
Abstract
Objective
To evaluate patterns of genetic testing among infants with CHD at a tertiary care center.
Study design
We conducted a retrospective observational cohort study of infants in the NICU with suspicion of a genetic disorder. 1075 of 7112 infants admitted to BCH had genetic evaluation including 329 with CHD and 746 without CHD. 284 of 525 infants with CHD admitted to CMHH had genetic evaluation. Patterns of testing and diagnoses were compared.
Results
The rate of diagnosis after testing was similar for infants with or without CHD (38% [121/318] vs. 36% [246/676], p = 0.14). In a multiple logistic regression, atrioventricular septal defects were most high associated with genetic diagnosis (odds ratio 29.99, 95% confidence interval 2.69–334.12, p < 0.001).
Conclusions
Infants with suspicion of a genetic disorder with CHD had similar rates of molecular diagnosis as those without CHD. These results support a role for genetic testing among NICU infants with CHD.
Similar content being viewed by others

Introduction
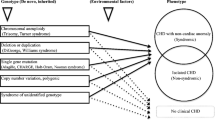
Congenital heart disease (CHD) comprises a broad spectrum of structural cardiac anomalies that account for nearly one-third of all major congenital anomalies and most infant deaths unrelated to infection [1,2,3,4]. Genetic variants play a significant role in the etiology of CHD: previous studies have described chromosomal aneuploidy in ~12% of people with CHD, pathogenic deletions or duplications in ~11%, and single gene disorders in 3–5% [5, 6]. However, many patients with CHD do not undergo genetic testing during infancy, reducing the ability of providers to consider genetic risk for comorbidities and complications when generating treatment plans. Developing a better understanding of which infants are most likely to have a molecular diagnosis can improve recommendations for genetic testing among infants with CHD, ultimately enabling personalized medicine to optimize infant health and long-term outcomes.
Evidence to guide the genetic evaluations of infants with critical illness is continuously evolving. For many infants, the recommendation of genetic testing is based on CHD type—for example, the utilization of multiplex PCR assay for infants with conotruncal defects due to the relatively high prevalence of 22q11.2 deletion syndrome [5, 7, 8]. Some centers employ a standardized genetic testing approach of karyotype and chromosomal microarray for first-tier evaluation of infants with CHD [9]. However, in larger cohorts of critically ill neonates who do not necessarily have CHD, approaches such as exome or genome sequencing (ES or GS) have led to molecular diagnostic rates ranging from 12–51% [10,11,12,13,14,15,16,17]. Infants with any CHD were a minority of those included in previous studies, and the use of clinical genetic testing for infants in this population is not standardized across institutions. Comparison of the molecular diagnostic yields of different genetic testing approaches between studies has proved challenging due to variation in phenotype, types of genetic testing available, and the method of evaluation for pathogenic variants. Furthermore, few studies have specifically explored the benefits of clinical ES or GS among infants with CHD [18].
To test our hypothesis that infants with CHD would have a molecular diagnostic rate similar to those reported among a general neonatal intensive care (NICU) population, we conducted a retrospective chart review of all infants admitted to the NICU at Boston Children’s Hospital (BCH) over a 10-year period, 2011–2021. Infants with CHD who underwent genetic evaluation at Children’s Memorial Hermann Hospital (CMHH) from 2013–2019 were also included to better capture the impact of genetic testing practices on diagnostic rates and provide external validation of the BCH results. We compared the patterns of genetic testing and molecular diagnoses among participants with and without CHD and determined which infant factors were associated with having a molecular diagnosis explaining their CHD identified during infancy.
Materials and methods
Cohort
The study was approved by the BCH and the University of Texas Health Science Center at Houston institutional review boards, and need for consent was waived for this retrospective study. The study was performed in accordance with the Declaration of Helsinki. No identifying information from an individual person is included in this manuscript. All infants admitted to the BCH NICU between 1 January 2011 and 31 October 2021 who had suspicion of a genetic disorder based on consultation of the genetics team were retrospectively identified in the research database. The data was divided into two cohorts: infants with CHD and without CHD. Infants with a prenatal or postnatal diagnosis of CHD at CMHH who were admitted to the NICU between 1 January 2013 and 30 March 2019 were retrospectively assessed for genetic testing.
Chart review and analysis
At BCH, demographics, clinical data such as genetic testing sent and results, and phenotypic information including presence of CHD, which was further subcategorized, and other congenital anomalies or phenotypic features were abstracted from the electronic medical records by at least two reviewers. Infants with cardiomyopathy or rhabdomyosarcoma without other structural CHD were excluded. From March 2017 to November 2018, rapid ES was available for qualifying infants with certain phenotypic characteristics via a research study [18]. CHD was not an automatically qualifying diagnosis, but some infants with CHD received rapid ES through the study due to other qualifying diagnoses such as the presence of multiple congenital anomalies or unexplained hypotonia. After the study period, rapid ES was routinely available on a clinical basis subject to institutional approval if sent while inpatient. At CMHH, retrospective chart review was performed on all admissions in the study period. CHD categories were similarly determined as at BCH, based on the agreement of two independent reviewers of the echocardiogram. Clinical data including extracardiac anomalies and neuroimaging were abstracted from the medical chart. Family history of genetic disorder as the indication for genetics consultation was determined from review of the initial genetics consultation note. Infants who had genetic testing sent only after NICU discharge were excluded from analysis. Deletions, duplications, or translocations identified on chromosomal microarray were considered a molecular diagnosis if they were previously associated with CHD or involved deletion of a known CHD gene. Regions of homozygosity identified by chromosomal microarray were not considered diagnostic, and none identified in this study overlapped the critical region for an imprinting disorder. ES/GS was available on a limited basis towards the end of the study period at CMHH.
Categorization
CHD types were categorized based on their anatomical phenotypes such as isolated septal defects, conotruncal, left or right-sided lesions, and laterality defects [19]. Presence of extracardiac anomalies in the following categories were assessed: retrognathia/micrognathia, omphalocele, gastroschisis, congenital diaphragmatic hernia, neural tube defect, congenital anomalies of the kidney and urinary tract, structural brain anomalies, esophageal atresia, and duodenal atresia. Renal findings of pelviectasis or hydronephrosis were not counted as congenital anomalies. Brain findings of intraventricular hemorrhage or simple cysts were not counted as congenital anomalies.
Statistical analysis and categorization
The eligible BCH cohort of 1075 infants, with 31% having CHD, provided 80% power to detect an odds ratio of at least 1.82. Analyses were completed in R version 4.2.2. Two-sided Fisher exact test was used to compare dichotomous or categorical outcomes. For continuous variables, the means of variables with normal distribution were compared using a student’s t-test while the means of variables with skewed distributions were compared using the Mann–Whitney test. A binomial multiple logistic regression model was used to identify infant characteristics associated with a molecular diagnosis using the R package stats. Adjusted odds ratios, confidence intervals, and likelihood ratio p-values were calculated using the epiDisplay package. All summary data is provided in manuscript tables. De-identified individual data and computational code is available upon email request.
Results
BCH cohort characteristics
Of the 7112 infants admitted to the BCH NICU during the study period, 1075 received a genetic evaluation of which 329 had CHD while 746 did not (Table 1). In all patients, indications for genetic evaluation were for diagnostic evaluation in setting of unexpectedly severe clinical presentation, multiple congenital anomalies, or features suggestive of a Mendelian genetic syndrome. In addition to the presence of CHD, facial dysmorphisms were the most common indication for genetics evaluation (119/269, 44%) among infants with CHD. Only eight infants with CHD who had genetic testing (3%) had a family history of a related genetic disorder as the indication for genetics consultation. Twelve percent (38/269) of infants with CHD neither a family history of a genetic disorder as their reason for genetics consultation nor another major congenital anomaly.
Infants with CHD were more likely to have multiple congenital anomalies (182/329 vs 107/746, p < 0.001), esophageal atresia (40/329 vs 15/746, p < 0.001), duodenal atresia (17/329 vs 8/746, p < 0.001), or congenital anomalies of the kidney and urinary tract (44/329 vs 56/746, p < 0.001) than those without CHD. There was no difference gestational age, birthweight, NICU admission duration, or mortality at 1-year between the CHD and non-CHD groups. Atrial and ventricular septal defects were the most prevalent types of CHD (33% and 21%, respectively).
Genetic testing at BCH
Testing was sent from the NICU with similar frequency in infants with or without CHD (269/329, 82% vs. 664/746, 86%; p = 0.06). Karyotype testing was more frequent in the CHD cohort (71/269 vs 109/644, p < 0.001). Chromosomal microarray was the most common test sent while in the NICU in the CHD cohort and non-CHD cohort (152/269, 56% and 334/746, 52%, respectively; Table 2). There was also an similar rate of molecular diagnosis among those who had genetic testing sent regardless of CHD status (121/318 CHD, 38%; 246/676 non-CHD, 36%; p = 0.14). Diagnosis rates were also similar among infants with CHD regardless of the presence of other congenital anomalies (14/38 isolated vs 98/231 non-isolated or familial, p = 0.60). The most common genetic diagnosis among infants with isolated CHD was 22q11 deletion (5/14, 36%). Karyotype was more likely to yield a molecular diagnosis in the CHD cohort (32/121 vs 21/246, p < 0.001), while gene panels were more likely to be diagnostic in the non-CHD cohort (52/246 non-CHD vs 7/121 CHD, p = 0.001). After removing the 29 infants with CHD and Trisomy 21, including 23 who had diagnosis by karyotype sent while in the NICU, there was no difference in karyotype yield between groups (9/92 CHD vs 21/246 non-CHD, p = 0.67).
Factors associated with molecular diagnosis among infants with CHD at BCH
In a multivariate model, there was no association between sex, or birthweight, and the likelihood of having a molecular diagnosis among the infants with CHD (Table 3). Diagnosis of atrioventricular septal defect or ventricular septal defect was associated with increased likelihood of a molecular diagnosis (odds ratio [OR] 29.99 (95% confidence interval [CI] 2.69–334.12, p < 0.001; OR 2.18, 95% CI 1.02–4.66, p = 0.042, respectively)). In analysis of the relationship between extracardiac anomalies and occurrence of molecular genetic diagnosis, esophageal atresia was associated with a decreased likelihood of having a molecular diagnosis (OR 0.11, 95% CI 0.02–0.06, p = 0.003). Within our cohort, karyotype testing was associated with an increased rate of molecular diagnosis (OR 3.67, 95% CI 1.41–9.54, p = 0.006), while molecular diagnoses were less frequent among infants who had gene panel testing (OR 0.25, 95% CI 0.10–0.68, p = 0.004) as part of their evaluation. When excluding the 29 infants with Trisomy 21 at BCH, gene panel testing and esophageal atresia remain associated with a decreased likelihood of a molecular genetic diagnosis (OR 0.31, 95% CI 0.11–0.87, p = 0.02; OR 0.07, 95% CI 0.01–0.60, p = 0.002, respectively), while VSD remains associated with an increased likelihood of a molecular genetic diagnosis (OR 2.38, 95% CI 1.07–5.31, p = 0.031). Assessment by karyotype and AVSD are no longer associated with a molecular diagnosis after excluding infants with T21.
Factors associated with diagnosis by CMA among infants in the combined CHD cohorts
Of the 525 infants with CHD admitted to the CMHH NICU, 284 had a genetic evaluation performed while in the NICU. Most genetic evaluations in infants with CHD at CMHH were with chromosomal microarray testing (271/284, 95%). Forty-six (17%) infants from CMHH received a molecular diagnosis from the chromosomal microarray. A multivariate model was used to determine the infant traits associated with molecular diagnosis by chromosomal microarray among the infants at CMHH and BCH who had chromosomal microarray testing (271 at UT, 152 at BCH). As with the multivariate model for any molecular diagnosis, there was no association with sex, birthweight, or infant mortality (Table 4). Further, no subtypes of CHD were associated with a difference in likelihood of molecular diagnosis by chromosomal microarray. Infants who died in the first year of life had a trend towards increased likelihood of molecular diagnosis (OR 6.84, 95% CI 0.92–51.08, p = 0.43). As with the BCH cohort, patients with esophageal atresia in the combined cohort were less likely to have a molecular diagnosis by chromosomal microarray (OR 0.08, 95% CI 0.01–0.75, p = 0.004).
Molecular diagnoses in combined CHD cohorts
An additional 10 infants at CMHH had a molecular diagnosis on a test other than chromosomal microarray: four with causal variants identified via single gene testing (3 CHD7, 1 EVC), two with causal variants identified by exome sequencing (1 CHD7 and 1 SMARCA4), two with Trisomy 21 diagnosed via karyotype, and two with unbalanced translocations found via karyotype. Among the 177 infants in the combined CHD cohort who had a molecular diagnosis, 48 (27%) had aneuploidies including 33 (69% of aneuploidies) with Trisomy 21, 5 (10% of aneuploidies) with Trisomy 13, 4 (8% of aneuploidies) with Trisomy 18, and two (4% of aneuploidies) with mosaic trisomies (Table 5). Eighty-six infants had deletions, duplications, or unbalanced translocations, including 36 with 22q11 deletion (52% of deletions). Single nucleotide variants were found in 40 infants, with CHD7 being the most affected gene, occurring in 14 infants (35% of single nucleotide variants). Among the other 26 infants with single nucleotide variants, only PTPN11 had variants in two infants. Finally, four infants had Beckwith–Wiedemann or Prader–Willi syndromes. Both Prader–Willi diagnoses were due to 15q11 deletions, while one Beckwith–Wiedemann diagnosis was due to a deletion and the other due to abnormal methylation patterns.
Discussion
Comprehensive genetic testing can have a significant impact on the health and long-term outcomes of infants with CHD by revealing an etiology for the CHD, helping care providers anticipate potential comorbidities, and ultimately enabling targeted therapies. We found that NICU infants with CHD are as likely to have a molecular diagnosis as infants without CHD who had a genetic consultation (121/318 vs 246/676, p = 0.14), despite the former group being enriched for congenital anomalies that are not commonly inclusion criteria for rapid genomic testing in the NICU when they occur in isolation [20, 21], such as esophageal atresia and micrognathia. Previous studies of neonates with CHD have also demonstrated that the prevalence of extracardiac anomalies, particularly those of the ear, nose, and throat or brain, are associated with an increased likelihood of receiving a molecular diagnosis after clinical genetic testing [22]. Karyotype and chromosomal microarray were the most common tests sent for infants with CHD during NICU admission. These findings confirmed our hypothesis that infants with CHD benefit from the approach to genetic testing that prioritizes use of rapid ES/GS which is becoming standard of care among a general NICU population, as has also been demonstrated in a recent study that focused on infants who underwent surgical repair for CHD [23]. Their cohort, though different regarding age and indication for hospital admission, also demonstrated a high rate of molecular diagnosis (42% overall).
Our multivariate analysis identified no particular subtype of CHD or extracardiac diagnosis as being associated with a molecular diagnosis. Across institutions, chromosomal microarray is one of the most utilized genetic tests due to its high diagnostic yield and relatively lower cost [24]. Previous studies have suggested that chromosomal microarray can provide a molecular diagnosis for 10–20% of children with CHD [19, 25, 26]. It is also known that the use of specific tests varies significantly across institutions, for example with 26–67% of infants undergoing surgical repair for CHD having a chromosomal microarray in one multi-institutional study [27]. However, in our multivariate model, chromosomal microarray was not positively associated with receiving a molecular diagnosis indicating that changes in utilization of genetic testing could improve overall diagnostic rates for infants with CHD, though a focus on improving the total proportion of infants with a molecular diagnosis would be most beneficial.
Of note, clinical ES became available during the study period at both of our institutions. 14% of infants with CHD and 15% of infants without CHD received a molecular diagnosis via ES. This finding aligns with previous recent research that found an incremental increase in diagnostic yield in infants with CHD who underwent testing with exome sequencing compared to chromosomal microarray [28]. Many of the larger deletions or duplications detected by chromosomal microarray would also be diagnosed by exome sequencing, often with a quicker time to result. In addition, only two out of the 48 exome diagnoses (variants in NOTCH1 and PTPN11) could have been found on the most common CHD panel [29]. Therefore, our results contribute to the growing body of evidence supporting the use of exome sequencing for NICU infants with CHD [29]. Furthermore, cost of testing and parental perspectives must be factored in when evaluating the use of exome/genome sequencing as part of the genetic evaluation of infants.
Our study is limited by the fact that the cohort reflects a level IV population that differs from the patient population of many NICUs. Infants with isolated CHD were a small proportion of those studied, so the diagnostic rates in that subset may not be generalizable to all infants with CHD in the NICU. In addition, the generalizability of our findings may be limited by the fact that our cohort consists of infants from only two centers and is an observational study in which the study interval overlapped a research study of rapid exome sequencing at BCH. Genetic testing practices at these centers were not standardized, and infants spent variable amounts of time in the NICU, which could have affected decisions regarding genetic consultations and testing. Prior studies have shown that variable testing modalities across different institutions can contribute towards significant differences in diagnostic yield [28]. Finally, our participants in the CHD and non-CHD subsets were not matched for clinical characteristics.
In conclusion, our study aimed to develop a better understanding of which infants are most likely to have a molecular diagnosis identified during early hospitalization to ultimately improve recommendations for genetic testing among infants with CHD. Genetic evaluations can optimize infant health and outcomes through changes in clinical care, targeted therapies, and early interventions. Our findings contribute to the growing body of evidence supporting the implementation of institutional genetic testing guidelines as well as utilization of newer sequencing methods in newborns with congenital heart defects to improve patient care. Our study has provided important findings that we hope contribute to the understanding of genetic knowledge in hopes of subsequently increasing access to testing.
Summary for social media if published
Congenital heart disease affects ~1% of infants. Most CHD is due to genetic risk. Of >1000 infants admitted to our NICU, 38% with CHD received a molecular diagnosis. Our results suggest that genetic testing should be considered for infants with CHD.
CHD affects ~1% of infants and is mostly genetic. Of >1000 infants, 38% with CHD received a genetic diagnosis. We suggest testing be considered for CHD.
Data availability
The datasets generated during and/or analysed during the current study are not publicly available due confidentiality but are available from the corresponding author on reasonable request.
References
Wu W, He J, Shao X. Incidence and mortality trend of congenital heart disease at the global, regional, and national level, 1990–2017. Medicine. 2020;99:e20593.
Giang KW, Mandalenakis Z, Fedchenko M, Eriksson P, Rosengren A, Norman M, et al. Congenital heart disease: changes in recorded birth prevalence and cardiac interventions over the past half-century in Sweden. Eur J Prev Cardiol. 2023;30:169–76.
Egbe A, Lee S, Ho D, Uppu S, Srivastava S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann Pediatr Cardiol. 2014;7:86.
Zimmerman MS. Global, regional, and national burden of congenital heart disease, 1990–2017: a systematic analysis for the Global Burden of Disease study 2017. Lancet Child Adolesc Health. 2020;4:185–200.
Pierpont ME, Brueckner M, Chung WK, Garg V, Lacro RV, McGuire AL, et al. Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation. 2018;138:e653–e711.
Morton SU, Quiat D, Seidman JG, Seidman CE. Genomic frontiers in congenital heart disease. Nat Rev Cardiol. 2022;19:26–42.
Peyvandi S, Lupo PJ, Garbarini J, Woyciechowski S, Edman S, Emanuel BS, et al. 22q11.2 deletions in patients with conotruncal defects: Data from 1,610 consecutive cases. Pediatr Cardiol. 2013;34:1687–94.
Vorstman JAS, Jalali GR, Rappaport AM, Scott C, Emanuel BS, et al. MLPA: a rapid, reliable, and sensitive method for detection and analysis of abnormalities of 22q. Hum Mutat. 2006;27:814–21.
Chaix MA. Genetic testing in congenital heart disease: a clinical approach. World J Cardiol. 2016;8:180.
Smith HS, Swint JM, Lalani SR, Otto MCO, Yamal J, Russell HV, et al. Exome sequencing compared with standard genetic tests for critically ill infants with suspected genetic conditions. Genet Med. 2020;22:1303–10.
Yang L, Wei Z, Chen X, Hu L, Peng X, Wang J, et al. Use of medical exome sequencing for identification of underlying genetic defects in <scp>NICU</scp>: experience in a cohort of 2303 neonates in China. Clin Genet. 2022;101:101–9.
Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, et al. Use of exome sequencing for infants in intensive care units. JAMA Pediatr. 2017;171:e173438.
Wang H, Xiao F, Dong X, Yulan L, Cheng G, Wang L, et al. Diagnostic and clinical utility of next‐generation sequencing in children born with multiple congenital anomalies in the China neonatal genomes project. Hum Mutat. 2021;42:434–44.
Wu B, Kang W, Wang Y, Zhuang D, Chen L, Pan X, et al. Application of full-spectrum rapid clinical genome sequencing improves diagnostic rate and clinical outcomes in critically Ill infants in the China neonatal genomes project. Crit Care Med. 2021;49:1674–83.
Lunke S, Eggers S, Wilson M, Patel C, Barnett C, Pinner J, et al. Feasibility of ultra-rapid exome sequencing in critically Ill infants and children with suspected monogenic conditions in the Australian Public Health Care system. JAMA. 2020;323:2503.
Dimmock D, Caylor S, Waldman B, Benson W, Ashburner C, Carmichael J, et al. Project Baby Bear: rapid precision care incorporating rWGS in 5 California children’s hospitals demonstrates improved clinical outcomes and reduced costs of care. Am J Hum Genet. 2021;108:1231–8.
Bowling KM, Thompson ML, Finnila CR, Hiatt SM, Latner DR, Amaral MD, et al. Genome sequencing as a first-line diagnostic test for hospitalized infants. Genet Med. 2022;24:851–61.
D’Gama AM, Del Rosario MC, Bresnahan MA, Yu TW, Wojcik MH, Agrawal PB. Integrating rapid exome sequencing into NICU clinical care after a pilot research study. NPJ Genom Med. 2022;7:51.
Findley TO, Crain AK, Mahajan S, Deniwar A, Davis J, Zavala AS, et al. Congenital heart defects and copy number variants associated with neurodevelopmental impairment. Am J Med Genet A. 2022;188:13–23.
Elliott AM, Souich C, Lehman A, Guella I, Evans DM, Candido T, et al. RAPIDOMICS: rapid genome-wide sequencing in a neonatal intensive care unit—successes and challenges. Eur J Pediatr. 2019;178:1207–18.
Williamson SL, Rasanayagam CN, Glover KJ, Baptista J, Naik S, Satodia P, et al. Rapid exome sequencing: revolutionises the management of acutely unwell neonates. Eur J Pediatr. 2021;180:3587–91.
Shikany AR, Landis BJ, Parrott A, Miller EM, Coyan A, Walters L. et al. A Comprehensive clinical genetics approach to critical congenital heart disease in infancy. J Pediatr. 2020;227:231–238.e14.
Durbin MD, Helvaty LR, Li M, Ware SM. A multicenter cross-sectional study in infants with congenital heart defects demonstrates high diagnostic yield of genetic testing but variable evaluation practices. Genet Med Open. 2023;1:100814.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64.
Gill K, Sasaki J, Jayakar P, Sosa L, Welch E. Chromosomal microarray detects genetic risks of neurodevelopmental disorders in newborns with congenital heart disease. Cardiol Young. 2021;31:1275–82.
Wu X, Li R, Fu F, Pan M, Han J, Yang X, et al. Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr. 2017;17:117.
Durbin MD, Fairman K, Helvaty LR, Huang M, Li M, Abreu D. et al. Genetic testing guidelines impact care in newborns with congenital heart defects. J Pediatr. 2023;260:113495.
Mone F, Eberhardt RY, Morris RK, Hurles ME, McMullan DJ, Maher ER, et al. COngenital heart disease and the diagnostic yield with exome sequencing (CODE) study: prospective cohort study and systematic review. Ultrasound Obstet Gynecol. 2021;57:43–51.
Paige SL, Saha P, Priest JR. Beyond gene panels: whole exome sequencing for diagnosis of congenital heart disease. Circ Genom Precis Med. 2018;11:e002097.
Acknowledgements
We would like to thank the infants, families, and providers in the BCH and UT NICUs.
Funding
SUM was supported by an American Heart Association Career Development Award and NIH/NHLBI R03HL150412 and K08HL157653. MW was supported by NIH/NICHD K23HD102589.
Author information
Authors and Affiliations
Contributions
TOF, PBA, MW and SUM conceptualized this work. EED, TOF, RH, ZSHK, RS, DS, AMD, HAF, MW and SUM participated in the acquisition and analysis of data. EED, TOF, RH, AMD, HAF, PBA, MW and SUM interpreted the data and results. EED, TOF, RH, ZSHD, AMD, PBA, MW and SUM drafted the manuscript. All authors critically reviewed the work for important intellectual content and approved the final version. Each author is accountable for their contribution to the manuscript, including related to the accuracy and integrity of the work.
Corresponding author
Ethics declarations
Competing interests
MW has received compensation for consultation to GeneDx, Illumina, and Sanofi. The other authors declare no competing financial interests in relation to the work described.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
D’Souza, E.E., Findley, T.O., Hu, R. et al. Genomic testing and molecular diagnosis among infants with congenital heart disease in the neonatal intensive care unit. J Perinatol 44, 1196–1202 (2024). https://doi.org/10.1038/s41372-024-01935-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41372-024-01935-1
- Springer Nature America, Inc.