et al. [67] showed that the genes re-expressed by HMAs were particularly enriched for IFN-gamma response pathway components. In a study, by Li et al. [69], exposure of epithelial cancer cell lines to azacitidine resulted in the upregulation of several immunomodulatory pathway genes including IFN signaling, antigen processing and presentation, and cytokines/chemokines.
Mice pre-treated with azacitidine demonstrated an enhanced response to anti-CTLA-4 therapy [80, 81]. The potentiating effect of azacitidine on the anti-tumoral activity of anti-CTLA-4 antibody involved the hypomethylation of human endogenous retroviral (ERV) elements, leading to the expression of viral double-stranded RNAs (dsRNAs) and the cytosolic sensing of these dsRNAs. Knock down of the dsRNA sensors, triggered in ovarian cells exposed to DNMTis, reduced this viral defense response by twofold [69]. The induction of human ERV transcripts facilitates the activation of viral defense associated genes and the cellular antiviral response. Induction of viral defense pathways includes upregulation of immune effector pathway signaling genes, thereby leading to sensitization and a favorable milieu for anti-CTLA-4 antibody therapy.
Hypomethylating agents appear to have a dual effect on anti-tumor immunity. In addition to the favorable effects on anti-tumor immunity discussed above, HMAs blunt anti-tumor immunity by upregulating the expression of the inhibitory checkpoint receptor PD-1 on T cells and inhibitory ligands PD-L2 and PD-L1 on the tumor cells. Immunologically, upregulation of PD-1/PD-L1 if left unchecked promotes T-cell exhaustion. Therapeutically, this phenomenon raises the possibility of enhanced sensitivity to PD-1/PD-L1 blockade. Wrangle et al. [67] first reported an immunomodulatory priming effect with HMA therapy that sensitized patients to subsequent PD-1/PD-L1 blockade. They noticed higher than expected response rates in patients with advanced non-small cell lung cancer (NSCLC) who had failed prior therapy with azacitidine and were subsequently treated on trials with anti-PD-1/PD-L1 antibodies: three of six patients achieved durable PRs lasting 14–26 months and an additional two patients had stable disease lasting approximately 8.5 months [67]. Azacitidine treatment of NSCLC cell lines showed alterations in innate and adaptive immunity related genes and pathways, including upregulation of genes involved in immune evasion such as PD-L1. It was suggested that increased PD-L1 expression may have been driven either by cell-intrinsic mechanisms or via adaptive resistance, through IFN-gamma signaling and subsequent STAT activation. Notably, CTLA-4 ligands namely CD80 and CD86, were not altered with azacitidine treatment in the cell line studies. In a study by Yang et al. [42], azacitidine upregulated PD-1 and PD-L1 (≥2-fold) mRNA expression on the PB mononuclear cells in approximately 50% of patients with AML or MDS. PD-1 and its ligands, PD-L1 and PD-L2, and to a lesser extent CTLA-4, were also aberrantly upregulated (≥2-fold) on BM CD34 + cells by mRNA expression, in approximately 30–40% of patients with AML or MDS. Patients resistant to azacitidine-based therapy had relative higher increments in gene expression on the PB mononuclear cells and BM CD34 + cells as compared with patients who achieved response, suggesting this could be a mechanism of resistance. Upregulation of PD-L1 and PD-L2 on mononuclear cells, but not PD-1 and CTLA-4 was associated with inferior OS. In MDS and AML cell lines treated with decitabine, the expression of PD-L1, PD-L2, PD-1, and CTLA-4 increased in a concentration-dependent manner. These changes were not observed after treatment with cytarabine [42].
Clinical trials of immune checkpoint inhibitors: hypomethylating agents in AML/MDS
A. Combinations of immune checkpoint inhibitors with HMAs in AML
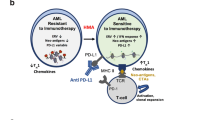
A number of trials combining HMAs with PD-1/PD-L1-based therapies have recently started enrollment for AML and MDS including azacitidine with the anti-PD-1 antibody nivolumab (NCT02397720), azacitidine with or without the anti-PD-L1 antibody durvalumab (NCT02775903), and azacitidine with or without the anti-PD-L1 antibody atezolizumab (NCT02508870) (Table 1) (Fig. 2). Among these, the nivolumab (Opdivo, BMS-936558, Bristol-Myers Squibb) and azacitidine combination is currently being evaluated in a phase I/II trial in patients with relapsed/refractory AML and in frontline therapy for older patients with AML (≥65 years) not fit for intensive chemotherapy (NCT02397720) [82]. The study included 53 patients with relapsed/refractory AML. The median age of the patients was 69 years; the median of prior salvage therapies was 2 (range, 1–7); 47% of the patients had poor-risk cytogenetics. Among 35 patients evaluable for response, the ORR was 34%, including six complete remissions (CRs)/complete remission with insufficient count recovery (CRi) (18%), and five hematologic improvements that were maintained >6 months (including two patients who had concomitant >50% blast reduction). The 8-week mortality was 6%. Notably, the response rates were higher in patients with a diploid karyotype. The outcomes compared favorably to a response rate of 15–18% in a historic control group of similar patients treated at the same institution with azacitidine alone or decitabine alone. At a median follow-up of 6 months, only one of 11 patients who achieved CR had lost response and two patients had died (1 relapse, 1 cardiac failure while in remission). This suggests a durable remission benefit among responders (Table 2). Grade 2–4 immune-related adverse events (irAEs) were observed in 12 patients (24%). There was a wide variation in the time to onset of irAEs, with irAEs observed as early as 4 days to 3.5 months from the initiation of the nivolumab therapy. The irAEs profile differed from that seen in solid tumors with more common irAEs of pneumonitis, nephritis, colitis, and dermatitis compared with endocrine insufficiencies, skin rash, and transaminitis described frequently in solid tumor trials. Patients who achieved CR/CRi had a higher total CD3 and higher CD8+ T cells infiltrate in the pre-therapy BM aspirate. Responders demonstrated progressive increase in BM CD8+ and CD4+ T-cell infiltrate. Both responders and non-responders had an increase in CTLA-4+ CD8+ cells on therapy, suggesting that combination blockade of these two major co-inhibitory pathways may improve response rates and durability of responses [82]. Dual combination of nivolumab (PD-1) and ipilimumab (CTLA-4) with azacitidine in relapsed and in frontline elderly AML therapy has recently begun enrollment (NCT02397720).

Immune check point inhibitors currently being tested in combination with azacitidine in ongoing clinical trials in AML/MDS are demonstrated in this figure. Important druggable targets include PD-1 (nivolumab, pembrolizumab) and CTLA-4 (ipilimumab, tremelimumab) receptors on T cells, PD-L1 (durvalumab, atezolizumab) on antigen-presenting and tumor cells, and inhibitor KIR (lirimumab) receptors on natural killer cells
B. Combinations of immune checkpoint inhibitors with HMAs in MDS
A phase II study is evaluating azacitidine in combination with nivolumab (N = 20), azacitidine with ipilimumab (N = 20), and azacitidine with nivolumab and ipilimumab (N = 20) in frontline intermediate 2/high-risk MDS by International Prognostic Scoring System (IPSS) as well as nivolumab alone, ipilimumab alone, and nivolumab with ipilimumab in patients with MDS who have failed prior therapy with a HMA (NCT02530463) (Table 2). The azacitidine combination with nivolumab was the first cohort to complete enrollment (N = 20) [65]. The median number of treatment courses was 4 (range, 2–11), and a response was noted in 13 of 16 (80%) patients (CR in six, marrow CR + hematologic improvement in seven). Three patients were too early to evaluate response and two patients have progressed. The cohort of nivolumab alone in patients with MDS who have failed prior therapy with an HMA has also completed enrollment (N = 15). Nivolumab as a single-agent in high-risk MDS after HMA therapy resulted in no responses in 15 evaluable patients resulting in termination of this cohort at 15 patients based on predefined stopping rules. This suggests, that similar to the AML experience, single-agent anti-PD-1 antibody may not be effective and that the combination of HMA with anti-PD-1 antibody may be a better approach [65]. The cohort of ipilimumab alone in patients with MDS who have failed prior therapy with an HMA is currently enrolling (N = 18 of 20 enrolled). Interestingly, ipilimumab as a single-agent demonstrated activity with responses in 33% of patients with high-risk MDS after HMA therapy. Ipilimumab has also demonstrated single-agent activity in relapsed AML post allogeneic stem cell transplant [66]. This suggests that there may be a differential efficacy profile for PD-1 vs. CTLA-4 inhibition in myeloid diseases, and that these may be acting via mutually exclusive pathways.
C. Recent trials of combination of immune checkpoint inhibitors with HMAs in AML/MDS
Durvalumab is an anti-PD-L1 antibody undergoing investigation in combination with azacitidine. Patients are randomized on this study to receive azacitidine alone or azacitidine with durvalumab in two independent cohorts: (1) frontline international prognostic scoring system-revised (IPSS-R) intermediate/high-risk MDS, and (2) frontline AML > / = 65 years of age who are not candidates for induction therapy (NCT02775903). Atezolizumab is another anti-PD-L1 antibody being evaluated in second line therapy in MDS either as single-agent atezolizumab or in combination with azacitidine in patients with MDS who have failed prior therapy with an HMA, and in frontline therapy with azacitidine and atezolizumab in untreated IPSS-R intermediate-high-risk MDS (NCT02508870).
Dual combination check point blockade with anti-CTLA-4 and anti-PD-1/PD-L1 antibodies with or without azacitidine have recently entered clinical trials in patients with MDS, including a phase II study of nivolumab and ipilimumab with azacitidine in frontline IPSS intermediate-2/high-risk MDS, a combination of nivolumab with ipilimumab in MDS post failure of HMA therapy (NCT02530463), and a phase I study evaluating durvalumab and azacitidine with or without tremelimumab (IgG2 anti-CTLA-4 antibody) in relapsed/refractory MDS (NCT02117219). Data from these studies are not yet available.
Conclusion
It is becoming increasingly clear that the heavy systemic disease burden, the immunosuppressive effect of the tumor microenvironment, and the moderate overexpression of immune checkpoints co-stimulatory receptors on T cells and ligands on tumor cells, result in low single-agent activity with immune checkpoint inhibitors in hematologic malignancies including follicular lymphoma, MM, AML, and MDS. The immune checkpoint inhibitors are best exploited in combination strategies with other standard therapies in these hematologic malignancies. Such rationally designed combination approaches have shown significant improvements in response rates and PFS. Induced expression of immune checkpoint pathway-related genes (PD-1, PD-L1, PD-L2) by HMA, leading to ‘‘exhaustion’’ of T cells may be restored by concomitant PD-1/PD-L1 blockade. At the same time, frequently described evasion mechanisms to immune checkpoint blockade therapy such as MHC downregulation, decreased tumor antigen expression, and loss or decreased co-stimulatory ligand expression may be abrogated by the anti-tumor immunity enhancing effects of HMAs. Consistent with these preclinical observations, anti-PD-1/PD-L1 antibodies in combination with HMAs are being evaluated in clinical trials in patients with AML and MDS with early encouraging results. Efforts at optimization of study designs, development of double checkpoint combinations, identification of biomarkers of response to facilitate selection of patients best suited for these therapies, and identification and management of immune-mediated toxicities are ongoing, and will hopefully further improve outcomes.
References
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–42.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34.
Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–17.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23.
Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185–202.
Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–77.
Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin. Cancer Res. 2012;18:1611–8.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9.
Moskowitz C, Ribrag V, Michot JM, Martinelli G, Zinzani PL, Gutierrez M, et al. (editors). PD-1 blockade with the monoclonal antibody pembrolizumab (MK-3475) in patients with classical hodgkin lymphoma after brentuximab vedotin failure: Preliminary results from a phase 1b study (KEYNOTE-013). 56th ASH Annual Meeting and Exposition. San Francisco, CA; 2014.
Diefenbach CS, H F, David KA, et al. A phase I study with an expansion cohort of the combination of ipilimumab and nivolumab and brentuximab vedotin in patients with relapsed/refractory hodgkin lymphoma: A trial of the ECOG-ACRIN Cancer Research Group (E4412 Arms D and E). Blood. 2016;128:1106.
Falchi L, Sawas A, Deng C, Amengual JE, Colbourn DS, Lichtenstein E, et al. Rate of complete metabolic responses to immune checkpoint inhibitors in extremely heavily pre-treated patients with classical Hodgkin’s lymphoma and immunoepigenetic priming. J Clin Oncol. 2016;34(15_suppl):e19012–e.
Zinzani PL, Ribrag V, Moskowitz CH, Michot JM, Kuruvilla J, Balakumaran A, et al. Safety & tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood. 2017;130:267–70.
Nayak L, Iwamoto FM, LaCasce A, Mukundan S, Roemer MGM, Chapuy B, et al. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood. 2017;129:3071–3.
Kwong YL, Chan TSY, Tan D, Kim SJ, Poon LM, Mow B, et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood. 2017;129:2437–42.
Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14:3044–51.
Westin JR, Chu F, Zhang M, Fayad LE, Kwak LW, Fowler N, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014;15:69–77.
Davis TA, Grillo-López AJ, White CA, McLaughlin P, Czuczman MS, Link BK, et al. Rituximab anti-CD20 monoclonal antibody therapy in non-hodgkin’s lymphoma: Safety and efficacy of re-treatment. J Clin Oncol. 2000;18:3135–43.
Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: Preliminary results of a phase Ib study. J Clin Oncol. 2016;34:2698–704.
Vincent Ribrag DEA, Martinelli G, DJ Green, T Wise-Draper, JG Posada, Vij R, et al. Pembrolizumab monotherapy for relapsed/refractory multiple myeloma (rrmm): Phase 1b keynote-013 study. EHA. 23 Jun 2017;181631;2017.
San Miguel J, Mateos M, Shah JJ, Ocio EM, Rodriguez-Otero P, Reece D, et al. (editors). Pembrolizumab in combination with lenalidomide and low-dose dexamethasone for relapsed/refractory multiple myeloma (RRMM): Keynote-023. American Society of Hematology Annual Meeting; 57th annual meeting, Orlando, Florida 2015.
Badros A, Hyjek E, Ma N, Lesokhin A, Dogan A, Rapoport AP, et al. Pembrolizumab, pomalidomide and low dose dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;130:1189–97.
San Miguel J, Weisel K, Moreau P, Lacy M, Song K, Delforge M, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14:1055–66.
Dimopoulos MA, Palumbo A, Corradini P, Cavo M, Delforge M, Di Raimondo F, et al. Safety and efficacy of pomalidomide plus low-dose dexamethasone in STRATUS (MM-010): a phase 3b study in refractory multiple myeloma. Blood. 2016;128:497–503.
FDA alerts healthcare professionals and oncology clinical investigators about two clinical trials on hold evaluating KEYTRUDA® (pembrolizumab) in patients with multiple myeloma [press release]; 2017. https://www.fda.gov/Drugs/DrugSafety/ucm574305.htm.
FDA halts multiple immunotherapy trials [press release]; 2017. http://www.ajpb.com/news/fda-halts-multiple-immunotherapy-trials
FDA places holds on several durvalumab combination trials [press release]; 2017. http://www.onclive.com/web-exclusives/fda-places-holds-on-several-durvalumab-combination-trials
Daver N, Basu S, Garcia-Manero G, Cortes J, Ravandi F, Ning J, et al. Defining the immune checkpoint landscape in patients (pts) with acute myeloid leukemia. American Society of Hematology. Blood 2016;128:2900.
Rifca Le Dieu DCT, Ramsay AG, Mitter R, Miraki-Moud F, Fatah R, Lee AM, Lister TA, et al. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood. 2009;114:3909–16.
Vidriales MB, Orfao A, Lopez-Berges MC, Gonzalez M, Hernandez JM, Ciudad J, et al. Lymphoid subsets in acute myeloid leukemias: increased number of cells with NK phenotype and normal T-cell distribution. Ann Hematol. 1993;67:217–22.
Van den Hove LE, Vandenberghe P, Van Gool SW, Ceuppens JL, Demuynck H, Verhoef GE, et al. Peripheral blood lymphocyte subset shifts in patients with untreated hematological tumors: evidence for systemic activation of the T cell compartment. Leuk Res. 1998;22:175–84.
Schnorfeil FM, Emmerig K, Neitz JS, Beck B, Draenert R, Hiddemann W, et al. Pseudo-exhaustion of CD8 + T cells in AML. Blood. 2013;122:2615.
Tan J, Chen S, Xu L, Lu S, Zhang Y, Chen J, et al. Increasing frequency of T cell immunosuppressive receptor expression in CD4 + and CD8 + T cells may related to T cell exhaustion and immunosuppression in patients with AML. Blood. 2016;128:5166.
Le Dieu R, Taussig DC, Ramsay AG, Mitter R, Miraki-Moud F, Fatah R, et al. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood. 2009;114:3909–16.
Gorgun G, Holderried TA, Zahrieh D, Neuberg D, Gribben JG. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J Clin Invest. 2005;115:1797–805.
Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114:1545–52.
Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8 + T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117:4501–10.
Williams P, Basu S, Garcia-Manero G, Cortes JE, Ravandi F, Al-Hamal Z, et al. Checkpoint Expression By Acute MyeloidLeukemia (AML) and the Immune Microenvironment Suppresses Adaptive Immunity. American Society of Hematology. Abstract (617), Georgia World Congress Center, Georgia, Atlanta, USA 2016.
Chen X, Liu S, Wang L, Zhang W, Ji Y, Ma X. Clinical significance of B7-H1 (PD-L1) expression in human acute leukemia. Cancer Biol Ther. 2008;7:622–7.
Goltz D, Gevensleben H, Grunen S, Dietrich J, Kristiansen G, Landsberg J, et al. PD-L1 (CD274) promoter methylation predicts survival in patients with acute myeloid leukemia. Leukemia. 2017;31:738–43.
Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28:1280–8.
Isidori A, Salvestrini V, Ciciarello M, Loscocco F, Visani G, Parisi S, et al. The role of the immunosuppressive microenvironment in acute myeloid leukemia development and treatment. Expert Rev Hematol. 2014;7:807–18.
Orleans-Lindsay JK, Barber LD, Prentice HG, Lowdell MW. Acute myeloid leukaemia cells secrete a soluble factor that inhibits T and NK cell proliferation but not cytolytic function—implications for the adoptive immunotherapy of leukaemia. Clin Exp Immunol. 2001;126:403–11.
Ustun C, Miller JS, Munn DH, Weisdorf DJ, Blazar BR. Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood. 2011;118:5084–95.
Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41.
Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101:3722–9.
Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619–21.
Croitoru-Lamoury J, Lamoury FM, Caristo M, Suzuki K, Walker D, Takikawa O, et al. Interferon-gamma regulates the proliferation and differentiation of mesenchymal stem cells via activation of indoleamine 2,3 dioxygenase (IDO). PLoS ONE. 2011;6:e14698.
Corm S, Berthon C, Imbenotte M, Biggio V, Lhermitte M, Dupont C, et al. Indoleamine 2,3-dioxygenase activity of acute myeloid leukemia cells can be measured from patients’ sera by HPLC and is inducible by IFN-gamma. Leuk Res. 2009;33:490–4.
Yuan Y, Lu X, Tao CL, Chen X, Shao HW, Huang SL. Forced expression of indoleamine-2,3-dioxygenase in human umbilical cord-derived mesenchymal stem cells abolishes their anti-apoptotic effect on leukemia cell lines in vitro. Vitr Cell Dev Biol Anim. 2013;49:752–8.
Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25 + T regulatory cells. Blood. 2007;109:2871–7.
Schnorfeil FM, Lichtenegger FS, Emmerig K, Schlueter M, Neitz JS, Draenert R, et al. T cells are functionally not impaired in AML: increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J Hematol Oncol. 2015;8:93.
Wendelbo O, Nesthus I, Sjo M, Paulsen K, Ernst P, Bruserud O. Functional characterization of T lymphocytes derived from patients with acute myelogenous leukemia and chemotherapy-induced leukopenia. Cancer Immunol Immunother. 2004;53:740–7.
Lamble A, Kosaka Y, Huang F, Sasser K, Adams H, Tognon C, et al. Mass cytometry as a modality to identify candidates for immune checkpoint inhibitor therapy within acute myeloid leukemia. American Society of Hematology. Blood 2016;128:2829.
Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory receptor expression depends more dominantly on differentiation and activation than “Exhaustion” of human CD8 T cells. Front Immunol. 2013;4:455.
Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14:824–37.
Kitagawa M, Yamaguchi S, Takahashi M, Tanizawa T, Hirokawa K, Kamiyama R. Localization of Fas and Fas ligand in bone marrow cells demonstrating myelodysplasia. Leukemia. 1998;12:486–92.
Chen X, Eksioglu EA, Zhou J, Zhang L, Djeu J, Fortenbery N, et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J Clin Invest. 2013;123:4595–611.
Wei Y, Chen R, Dimicoli S, Bueso-Ramos C, Neuberg D, Pierce S, et al. Global H3K4me3 genome mapping reveals alterations of innate immunity signaling and overexpression of JMJD3 in human myelodysplastic syndrome CD34 + cells. Leukemia. 2013;27:2177–86.
Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–65.
Kotsianidis I, Bouchliou I, Nakou E, Spanoudakis E, Margaritis D, Christophoridou AV, et al. Kinetics, function and bone marrow trafficking of CD4 + CD25 + FOXP3 + regulatory T cells in myelodysplastic syndromes (MDS). Leukemia. 2009;23:510–8.
Kordasti SY, Ingram W, Hayden J, Darling D, Barber L, Afzali B, et al. CD4 + CD25high Foxp3 + regulatory T cells in myelodysplastic syndrome (MDS). Blood. 2007;110:847–50.
Epling-Burnette PK, Bai F, Painter JS, Rollison DE, Salih HR, Krusch M, et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood. 2007;109:4816–24.
Garcia-Manero G, Daver NG, Montalban-Bravo G, Jabbour EJ, DiNardo CD, Kornblau SM, et al. A phase II study evaluating the combination of nivolumab or ipilimumab with azacitidine in patients with previously treated or untreated myelodysplastic syndromes. American Society of Hematology; Blood 2016;128:344.
Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375:143–53.
Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget. 2013;4:2067–79.
Heninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol. 2015;6:29.
Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–98.
Srivastava P, Paluch BE, Matsuzaki J, James SR, Collamat-Lai G, Blagitko-Dorfs N, et al. Induction of cancer testis antigen expression in circulating acute myeloid leukemia blasts following hypomethylating agent monotherapy. Oncotarget. 2016;7:12840–56.
Coral S, Sigalotti L, Altomonte M, Engelsberg A, Colizzi F, Cattarossi I, et al. 5-aza-2’-deoxycytidine-induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications. Clin Cancer Res. 2002;8:2690–5.
Coral S, Sigalotti L, Gasparollo A, Cattarossi I, Visintin A, Cattelan A, et al. Prolonged upregulation of the expression of HLA class I antigens and costimulatory molecules on melanoma cells treated with 5-aza-2’-deoxycytidine (5-AZA-CdR). J Immunother. 1999;22:16–24.
Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, et al. Induction of a CD8 + T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010;116:1908–18.
Costantini B, Kordasti SY, Kulasekararaj AG, Jiang J, Seidl T, Abellan PP, et al. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica. 2013;98:1196–205.
Catakovic K, Klieser E, Neureiter D, Geisberger R. T cell exhaustion: from pathophysiological basics to tumor immunotherapy. Cell Commun Signal. 2017;15:1.
Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011;35:400–12.
Orskov AD, Treppendahl MB, Skovbo A, Holm MS, Friis LS, Hokland M, et al. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget. 2015;6:9612–26.
Perez-Gracia JL, Labiano S, Rodriguez-Ruiz ME, Sanmamed MF, Melero I. Orchestrating immune check-point blockade for cancer immunotherapy in combinations. Curr Opin Immunol. 2014;27:89–97.
Karpf AR, Peterson PW, Rawlins JT, Dalley BK, Yang Q, Albertsen H, et al. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc Natl Acad Sci USA. 1999;96:14007–12.
Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162:961–73.
Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2017;169:361.
Daver N, Basu S, Garcia-Manero G, Cortes JE, Ravandi F, Jabbour EJ, et al. Phase IB/II study of nivolumab in combination with azacytidine in patients with relapsed acute myeloid leukemia. American Society of Hematology (616); Blood. 2016;128:763.
Author contributions
ND, PB performed the research for the review, were involved in writing the paper and share co-authorship. GGM, SSY, JA, PS, HK involved in providing intellectual content, writing and reviewing the manuscript.
Funding
This manuscript was supported in part by the MD Anderson Cancer Center Leukemia Support Grant (CCSG) CA016672, the MD Anderson Cancer Center Leukemia SPORE CA100632, the Charif Souki Cancer Research Fund, and generous philanthropic contributions to the MD Anderson Moon Shots Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
ND has received research funding from BMS, Pfizer, Merck, and served as a consultant for BMS, Pfizer, and Celgene. HK has received research funding from BMS, Pfizer, and served as a consultant for Pfizer. PS and JA have served as consultants for BMS, EMD Serrono, and AstraZeneca. The remaining authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Daver, N., Boddu, P., Garcia-Manero, G. et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia 32, 1094–1105 (2018). https://doi.org/10.1038/s41375-018-0070-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0070-8
- Springer Nature Limited
This article is cited by
-
Identification of a novel monocyte/macrophage-related gene signature for predicting survival and immune response in acute myeloid leukemia
Scientific Reports (2024)
-
Comparing Arsenic-Containing Qinghuang Powder and Low-Intensity Chemotherapy in Elderly Patients with Acute Myeloid Leukemia
Chinese Journal of Integrative Medicine (2023)
-
Therapeutic Targets in Myelodysplastic Neoplasms: Beyond Hypomethylating Agents
Current Hematologic Malignancy Reports (2023)
-
Management of patients with lower-risk myelodysplastic syndromes
Blood Cancer Journal (2022)
-
Neoantigen reactive T cells correlate with the low mutational burden in hematological malignancies
Leukemia (2022)