
Abstract
Over the last few years, the complexity and diversity of gut microbiota within and across individuals has been detailed in relation to human health. Further, understanding of the bidirectional association between gut microbiota and metabolic disorders has highlighted a complimentary, yet crucial role for microbiota in the onset and progression of obesity-related cancers. While strategies for cancer prevention and cure are known to work efficiently when supported by healthy diet and lifestyle choices and physical activity, emerging evidence suggests that the complex interplay relating microbiota both to neoplastic and metabolic diseases could aid strategies for cancer treatment and outcomes. This review will explore the experimental and clinical grounds supporting the functional role of gut microbiota in the pathophysiology and progression of cancers in relation to obesity and its metabolic correlates. Therapeutic approaches aiding microbiota restoration in connection with cancer treatments will be discussed.
Similar content being viewed by others


Introduction
Noncommunicable diseases (NCDs) are responsible for over 70% of all deaths every year [1]. NCDs mainly include obesity and metabolic disorders, cardiovascular, and cerebrovascular diseases, as well as cancers. Overweight and obesity are estimated to cause 4.5 million deaths annually, and they significantly contribute to the cancer burden in North America, Europe, and the Middle East [2]. Indeed, an association between obesity and increased risk of cancer has long been recognized [3] and, according to an eminent US-based study, the proportion of cancer-related death attributed to obesity is 14% for men and 20% for woman [4]. Moreover, a dose- and time-response relationship links obesity to cancer risk [2, 4]. Growing awareness on the intricate relationship linking obesity and cancer to changes in microbiota has improved our understanding of the underlying causal mechanisms, and prompted investigations on microbiota manipulation to aid antineoplastic treatments. This review aims to explore the main findings supporting a functional role for gut microbiota in the pathophysiology and progression of cancers, with a special focus on obesity-associated neoplasms. Insights on therapeutic approaches aiding microbiota restoration in connection with cancer treatment will also be discussed.
Obesity-related low-grade inflammation and cancer risk
It is recognized that obesity increases susceptibility for 13 different cancers, with a potential association with 3 other tumors (Table 1) [5].
The underlying mechanisms are multiple and include pro-inflammatory processes generated by excessive white adipose tissue (WAT) expansion, which stimulates the secretion of cytokines and chemotactic factors [6]. These include platelet-derived growth factor, transforming growth factor-β, monocyte chemoattractant protein-1, interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α. Such pro-inflammatory mediators can activate endothelial precursor cells, immune cells and preadipocytes accompanying macrophage infiltration in stromal tissue and vascular bed of AT [6]. In the absence of adequate neo-angiogenetic support, AT undergoes hypoxia with overexpression of hypoxia-induced factor-α, adipocyte stress, and death. The inherent phenotypical switch in macrophage polarization, from the antinflammatory M2 to the pro-inflammatory M1 conformation, engages a process of phagocytosis of dead adipocytes in the context of a circle configuration termed crown-like structure [7, 8]. Lipolytic release of free fatty acids (FFAs) from entrapped adipocytes can activate toll-like receptor-4 on the macrophage plasma membrane and increase nuclear factor kappa B expression through activation of pro-inflammatory genes, like those coding for TNF-α, IL-1β, and cyclooxygenase-2 (COX-2) [9]. FFAs release is further stimulated by TNF-α and other cytokines sustaining inflammation and, in conjunction with angiogenesis, production of extracellular matrix. Leptin, a cognate product of adipocytes, is also capable of pro-inflammatory, proliferative, and proangiogenic actions through stimulation of TNF-α and IL-6 production. Conversely, the insulin-sensitizing hormone adiponectin is anti-inflammatory, antiproliferative, and antiangiogenic through activation of AMP-kinase [10].
The notion of inflammation as a tumor-promoting event involves an intricate grid of relations between innate resistance and acquired immunity. Obesity can impair innate and adaptative immunity with an increase of neutrophils, Th1 cells, CD8 cytotoxic T cells, natural killer (NK) cells and a decrease of regulatory T cells [11]. There is also an involvement of inflammasomes, i.e. cytoplasmic multi-protein complexes consisting of caspases able to activate several pro-inflammatory interleukins in response to endogenous or exogenous pathogen- or damage-associated molecular pattern. Inflammasome activation has been shown to be associated with insulin resistance [12].
Insulin resistance plays a key role in low-grade inflammation related to obesity and carcinogenesis. Chronic calorie excess promotes tissue desensitization to insulin with consequent insulin resistance. To achieve peripheral insulin actions, the pancreas responds with compensatory hyperinsulinemia, which has been associated with multiple cancers, like breast, endometrial, colonic, liver, esophageal, kidney, and pancreatic cancers [5]. The mitogenic effects of insulin signaling involve insulin receptor A and the proliferative pI3K/AKT/mTOR pathways [13]. Moreover, insulin can activate several downstream components that are instrumental for insulin-like growth factors (IGFs)-dependent mitogenic actions. Hyperinsulinemia increases liver production of IGF-I and, through downregulation of IGFs carriers, enhances local IGF-I bioavailability [14]. Stimulation of IGF-I receptor and hybrid insulin/IGFs receptors can elicit mitogenic, proliferative, and antiapoptotic actions in peripheral tissues.
Hormonal consequences of hyperinsulinemia encompass blunted liver production of sex hormone-binding globulin, which allows for increased local bioavailability of free sex hormone, together with increased androgen production from ovaries and adrenals [15]. In parallel, the aromatase activity of AT enhances estrogen production and further increases local bioavailability of unbound estrogens, with consequent increased risk of carcinogenetic effects of estrogens on estrogen-sensitive tissues. With regards to breast cancer, although obesity is inversely associated with progesterone receptor (PR)- and ER-positive premenopausal breast cancer, it is a recognized risk factor for postmenopausal hormone receptor-positive breast cancer. Hence, the risk of breast cancer is greater in postmenopausal women with obesity, especially for BMIs >35.0 kg/m2 [16]. A significant association has been highlighted between breast cancer risk and insulin and IGF-I levels. Moreover, breast cancer risk is associated with leptin levels, and mice with deficient peripheral leptin receptor exhibited a decreased mammary tumor growth and progression as compared to wild-type mice [17], while an increased tumor incidence and invasiveness were found in mammary tissues of mice with diet-induced obesity through transcriptional programming regulated by leptin [18]. In vitro studies further supported a role for leptin in cell proliferation, migration, angiogenesis of breast cancer stem cells, involving in a complex signaling network with Notch and IL-1 [19].
Evidence of a relationship between obesity and certain cancers of the digestive tract emerged in experimental and epidemiological studies. Insulin elicits a proliferative, antiapoptotic, and pro-inflammatory effects on cell lines, tumor tissue, and animal models of colorectal cancer (CRC) [20]. In CRC, the anticarcinogenetic effects of adiponectin [21], as well as the promitogenic effects of TNF-α, IL-6, IL‐13, and IL-1β have also been documented [22, 23]. At the epidemiological level, an obese BMI is exposed to an increased risk of early-onset CRC, with a significant age × BMI interaction observed for ages <50 years in both sexes [24]. With regards to hepatocellular carcinoma (HCC), its relation to obesity is essentially mediated by metabolic syndrome and nonalcoholic fatty liver disease (NAFLD) [25]. Insulin resistance amplifies de novo lipogenesis and increases the flux of FFAs, which leads to mitochondrial dysfunction with consequent oxidative stress, endoplasmic reticulum (ER) stress, and activation of unfolded protein response, all of which contribute to liver inflammation. Important contributors of increased HCC risk in obesity are represented by dysregulated adipokine secretion and amplified expression of signaling pathways associated with hepatocarcinogenesis, such as nuclear factor erythroid 2 related factor 1 (Nrf-1), NF-κB, mammalian target of rapamycin (mTOR), PI3K/phosphatase and tensin homolog (PTEN)/Akt, and Janus kinase/signal transducer and activator of transcription (JAK/STAT) [26]. Epidemiological studies and meta-analyses showed a 17% and 89% higher risk of HCC in the presence of overweight and obesity [27], respectively, and pinpointed a 25% increased risk of HCC for each 5 kg/m2 increase in BMI [28]. Finally, obesity and gastroesophageal reflux disease (GERD) are main risk factors for oesophageal adenocarcinoma (EAC). In epidemiological studies obesity is associated with EAC [28]. The link is particularly robust for abdominal obesity, Barrett’s oesophagus and EAC, and persists after adjusting for GERD [29]. In EAC, the IGF pathway seems to play a more dominant role than insulin, and associations have also been found between leptin/adiponectin, Barrett’s oesophagus and progression to EAC [30].
Intestinal microbiota
Microbiota encompass ~1014 microorganisms composed by bacteria, eukaryotes, viruses, and archaea positioned in body’s interfaces with the environment, i.e. skin, respiratory tract, gastrointestinal system, and urogenital apparatus [31]. Human microbiome is associated with systemic health in relation to substrate metabolism, energy balance, nervous and cardiovascular functions, immunity and inflammation, and it is influenced by early life events, feeding and dietary habits, growth processes, life-style, metabolic external and internal stressors factors, aging and diseases [32]. Gut microbiota contribute to absorption of dietary fats and fat-soluble vitamins, digestion of carbohydrates and polysaccharides, bile acid-related metabolism and fermentation of nondigestible food components that are ultimately metabolized in components capable of intercellular signaling such as short-chain fatty acids (SCFAs), trimethylamine-N-oxide, bile acids, incretin hormones, polyamines, polyphenols and vitamins [33]. In the gut, microbiota preserve the integrity and permeability of the intestinal epithelial barrier through tight junction proteins, a normal endocannabinoid system tone and lipopolysaccharide (LPS) detoxification by intestinal alkaline phosphatase [34]. LPS is a microorganism-derived pro-inflammatory component released in the colon upon death of gram-negative bacteria. High-fat diet is particularly effective in altering microbiota composition and favouring LPS absorption across the intestinal barrier through chylomicrons, thereby stimulating a pro-inflammatory condition [35].
With regards to energy states, gut microbiota is addressed as one of the leading factors accompanying and pathogenetically contributing to obesity and its metabolic associates, e.g. diabetes and dyslipidaemia, NAFLD, atherosclerosis and cardiovascular diseases, as well as kidney disorders [36]. Causal mechanisms involve regulation of energy extraction from nutrients, the ability of microorganisms to ferment undigested dietary polysaccharides generating SCFAs with their lipogenetic actions, the promotion of transcription factor-1 binding expression, and a decreased liver fatty acid oxidation [36]. In animal and human studies, main changes in gut microbial populations associated with obesity and metabolic diseases involve a low diversity in phyla and a rise in the Firmicutes:Bacteroidites ratio, which are partly reverted upon dietary/caloric manipulation [37]. The severity of obesity has an additional detrimental impact on gut microbiota, especially in terms of microbial gene richness, which is poorly improved after weight loss due to bariatric surgery, regardless of metabolic improvements [36]. Additional changes in gut microbiota composition have been related to age and gender. In elderly subjects, a lower number of anaerobic bacteria, including Bifidobacteria, and a higher number of enterobacteria, have been documented [38]. Age-related modifications of intestinal microbiota recognize multiple causes, such as tissue aging, pathophysiological abnormalities, alterations in taste and smell perception, gastritis and achlorhydria, and dietary modifications. Gender-specific differences in gut microbiota have also been documented, particularly in the Bacteroides-Prevotella microbial group, which are higher in males than females [39]. These gender differences are likely explained by actions of sex-related hormones on environment-microbiota interaction [40], as shown in experimental gonadectomy and subsequent sex steroid restoration [40, 41]. Noticeably, richness and diversity of gut microbiota affects systemic estrogens through activities of beta-glucuronidase and beta-glucurosidase enzymes, which are responsible of estrogen deconjugation and conjugation [42]. A switch in the enzymatic activities associated with the estrobolome, i.e. the entire genetic set of bacteria that process estrogens, can have an impact on the development of estrogen-dependent cancers.
Microbiota and cancer
Microbiota can influence the risk of cancer both directly, through dysbiotic actions in the developing cancer site, or indirectly, through predisposition to metabolic disorders and influence on sex hormone status [43]. In obesity, chronic pro-inflammatory state is linked to dysfunctional gut barrier and LPS leakage, which leads to metabolic endotoxaemia [33,34,35]. Subsequent exposure of peripheral tissues to immunogenic bacterial components can promote a pro-inflammatory status linked to the promotion of self-renewing tumor growth factors. Obesity, insulin resistance and unhealthy dietary factors can, therefore, act to impair commensal microbiota homeostasis, promote the release of growth factors, alter the immune surveillance, and stimulate cancer cell proliferation and invasion [44, 45]. The microbiota is separated from the host’s epithelial cells, which is important for regulating immune activity and supporting the host-microbe association. The mucus produced by the intestinal cells of the calyx, together with antimicrobial peptides produced by the Paneth cells, limit the interaction between the microbiota and the host’s immune system. Altering the intestinal barrier and functional biofilms that protect against pathogens invasion can contribute to enhancing the inflammation-mediated proliferative stimulus, hence creating a fitness interdependence between harmful microbes and cancer cells [46]. While cancer is considered a disease mainly caused by environmental and genetic factors, microorganisms are implicated in up to 20% of cases of human neoplasms [47]. Microbes can become part of the tumor microenvironment by influencing the growth and spread of cancer and several examples of an association between bacteria and carcinogenesis exist to date. A such, Helicobacter pylori (H. pylori) has been associated with noncardiac gastric cancer and lymphoma [48], and its virulence is reportedly connected with the A gene (CagA) associated with cytotoxin and the secretion of virulence factors to promote chronic inflammation, oxidative stress and host DNA damage [49]. Also relevant is Fusobacterium nucleatum, a member of oral microbiota, which expresses FadA that is capable of modulating the host’s E-cadherin and activating β-catenin, thus amplifying expression of transcription factors, oncogenes, Wnt genes and inflammatory genes, associated with CRC [50]. Another bacterial species associated with the carcinogenic process is Escherichia Coli (E. Coli), that expresses the pathogenicity island pks coding for the genotoxin colibactin, which alters p53 expression [51]. Further, Bacteroides fragilis produces the BFT toxin which activates the Wnt/β-catenin pathway and the nuclear-κB factor promoting cell proliferation, inducing the production of inflammatory mediators thus inducing carcinogenesis [52]. Moreover, the microbiota can induce carcinogens through metabolites that are capable of modulating inflammation, carcinogenesis, immune response, and DNA damage. Further, pancreatic cancer has been associated with oral dysbiosis mediated by Firmicutes, Proteobacteria, bacteria of the Cytophaga-Flavobacterium-Bacteroides group, and Actinobacteria [53]. Periodontitis has also been related to lung carcinogenesis [54].
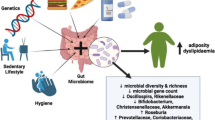
A schematic representation of the links between obesity, microbiota, and cancer is presented in Fig. 1.

NAFLD nonalcoholic fatty liver disease. Estrobolome: the aggregate of the enteric bacterial genes whose products are capable of metabolizing estrogens.
Among the obesity-associated cancers, a direct or indirect involvement for microbiota has been identified for several cancers that are followingly summarized.
Breast cancer
The human breast is not a sterile tissue, as it contains microorganisms organized in a specific microbiome niche which is distinct from that of overlying skin tissue [55]. This microbiome works to preserve healthy breast status both directly by inactivating harmful metabolic substances and indirectly via stimulation of resident immune response [56]. Although the bacterial composition of breast does not seem to vary between tumor tissue and adjacent normal tissue, both at the population and individual level [57], variations in abundance of E. Coli and other bacterial profiles have been documented between healthy and breast cancer women [57]. A discriminatory role for Methylobacterium abundance between normal and adjacent breast tumor tissue has also been reported, suggesting a potential local contribution of this bacterium in breast cancer development [58]. Gut dysbiosis associated with metabolic disorders has been hypothesized to influence the risk of postmenopausal breast cancer by altering enteric regulation of estrogen metabolism modulated by deconjugation reactions [59]. In this context, dietary components, such as fats, play a relevant role in promoting the overgrowth of Proteobacteria species, through the synthesis and excretion of bile acids, which can be catabolized by gut commensal bacteria into metabolites. Beta-glucuronidases synthesized by these organisms can deconjugate estrogens and increase circulating estrogens thereby contributing to modifications in the estrobolome [60]. Alternatively, the phytoestrogen enterolactone, which interacts with the gut microbiome and can be converted into enterolactone glucuronide has been recently associated with a protective role on breast cancer in postmenopausal women [61].
Colorectal cancer
A role for microorganisms in CRC development has long been known. Escherichia, Enterococcus, Bacteroides, and Clostridium genera influence the induction of aberrant crypt foci by 1,2-dimethylhydrazine in rats [62]. In humans, CRC-associated microbiota is different compared with healthy gut microbiota, in terms of species richness, abundance of protective species such as Roseburia, and increases in procarcinogenic species such as Bacteroides, Escherichia, Fusobacterium, and Porphyromonas [63]. CRC is associated with dysbiosis and a meta-analysis of metagenomic studies highlighted a direct association between CRC and seven enriched bacteria with enterotoxigenic activities encompassing, among the others, B. fragilis and Prevotella intermedia, as well as negative correlations with probiotic bacteria such as the butyrate-producer Clostridium butyicum and the lactic acid bacterium Streptococcus thermophiles [64]. Substantial changes in abundance of specific bacteria can be detected in patients with CRC and might serve as biomarkers for disease screening, prognostication and prediction of treatment response [65].
Hepatocellular carcinoma
In the liver, not containing a known microbiota, HCC has been linked to intestinal bacteria through metabolites [66] such as Pattern Recognition Receptors ligands, which recognize pathogen-associated molecular patterns (PAMPs), the latter involved in activating innate immune responses and protecting the host from infection. LPS, which belongs to a prototypical class of PAMPs, is capable of promoting liver carcinogenesis when bacteria cross the intestinal barrier and enter the systemic circulation [67]. Deoxycholic acid, a secondary bile acid, promotes the secretion of inflammatory cytokines in hepatic stellate cells, supporting the development of HCC [68]. Moreover, secondary bile acids, through the signaling of β-catenin, kinases 1 and 2 (ERK 1/2) regulated by the signaling of the activator of protein 1 (AP1), stimulate the invasiveness and proliferation of colon cancer cells [69]. They also damage the architecture and function of the colon epithelium through multiple oxidative DNA damage, inflammation and activation of the NF-κB pathway [70].
Prostate cancer
In healthy conditions, the prostate contains only a few microbial organisms, which remain indolent if the luminal epithelial barrier and antimicrobial prostate secretions are normal. Prostate disorders, impaired antimicrobial activity and loss of epithelial barrier integrity can promote the overgrowth of pathogenic urinary microbiome in the prostate ducts [71], enabling microorganisms to penetrate through the epithelial barrier and reach the stromal prostate component, where they promote local inflammation [71]. In chronic prostatitis/chronic pelvic pain syndrome, the urinary microbiome shows a higher representation of Clostridia and Bacteroides and a higher alpha-diversity, as compared to controls with higher prevalence of Bacilli bacteria such as Lactobacillus and Staphylococcus [72]. Similar alterations occur in seminal fluids of men with prostatitis, confirming the pro-inflammatory role of male urinary dysbiosis. While men with and without biopsy-proven prostate cancer showed no variation in the load and diversity of urinary microbiome, a greater representation of pro-inflammatory and pro-carcinogenetic bacteria, such as Streptococcus anginosus, Anaerococcus lactolyticus, A. obesiensis, Varibaculum cambriense and Propionimicrobium lymphophilum, was noticed in a subset of patients with prostate cancer in relation to cancer grading and coexistent prostate inflammation [73]. This pro-inflammatory microenvironment could facilitate the risk of chronic prostatitis and predispose to the occurrence and progression of prostate cancer [74]. Less clear is the involvement for gut dysbiosis in the risk of prostate cancer of persons with obesity and metabolic impairment. Alterations in sex steroids (e.g., decreased testosterone, elevated oestrogen levels), oxidized LDL cholesterol and low-grade inflammation are related to the progression of BPH and chronic prostate inflammation, and could promote a pro-inflammatory microenvironment with a higher risk of prostate cancer development [75].
Microbiota and antineoplastic treatments
Chemotherapy
Studies have focused on the ability of gut microbiota on influencing the course and/or outcome of different antineoplastic therapies, such as chemotherapy, radiotherapy, and immunotherapy. Several antitumor compounds have been studied in relation to the metabolic modifications generated by gut microbiota. However, only a few have been found to be directly metabolized by gut microbiota to date, such as the antimetabolite methotrexate, the radiation-sensitizer misonidazole, and the topoisomerase-I inhibitor irinotecan. This latter represents a parenteral anticancer therapy that is used for CRC. Its metabolism first consists of liver metabolism and subsequent biliary excretion into the gastrointestinal tract, where beta-glucuronidases of microbial organisms, particularly of Firmicutes phylum, exert a conversion into the active metabolite SN38, which is responsible for local mucosal toxicity associated with diarrhea [76]. This mechanism has been exploited to investigate the potential effect of antibiotics or specific glucuronidase inhibitors in animals, as well as of probiotics in humans to control irinotecan-induced diarrhea and gut toxicity, with partial benefits [77]. In addition to the actions of gut microbiota on anticancer drugs metabolism and absorption, an indirect influence of gut microbiota involves modification of cytochrome P450 gene expression that is associated with a faster metabolism of oral xenobiotics, suggesting an indirect role of gut microbiome in the modulation antineoplastic drug responses and susceptibility to adverse events [78]. A modulatory effect of gut microbiota has also been identified for gemcitabine through the action of E. Coli even if the underlying mechanisms remain unclear. Conversely, a synergic effect of gut microbiota with platinum-derived anticancer drugs, such as cisplatin and oxaliplatin, has been demonstrated on their ability to inhibit DNA replication and intra-strand platinum–DNA adducts, thereby preventing formation of DNA double-strand breaks [79]. In particular, the underlying mechanism could be explained by the evidence that gut microbiota-depleted mouse models have shown a decreasing of ROS-mediated effect of platinum-derived compounds on tumor cells, due to an altered activation of myeloid differentiation primary response 88-associated innate immune response [79]. Experimental evidence also exists that the response to the alkylating agent cyclophosphamide interacts with the gram-negative species Barnesiella intestinihominis (B. intestinihominis) to modulate infiltration of interferon-γ producing T cells [80]. This agent selectively impacts the diversity of gut microbiota and increases gram-positive bacteria such as Enterococcus hirae (E. hirae), which translocate into mesenteric lymph nodes and induce a significant increase of the intra-tumoral CD8 + T cell/T regulatory cell ratio as well as T-helper (TH) 17 and memory TH1 lymphocytes, thereby promoting an antineoplastic adaptive immune response in the spleen [80]. By reverse, synergistic actions of cyclophosphamide and gut microbiota are confirmed in mice depleted of Gram-positive bacteria [81]. An immune response related to the presence of B. intestinihominis and E. hirae has been associated with a more favorable prognosis in patients with advanced lung or ovarian cancer treated with chemo-immunotherapy [82].
Antineoplastic immunotherapy
Anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA4) immunotherapy induces a mucosal damage in the gut by compromising barrier integrity and facilitating the access of bacteria to the lamina propria, with activation of local innate immune response and subsequent systemic inflammation by translocated bacteria [83]. In particular, the antineoplastic immune response induced by anti-CTLA4 immunotherapy involves an anti-commensal effect against Burkholderiales and Bacteroidales (Bacteroides thetaiotaomicron and Bacteroides fragilis), which are responsible for gut mucosal disruption, translocation of bacterial species, and adjuvant support for antitumour immune response [84]. The relevance of this mechanism is supported by clinical evidence that patients with metastatic melanoma treated with ipilimumab exhibit greater survival and progression-free survival in relation to abundance of Fecalibacterium and other Firmicutes as compared to microbiota enriched in Bacteroides [85]. In melanoma patients, an association has also been identified between gut microbiota profile, particularly abundance of the Ruminococcaceae family, and response to treatment with anti-programmed cell death protein-1 (anti-PD1) antibodies [86]. This effect is corroborated in experimental melanoma models, where abundance and/or treatment with Bifidobacterium have been associated with anti-PD1 antitumor activity, supporting the possibility that this bacterial species represent an interesting adjuvant therapy to checkpoint inhibitors such as anti-PD1 drugs [87]. On the other side, antibiotic treatments have been found to significantly reduce the effectiveness of anti-PD1 immunotherapy in patients with advanced colorectal carcinoma and non-small cell lung cancer in terms of primary progressive disease, shorter overall survival, and progression-free survival [88].
Modification of microbiota through weight loss and cancer risk
Randomized controlled trials and intervention studies suggested that intentional weight loss, either through dietary modification and/or physical activity, are beneficial for cancer risk. A 3-year follow-up in a large female cohort found significant reductions in the risk of obesity-related cancer in women who intentionally lost more than 5% of their body weight [89]. In a meta-analysis on mortality risk among breast and CRC survivors, an inverse association with physical activity before cancer diagnosis was reported [90]. Conversely, observational studies showed that weight gain after breast cancer diagnosis was inversely associated with disease-free survival [91].
Bariatric surgery, the most effective treatment for sustainable weight loss in patients with obesity, elicits short and long-term weight-reducing effects due both to caloric restriction and modification of metabolic regulators acting at the gastrointestinal and central level [92]. Bariatric surgery has exhibited the ability to reduce the risk of obesity-related mortality and morbidities when compared with intensive medical and lifestyle interventions [93], as well as it is associated with an overall reduced incidence in cancer from different sites [94]. However, a site-specific cancer preventive effect of bariatric surgery seems to exist. For example, growing interest has focused on CRC risk after bariatric surgery and, while a systematic review and meta-analysis suggested a beneficial effect of bariatric surgery on CRC risk [95], a prospective study showed a time-dependent increase in CRC risk 10 years or more after bariatric surgery [96]. In line with this finding, a large case-control study using a propensity score-matching methodology found an elevated risk of CRC in people with obesity subjected to bariatric surgery [97]. In the case of hormone-related cancers, bariatric surgery may act favorable on cancer prevention. Particularly, a study in women with BMI > 35 kg/m2 found that bariatric surgery reduced the risk both of premenopausal and postmenopausal breast cancer, with an effect that was stronger among ER− premenopausal cases and ER+ postmenopausal cases [98], although data analysis robustness of this study has been questioned. To explain the previously reported discrepancies, it has been hypothesized that persistent gut dysbiosis could play a role. Bariatric surgery induces changes in gut microbiota diversity, with relative increases in Bacteroidetes, Proteobacteria, and Gammaproteobacteria, and a decrease in the abundance of Firmicutes [99, 100]. These functional and taxonomic changes could originate from postsurgical gastrointestinal rearrangements, as well as changes in eating habits and macronutrients consumption. Whether these changes promote a shift towards a lean microbiome phenotype is, however, uncertain. In fact, a study on changes of gut microbiota in obese persons subjected to bariatric surgery revealed an improved but still impaired postsurgical microbial diversity, which is associated with the risk of CRC [101]. Noticeably, postsurgical dietary modifications could even favor the production of harmful metabolites and increase the risk of DNA damage, leaky gut and inflammation, as exemplified by a study on gastric bypass showing an increase in bile acids, which can induce cytotoxic effects and the proliferation of malignant cells [102]. Hence, persistent intestinal dysbiosis, ongoing excess body weight after bariatric surgery, postsurgical modifications in diet composition and digestion, and exposure to proliferative agents like bile acid could play a role on carcinogenesis that awaits further clarification.
Microbiota manipulation and cancer
Prebiotics
Manipulation of intestinal microbiota through prebiotics has beneficial effects on their abundance and diversity, microbial production of SCFA, metabolic dysfunction, WAT accumulation, and immune functions [103]. Prebiotics are nutrients not easily digested by humans, which are capable of stimulating the growth and/or activity of beneficial bacterial species in the gut. Examples of prebiotics include fructans, inulin, fructoligosaccharides (FOS), galactoligosaccharides (GOS), and lactulose. Inulin modulates inflammation and metabolic endotoxemia, causes a relative intestinal increase in Bifidobacterium and Faecalibacterium prausnitzii, is negatively correlated with serum LPS and, through effects on gastrointestinal hormones like glucagon-like peptide-1 (GLP-1), peptide YY (PYY), and ghrelin, can influence food intake, glucose homeostasis and energy balance [104]. It has also been shown that oligofructose, like inulin, elicits adjuvant effects on cytotoxic treatment in cancer-bearing mice [105]. Interestingly, metformin has been found to promote the growth of Caenorhabditis elegans (C. Elegans), inhibiting the metabolism of methionine, and influencing the metabolism of folate [106]. Collectively, prebiotics show promising effects in preventing carcinogenesis by promoting growth and/or activity of health-promoting microorganisms.
Probiotics
Living microorganisms in a state of cryptobiosis confer health advantages for the host if administered in adequate quantities [107]. In experimental conditions, several Lactobacilli strains have shown the ability to attenuate diet-induced weight gain, improve insulin sensitivity and glucose homeostasis, reduce liver steatosis, and decrease pro-inflammatory macrophage infiltration in WAT and liver. In humans, bacterial strains like Lactobacillus acidophilus, Bifidobacterium lactis, and Akkermansia muciniphila (A. muciniphila), have displayed anti-obesity effects [108]. Immunomodulatory and anticancer activities of probiotics such as Lactobacilli (Lactobacillus casei, Lactobacillus plantarum, Lactobacillus rhamnosus GG) involve activation of NK cells or maturation of dendritic cells (DC) [109], or induction of ferrichrome, which promotes cancer cell apoptosis through JNK signaling pathway ([110]. In mice with HCC, a mixture of L. rhamnosus GG, E. Coli Nissle 1917 and VSL#3 shifted the gut microbial community toward beneficial bacteria like Prevotella and Oscillibacter, and exerted antiangiogenic, anti-inflammatory and antiproliferative effects [111]. Likewise, transfer of Bifidobacterium short or Bifidobacterium longum in mice lacking Bifidobacteriales has been shown to reduce melanoma growth and restore anti-melanoma CTL responses [87].
Fecal transplant
Fecal microbiota transplantation technique (FMT) involves the exchange of the intestinal microbiota between individuals and has been used to treat bacterial infections, particularly from Clostridium difficile. FMT is showing promising results for the treatment of obesity, metabolic syndrome and insulin resistance [112]. In terms of microbial composition, FMT recipients develop a relative abundance of Ruminococcus bromii and Roseburia intestinalis, or A. muciniphila species, all of which are associated with improvements in insulin sensitivity. However, while the movement of specific microbes improves insulin sensitivity, the nature of the microbiome manipulated by FMT appears to be unstable. Notwithstanding the advantage conferred by FMT on dysglycaemia in short-term term studies, i.e. lasting <6 weeks, effects reportedly vanish in long-term studies [113]. This could be due to the fact that obesity is associated with chronic low-grade inflammation, which in itself causes intestinal dysbiosis [114]. The rationale of FMT for cancer management includes reconstruction of intestinal microbiota, amelioration of bile acid metabolism, and modulation of immunotherapy efficacy [115]. In mice, FMT could alleviate the gut dysbiosis caused by the microbiota, and restore the eubiosis, thus reducing inflammation and proliferative and carcinogenic pathways [43]. Preclinical evidence of an advantageous cancer-related utilization of FMT involves CRC, pancreatic cancer, HCC, breast cancer, and melanoma [115]. Whether FMT can reduce carcinogenesis and tumor progression in humans remains a potential area of investigation.
Ketogenic diet
Excessive consumption of refined sugars elicits harmful effects on human health in terms of susceptibility to obesity, microbial diversity, and proliferation of pathogenic species, such as C. difficile and C. perfringens [116]. Moreover, refined sugars are an energy source for cancer cells [117]. In recent years, the low-calorie ketogenic diet (KD) approach is becoming increasingly popular for the treatment of cardiometabolic disorders and cancer. Low-calorie KD provides <800 kcal/day and a carbohydrate amount <30 g/day, consisting of about 13% of the total energy, while fats and proteins account for 44% and 43%, respectively [118]. KD is capable of producing physiological ketosis, i.e. an increase in ketone bodies, acetoacetate, and β-hydroxybutyrate [118]. Ketone bodies act as energy substrates, control mitochondrial metabolism and energy, have favorable effects on microbiota. KD show positive metabolic effects in obesity, in terms of oxidative stress, ROS/superoxide production, lipid peroxidation, protein oxidation, inflammation and immune intestinal cell function, and microbiota diversity. Recently, KD has proven able to decrease growth of cancer in animal models of malignant glioma, prostate cancer, CRC and gastrointestinal cancer, as well as in humans with brain or prostate cancer, although the inhomogeneous design of available studies warrants caution [119]. When used as an adjuvant therapy, KD is capable of sensitizing cancer cells to chemotherapy and radiotherapy treatments [120]. Microbiota modification induced by KD is thought to play a key role in some cancers and the crosstalk between different organs also via tumor suppressor metabolites. Like microbiota, KD is responsible for the production of SCFA, which can aid cancer treatment and cancer prevention. Therefore, KD and KD-induced microbiota could synergistically contribute to prevent tumorigenesis and constitute promising strategies to slow down carcinogenesis and increase the effectiveness of cancer therapies.
Conclusions
The symbiotic relationship between the microbial community and the host is not just an innocent bystander in metabolic alterations predisposing to cancer development. Dysbiosis is, both locally and systemically, an integral part of carcinogenesis that intervenes to modulate responsiveness and tolerance of antineoplastic therapies, particularly immunotherapy. Influencing the microbial community could aid the therapeutic approach to cancer management, in relation to reorganization of intestinal microbiota, improvement of harmful metabolites, antitumoral immune response, and modulation of anticancer therapy. A crucial step towards implementation of anticancer strategies is represented by exogenous approaches capable of modulating gut microbiota to improve intestinal dysbiosis and, potentially, influence cancer outcomes in different sites. Nevertheless, there is a critical need to translate emerging experimental evidence into robust clinical results, and to support the attained clinical findings with randomized controlled trial outcomes. This strategic course would not only help to recollect and consolidate current evidence to legitimate the validity of studies presented, but could also represent a utility prediction tool for microbiota manipulation in adjuvant therapeutic management of neoplastic disorders.
References
World Health Organization (WHO). Fact-sheets on cardiovascular disease. 2017. Available at: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
Arnold M, Leitzmann M, Freisling H, Bray F, Romieu I, Renehan A, et al. Obesity and cancer: an update of the global impact. Cancer Epidemiol. 2016;41:8–15.
Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Canc Inst. 1981;66:1191–308.
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38.
Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K, et al. Body Fatness and Cancer–Viewpoint of the IARC Working Group. N Engl J Med. 2016;375:794–8.
Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Investig. 2011;121:2094–101.
Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Investig. 2007;117:175–84.
Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55.
Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–9.
Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leuk Biol. 2000;68:437–46.
Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta. 2014;1842:446–62.
Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038–48.
Poloz Y, Stambolic V. Obesity and cancer, a case for insulin signaling. Cell Death Dis. 2015;6:e2037.
Clemmons DR, Underwood LE. Nutritional regulation of IGF-I and IGF binding proteins. Ann Rev Nutr. 1991;11:393–412.
Plymate SR, Matej LA, Jones RE, Friedl KE. Inhibition of sex hormone-binding globulin production in the human hepatoma (Hep G2) cell line by insulin and prolactin. J Clin Endocrinol Metabol. 1988;67:460–4.
Neuhouser ML, Aragaki AK, Prentice RL, Manson JE, Chlebowski R, Carty CL, et al. Overweight, obesity, and postmenopausal invasive breast cancer risk: a secondary analysis of the women’s health initiative randomized clinical trials. JAMA Oncol. 2015;1:611–21.
Park J, Kusminski CM, Chua SC, Scherer PE. Leptin receptor signaling supports cancer cell metabolism through suppression of mitochondrial respiration in vivo. Am J Pathol. 2010;177:3133–44.
Chang CC, Wu MJ, Yang JY, Camarillo IG, Chang CJ. Leptin-STAT3-G9a signaling promotes obesity-mediated breast cancer progression. Cancer Res. 2015;75:2375–86.
Lipsey CC, Harbuzariu A, Daley-Brown D, Gonzalez-Perez RR. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World J Methodol. 2016;6:43–55.
Gunter MJ, Leitzmann MF. Obesity and colorectal cancer: epidemiology, mechanisms and candidate genes. J Nutr Biochem. 2006;17:145–56.
Mutoh M, Teraoka N, Takasu S, Takahashi M, Onuma K, Yamamoto M, et al. Loss of adiponectin promotes intestinal carcinogenesis in Min and wild-type mice. Gastroenterology. 2011;140:2000–8.
Liu Z, Brooks RS, Ciappio ED, Kim SJ, Crott JW, Bennett G, et al. Diet-induced obesity elevates colonic TNF-alpha in mice and is accompanied by an activation of Wnt signaling: a mechanism for obesityassociated colorectal cancer. J Nutr Biochem. 2012;23:1207–13.
Matsui S, Okabayashi K, Tsuruta M, Shigeta K, Seishima R, Ishida T, et al. Interleukin-13 and its signaling pathway is associated with obesity-related colorectal tumorigenesis. Cancer Sci. 2019;110:2156–65.
Sanford NN, Giovannucci EL, Ahn C, Dee EC, Mahal BA. Obesity and younger versus older onset colorectal cancer in the United States, 1998-2017. J Gastrointest Oncol. 2020;11:121–6.
European Association for the Study of the Liver (EASL); European Association for the Study of547 Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64:1388–402.
Rajesh Y, Sarkar D. Molecular Mechanisms Regulating Obesity-Associated Hepatocellular Carcinoma. Cancers. 2020;12:1290.
Larsson SC, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer. 2007;97:1005–8.
Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–78.
Singh S, Sharma AN, Murad MH, Buttar NS, El-Serag HB, Katzka DA, et al. Central adiposity is associated with increased risk of esophageal inflammation, metaplasia, and adenocarcinoma: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2013;11:1399–412.
Rubenstein JH, Shaheen NJ. Epidemiology, diagnosis, and management of esophageal adenocarcinoma. Gastroenterology. 2015;149:302–17.e1.
Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, et al. An estimation of the number of cells in the human body. Ann Hum Biol. 2013;40:463–71.
Sommer F, Anderson JM, Bharti R, Raes J, Rosenstiel P. The resilience of the intestinal microbiota influences health and disease. Nature Rev Microbiol. 2017;15:630–8.
Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–7.
Everard A, Geurts L, Van Roye M, Delzenne NM, Cani PD. Tetrahydro Iso-alpha Acids from Hops Improve Glucose Homeostasis and Reduce Body Weight Gain and Metabolic Endotoxemia in High-fat Diet-fed Mice. PLoS ONE. 2012;7:e33858.
Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007;50:2374–83.
Marzullo P, Di Renzo L, Pugliese G, De Siena M, Barrea L, Muscogiuri G, et al. From obesity through gut microbiota to cardiovascular diseases: a dangerous journey. Int J Obes Suppl. 2020;10:35–49.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4.
Mitsuoka T, Hayakawa K. Die Faecalflora bei Menschen. I. Die Zusammensetzung der Faecalflora der verschiedenen Altergruppen [The fecal flora in man. I. Composition of the fecal flora of various age groups]. Zentralbl Bakteriol Orig A. 1973;223:333–42.
Mueller S, Saunier K, Hanisch C, Norin E, Alm L, Midtvedt T, et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol. 2006;72:1027–33.
Org E, Mehrabian M, Parks BW, Shipkova P, Liu X, Drake TA, Lusis AJ. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes. 2016;7:313–22.
Bolnick DI, Snowberg LK, Hirsch PE, Lauber CL, Org E, Parks B, et al. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat Commun. 2014;5:4500.
Fuhrman BJ, Feigelson HS, Flores R, Gail MH, Xu X, Ravel J, Goedert JJ. Associations of the fecal microbiome with urinary estrogens and estrogen metabolites in postmenopausal women. J Clin Endocrinol Metab. 2014;99:4632–40.
Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800–12.
Gérard P. Gut microbiota and obesity. Cell Mol Life Sci. 2016;73:147–62.
Lazar V, Ditu LM, Pircalabioru GG, Gheorghe I, Curutiu C, Holban AM, et al. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front Immunol. 2018;9:1830.
Yu LC. Microbiota dysbiosis and barrier dysfunction in inflammatory bowel disease and colorectal cancers: exploring a common ground hypothesis. J Biomed Sci. 2018;25:79.
de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–15.
Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1-241.
Wroblewski LE, Peek RM Jr. Helicobacter pylori in gastric carcinogenesis: mechanisms. Gastroenterol Clin North Am. 2013;42:285–98.
Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306.
Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes. 2014;5:675–80.
Sears CL, Geis AL, Housseau F. Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. J Clin Investig. 2014;124:4166–72.
Farrell JJ, Zhang L, Zhou H, Chia D, Elashoff D, Akin D, et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut. 2012;61:582–8.
Zeng XT, Xia LY, Zhang YG, Li S, Leng WD, Kwong JS. Periodontal disease and incident lung cancer risk: a meta-analysis of cohort studies. J Periodontol. 2016;87:1158–64.
Hieken TJ, Chen J, Hoskin TL, Walther-Antonio M, Johnson S, Ramaker S, et al. The microbiome of aseptically collected human breast tissue in benign and malignant disease. Sci Rep. 2016;6:30751.
Xuan C, Shamonki JM, Chung A, Dinome ML, Chung M, Sieling PA, et al. Microbial dysbiosis is associated with human breast cancer. PLoS One. 2014;9:e83744.
Urbaniak C, Gloor GB, Brackstone M, Scott L, Tangney M, Reid G. The microbiota of breast tissue and its association with breast cancer. Appl Environ Microbiol. 2016;82:5039–48.
Wang H, Altemus J, Niazi F, Green H, Calhoun BC, Sturgis C, et al. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget. 2017;8:88122–38.
Yang J, Tan Q, Fu Q, Zhou Y, Hu Y, Tang S, Zhou Y, et al. Gastrointestinal microbiome and breast cancer: correlations, mechanisms and potential clinical implications. Breast Cancer. 2017;24:220–8.
Shapira I, Sultan K, Lee A, Taioli E. Evolving concepts: how diet and the intestinal microbiome act as modulators of breast malignancy. ISRN Oncol. 2013;2013:693920.
Zaineddin AK, Vrieling A, Buck K, Becker S, Linseisen J, Flesch-Janys D, et al. Serum enterolactone and postmenopausal breast cancer risk by estrogen, progesterone and herceptin 2 receptor status. Int J Cancer. 2012;130:1401–10.
Onoue M, Kado S, Sakaitani Y, Uchida K, Morotomi M. Specific species of intestinal bacteria influence the induction of aberrant crypt foci by 1,2-dimethylhydrazine in rats. Cancer Lett. 1997;113:179–186.
Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66:70–78.
Dai Z, Coker OO, Nakatsu G, Wu WKK, Zhao L, Chen Z, et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome. 2018;6:70.
Wong SH, Yu J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat Rev Gastroenterol Hepatol. 2019;16:690–704.
Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–16.
Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology. 2010;52:1322–33.
Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101.
Pai R, Tarnawski AS, Tran T. Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell. 2004;15:2156–63.
Gadaleta RM, Oldenburg B, Willemsen EC, Spit M, Murzilli S, Salvatore L, et al. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-κB signaling in the intestine. Biochim Biophys Acta. 2011;1812:851–8.
Sfanos KS, Canene-Adams K, Hempel H, Yu S-H, Simons B, Schaeffer A, et al. Bacterial prostatitis enhances 2-amino-1-methyl-6-phenylimidazo[4,5-β]pyridine (PhIP)-induced cancer at multiple sites. Cancer Prev Res. 2015;8:683–92.
Shoskes DA, Altemus J, Polackwich AS, Tucky B, Wang H, Eng C. The urinary microbiome differs significantly between patients with chronic prostatitis/chronic pelvic pain syndrome and controls as well as between patients with different clinical phenotypes. Urology. 2016;92:26–32.
Shrestha E, White JR, Yu S-H, Kulac I, Ertunc O, De Marzo AM, et al. Profiling the urinary microbiome in men with positive versus negative biopsies for prostate cancer. J Urol. 2018;199:161–71.
Sfanos KS, Yegnasubramanian S, Nelson WG, De Marzo AM. The inflammatory microenvironment and microbiome in prostate cancer development. Nat Rev Urol. 2018;15:11–24.
Vignozzi L, Gacci M, Maggi M. Lower urinary tract symptoms, benign prostatic hyperplasia and metabolic syndrome. Nat Rev Urol. 2016;13:108–19.
Stringer AM, Gibson RJ, Logan RM, Bowen JM, Yeoh AS, Keefe DM. Faecal microflora and beta-glucuronidase expression are altered in an irinotecan-induced diarrhea model in rats. Cancer Biol Ther. 2008;7:1919–25.
Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–5.
Kang MJ, Kim HG, Kim JS, Oh DG, Um YJ, Seo CS, et al. The effect of gut microbiota on drug metabolism. Expert Opin Drug Metab Toxicol. 2013;9:1295–308.
Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–70.
Daillère R, Vétizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, et al. Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity. 2016;45:931–43.
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–6.
Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature. 2010;467:967–71.
Zitvogel L, Ayyoub M, Routy B, Kroemer G. Microbiome and anticancer immunosurveillance. Cell. 2016;165:276–87.
Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–84.
Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28:1368–79.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–9. https://doi.org/10.1126/science.aac4255.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97.
Luo J, Hendryx M, Manson JE, Figueiredo JC, LaBlanc ES, Barrington W, et al. Intentional weight loss and obesity-related cancer risk. JNCI Cancer Spectr. 2019;3:pkz054.
Schmid D, Leitzmann MF. Association between physical activity and mortality among breast cancer and colorectal cancer survivors: a systematic review and metanalysis. Ann Oncol. 2014;25:1293–311.
Vance V, Mourtzakis M, McCargar L, Hanning R. Weight gain in breast cancer survivors: prevalence, pattern and health consequences. Obes Rev. 2011;12:282–294.
Belligoli A, Bettini S, Busetto L. Bariatric surgery: Is a matter of cutting calories or cutting metabolic regulators? Curr Opin Endocr Metab Res. 2019;4:83–8.
Carlsson LM, Peltonen M, Ahlin S, Anveden A, Bouchard C, Carlsson B, et al. Bariatric surgery and prevention of type 2 diabetes in Swedish obese subjects. N Engl J Med. 2012;367:695–704.
Sjöström L, Gummesson A, Sjöström CD, Narbro K, Peltonen M, Wedel H, et al. Swedish Obese Subjects Study. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects study): a prospective, controlled intervention trial. Lancet Oncol. 2009;10:653–62.
Afshar S, Kelly SB, Seymour K, Lara J, Woodcock S, Mathers JC. The effects of bariatric surgery on colorectal cancer risk: systematic review and meta-analysis. Obes Surg. 2014;24:1793–9.
Derogar M, Hull MA, Kant P, Östlund M, Lu Y, Lagergren J. Increased risk of colorectal cancer after obesity surgery. Ann Surg. 2013;258:983–8.
Mackenzie H, Markar SR, Askari A, Faiz O, Hull M, Purkayastha S, et al. Obesity surgery and risk of cancer. Br J Surg. 2018;105:1650–7.
Feigelson HS, Caan B, Weinmann S, Leonard AC, Powers JD, Yenumula PR. et al. Bariatric surgery is associated with reduced risk of breast cancer in both premenopausal and postmenopausal women. Ann Surg. 2020;272:1053–1059. https://doi.org/10.1097/SLA.0000000000003331.
Tremaroli V, Karlsson F, Werling M, Stahlman M, Kovatcheva-Datchary P, Olbers T, et al. Roux-en-Y gastric bypass and vertical banded gastroplasty induce long-term changes on the human gut microbiome contributing to fat mass regulation. Cell Metabolism. 2015;22:228–38.
Palleja A, Kashani A, Allin KH, Nielsen T, Zhang C, Li Y, et al. Roux-en-Y GAstric Bypass Surgery of Morbidly Obese Patients Induces Swift and Persistent Changes of the Individual Gut Microbiota. Genome Med. 2016;8:67.
Aron-Wisnewsky J, Prifti E, Belda E, Ichou F, Kayser BD, Dao MC, et al. Major microbiota dysbiosis in severe obesity: fate after bariatric surgery. Gut. 2019;68:70–82.
Tarashi S, Siadat SD, Ahmadi Badi S, Zali M, Biassoni R, Ponzoni M, et al. Gut bacteria and their metabolites: which one is the defendant for colorectal cancer? Microorganisms. 2019;7:561.
Scott KP, Antoine JM, Midtvedt T, van Hemert S. Manipulating the gut microbiota to maintain health and treat disease. Microb Ecol Health Dis. 2015;26:25877.
Bodnaruc AM, Prud’homme D, Blanchet R, Giroux I. Nutritional modulation of endogenous glucagon-like peptide-1 secretion: a review. Nutr Metab. 2016;13:92.
Taper HS, Roberfroid MB. Nontoxic potentiation of cancer chemotherapy by dietary oligofructose or inulin. Nutr Cancer. 2000;38:1–5.
Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cochemé HM, Noori T, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–39. https://doi.org/10.1016/j.cell.2013.02.035
Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11:506–14.
Gomes AC, de Sousa RG, Botelho PB, Gomes TL, Prada PO, Mota JF. The additional effects of a probiotic mix on abdominal adiposity and antioxidant status: a double-blind, randomized trial. Obesity. 2017;25:30–38.
Cai S, Kandasamy M, Rahmat JN, Tham SM, Bay BH, Lee YK, et al. Lactobacillus rhamnosus GG ACtivation of Dendritic Cells and Neutrophils Depends on the Dose and Time of Exposure. J Immunol Res. 2016;2016:7402760.
Konishi H, Fujiya M, Tanaka H, Ueno N, Moriichi K, Sasajima J, et al. Probiotic-derived ferrichrome inhibits colon cancer progression via JNK-mediated apoptosis. Nat Commun. 2016;7:12365.
Li J, Sung CY, Lee N, Ni Y, Pihlajamäki J, Panagiotou G, et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc Natl Acad Sci USA. 2016;113:E1306–15.
Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–6.e7.
Kootte RS, Levin E, Salojärvi J, Smits LP, Hartstra AV, Udayappan SD, et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab. 2017;26:611–9.e6.
Li J, Sung CY, Lee N, Ni Y, Pihlajamäki J, Panagiotou G, et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc Natl Acad Sci USA. 2016;113:E1306–15.
Chen D, Wu J, Jin D, Wang B, Cao H. Fecal microbiota transplantation in cancer management: current status and perspectives. Int J Cancer. 2019;145:2021–31.
Sinh P, Barrett TA, Yun L. Clostridium difficile Infection and Inflammatory Bowel Disease: A Review. Gastroenterol Res Pract. 2011;2011:136064.
Healy ME, Lahiri S, Hargett SR, Chow JD, Byrne FL, Breen DS, et al. Dietary sugar intake increases liver tumor incidence in female mice. Sci Rep. 2016;6:22292.
Caprio M, Infante M, Moriconi E, Armani A, Fabbri A, Mantovani G, et al. Very-low-calorie ketogenic diet (VLCKD) in the management of metabolic diseases: systematic review and consensus statement from the Italian Society of Endocrinology (SIE). J Endocrinol Investig. 2019;42:1365–86.
Chung HY, Park YK. Rationale, feasibility and acceptability of ketogenic diet for cancer treatment. J Cancer Prev. 2017;22:127–34.
Seyfried TN, Kiebish MA, Marsh J, Shelton LM, Huysentruyt LC, Mukherjee P. Metabolic management of brain cancer. Biochim Biophys Acta. 2011;1807:577–94.
Acknowledgements
Obesity Programs of nutrition, Education, Research and Assessment (OPERA) group members served as collaborators and approved the final version of the paper: Annamaria Colao, Carlo Alviggi, Sara Aprano, Rocco Barazzoni, Luigi Barrea, Francesco Beguinot, Annamaria Belfiore, Giuseppe Bellastella, Silvia Bettini, Giuseppe Bifulco, Maurizio Bifulco, Caterina Brasacchio, Filomena Bottiglieri, Luca Busetto, Brunella Capaldo, Massimiliano Caprio, Felipe Casanueva, Luigi Di Luigi, Andrea Di Nisio, Laura Di Renzo, Carolina Di Somma, Lorenzo Maria Donini, Katherine Esposito, Massimo Federici, Dario Giugliano, Lucio Gnessi, Gianluca Gortan Cappellari, Brunella Guida, Maria Angela Guzzardi, Daniela Laudisio, Andrea Lenzi, Alessia Liccardi, Carla Lubrano, Paolo Emidio Macchia, Silvia Magno, Paolo Marzullo, Davide Menafra, Silvia Migliaccio, Fabrizio Muratori, Giovanna Muscogiuri, Raffaele Napoli, Caterina Pelosini, Francesca Pivari, Rosario Pivonello, Eleonora Poggiogalle, Gabriella Pugliese, Gabriele Riccardi, Alberto Ritieni, Fiammetta Romano, Domenico Salvatore, Alessandro Sanduzzi, Ferruccio Santini, Silvia Savastano, Paolo Sbraccia, Giovanni Scambia Laura Soldati, Giovanni Spera, Maria Grazia Tarsitano, Dario Tuccinardi, Olga Vaccaro, Mary Venneri, Samir Sukkar, Roberto Vettor.
Funding
This article is published as part of a supplement funded by the scientific assistance of Panta Rei Impresa Sociale srl (https://www.panta-rei.eu/pantarei/).
Author information
Authors and Affiliations
Consortia
Contributions
The authors’ responsibilities were as follows: PM, SB, DM, and SA concept of this paper and drafted the manuscript. GM, LB, SS, and AC revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A full list of author and their affiliation appear at the end of the paper.
Rights and permissions
About this article

Cite this article
Marzullo, P., Bettini, S., Menafra, D. et al. Spot-light on microbiota in obesity and cancer. Int J Obes 45, 2291–2299 (2021). https://doi.org/10.1038/s41366-021-00866-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-021-00866-7
- Springer Nature Limited