
Abstract
Background
Fat mass and lean mass are two biggest components of body mass. Both fat mass and lean mass are under strong genetic determinants and are correlated.
Methods
We performed a bivariate genome-wide association meta-analysis of (lean adjusted) leg fat mass and (fat adjusted) leg lean mass in 12,517 subjects from 6 samples, and followed by in silico replication in large-scale UK biobank cohort sample (N = 370 097).
Results
We identified four loci that were significant at the genome-wide significance (GWS, α = 5.0 × 10−8) level at the discovery meta-analysis, and successfully replicated in the replication sample: 2q36.3 (rs1024137, pdiscovery = 3.32 × 10−8, preplication = 4.07 × 10−13), 5q13.1 (rs4976033, pdiscovery = 1.93 × 10−9, preplication = 6.35 × 10−7), 12q24.31 (rs4765528, pdiscovery = 7.19 × 10−12, preplication = 1.88 × 10−11) and 18q21.32 (rs371326986, pdiscovery = 9.04 × 10−9, preplication = 2.35 × 10−95). The above four pleiotropic loci may play a pleiotropic role for fat mass and lean mass development.
Conclusions
Our findings further enhance the understanding of the genetic association between fat mass and lean mass and provide a new theoretical basis for their understanding.
Similar content being viewed by others


Introduction
Obesity is a chronic metabolic disease, characterized by accumulation of body fat tissue [1]. It is a serious public health problem and the age-adjusted estimates of global obesity prevalence was reported to be 30% for men and 35% for women [2, 3]. Body mass index (BMI), defined as body weight divided by height squared, is the currently standard measure of obesity. But it is not an ideal phenotype to measure obesity. The reason is that body mass is a composite combination of various types of body composition, including bone mass, fat mass, lean mass and other soft tissues. Among them, lean mass takes up the majority of body mass. But it is the fat mass that is responsible for obesity [4]. Leg fat mass (LFM), composed of only subcutaneous fat mass, is one of the primary independent determinants of resting energy expenditure [5]. Compared to other fat compartments, LFM may be ‘metabolically protective’ and may mitigate insulin resistance [6]. According to a previous study [7], greater subcutaneous thigh adipose tissue was associated with a lower prevalence of the metabolic syndrome in obese men and women. Van et al. [8] suggested that the independent association of LFM with risk factors for insulin resistance and dyslipidemia exists in both women and men. Health ABC conducted a separate assessment of older men and women, indicated that there was a favorable association of subcutaneous LFM with glucose and lipid concentrations in both sexes [9].
Lean body mass (LBM) is a vital component in metabolism and ambulation, which is significantly correlated to basal metabolic rate [10]. LBM have a considerable heritability ranging from 52 to 84% [11]. Leg lean mass (LLM) is part of the appendicular lean mass. Probably because of the load bearing function, legs have greater lean mass than arms. LLM is a reliable proxy measure of muscle strength [12]. The heritability of LLM is reported to be up to 69% [13]. Lower LLM is associated with mobility disability in older men and women [14]. Venturelli et al. [15] reported a strong positive correlation between muscle telomere length measured in muscle biopsies by quantitative polymerase chain reaction [6] and LLM.
Fat mass and lean mass have important implications for health and physical functioning. From a biological perspective the fat mass and lean mass may have their own distinct developmental mechanisms that may overlap with each other. Fat mass is associated with metabolic syndrome, insulin resistance and dyslipidemia [7, 8], and loss of muscle mass may have an impact on the body’s ability to metabolise glucose [16]. The prevalence of muscle loss has been estimated to be between 15 and 25% in those under 70-years [17]. Women experience an increase in weight until their mid 50s, with an earlier loss of muscle mass, followed by a subsequent loss of fat mass [18].
In this study, aiming to identify pleiotropic loci for fat mass and lean mass, we performed a bivariate genome-wide association meta-analysis of LFM and LLM in 12,517 subjects from six samples. We then replicated the results in the large-scale UK Biobank (UKB) cohort.
Materials and methods
Discovery sample
We collected six GWAS samples for meta-analysis. Three samples were from the in-house studies, the other three were accessed through the database of genotype and phenotype (dbGAP). All samples have been approved by their respective institutional ethics review boards, and all participants provided written informed consent before entering the study. Details of the samples have been described previously [19]. Briefly, the first sample comprised 878 unrelated subjects of European ancestry form the Omaha Osteoporosis Study. The second sample comprised 2219 unrelated subjects of European ancestry from the Kansas City Osteoporosis Study. And the third sample comprised 1541 unrelated subjects of Chinese Han ancestry form the China Osteoporosis Study (COS). The fourth and fifth samples were from the Women’s health initiative (WHI) observational study. The fourth sample comprised 445 subjects of Hispanic ancestry (WHI-HIS), and the fifth sample comprised 847 subjects of African-American ancestry (WHI-AA). The sixth sample was from the Framingham heart study (FHS). FHS is a longitudinal and prospective cohort comprising >16,000 participants spanning three generations of European ancestry. We identified a total of 6 587 genotyped and phenotyped FHS participants, of which 693, 2749, and 3145 were from the original, offspring, and third generations, respectively.
Phenotype measurements
LFM and LLM were measured by dual-energy X-ray absorptiometry (DEXA, Lunar Corp., Madison, WI, USA or Hologic Inc., Bedford, MA, USA), according to the manufacture’ protocol. In the FHS sample, LLM was quantified by subtracting LFM from leg soft tissue mass.
Both LFM and LLM were adjusted for significant covariates among gender, age, age squared, height, height squared and the first five principal components derived from genome-wide genotype data. In addition, LFM was adjusted by LLM, and LLM was adjusted by LFM. A step-wise linear regression model was used to screen significant covariates using the R function stepAIC. The residuals were normalized by the inverse quantiles of standard normal distribution.
Genotyping and quality control
According to the manufacturer’s protocol, all GWAS samples were genotyped by high-throughput SNP genotyping arrays (Affymetrix Inc, Santa Clara, CA, USA; or Illumina Inc., San Diego, CA, USA within individual samples). Strict quality control (QC) was implemented at both the individual and SNP level within each sample. At the individual level, PLINK [20] was used to check the individual’s sex based on genotype data from X-chromosome, and compared to the sex recorded in the questionnaire. Individuals whose imputed sex was ambiguous or inconsistent with the reported sex were removed. At the SNP level, SNPs that violates the Hardy–Weinberg Equilibrium rule (p < 1.0 × 10−5) were removed. Genotype-derived principle components were used to monitor population outliers and outliers were removed. In the familial sample FHS, genotypes presenting the Mendel error were set to missing.
Genotype imputation
Each individual GWAS sample was imputed into the 1000 genomes project phase 3 sequence variants (as of May 2013) [21]. Haplotypes representing 170 individuals of admixed American ancestry, 240 individuals of European ancestry, 244 of East Asian ancestry and 319 of African ancestry, were downloaded from the project website. The haplotypes of bi-allelic variants, including SNPs and bi-allelic insertions/deletions (indels), were extracted to form reference panel for imputation. Variants with zero or one copy of minor alleles were removed in QC procedures.
Each GWAS sample was imputed by the respective reference panel of the closest ancestry. Chi-square test was used to examine the consistency of allele frequency between the GWAS sample and the reference sample before imputation. GWAS SNPs which failed the consistency test (p < 1.0 × 10−6) were transformed into the inverse strand to correct potential mis-strandedness. SNPs that again failed the consistency test were removed from the GWAS sample. We used the fast and accurate diploid genotype imputation algorithm FISH [22] to perform imputation.
Association analysis in individual studies
In each GWAS sample, the association between normalized phenotype residuals and genotyped and imputed genotypes was tested under an additive inheritance mode. Both univariate and bivariate association tests were examined. The univariate/bivariate linear regression model was used to examine the association in unrelated samples. In the familial FHS sample, the univariate/bivariate mixed linear models accounting for genetic relatedness within each pedigree were used [23, 24].
Univariate association signals of the FHS sample were validated by the method of Chen and Abecasis [25] in the software MERLIN.
Meta-analysis
Summary association statistics from individual GWAS samples were combined to perform univariate/bivariate meta-analysis. As QC steps, only well imputed, which was defined as SNPs having imputation certainty measure r2 > 0.3 in at least two of the six samples, and common (minor allele frequency, MAF > 0.05 in the European population) SNPs were included.
Univariate and bivariate meta-analysis were examined by the fixed-effects model [26]. For a particular SNP, let (β1i, β2i) be the vector of regression coefficient for the two traits in the ith study (i = 1,…, n, n = 6), \({\mathbf{V}}_i = \left[ {\begin{array}{*{20}{c}} {v_{11}} & {v_{12}} \\ {v_{21}} & {v_{22}} \end{array}} \right]\) be the corresponding symmetric variance-covariance matrix for the two regression coefficients. The β1i, β2i and Vi are obtained from individual study analysis. Define the data structure as follows:
B = (β11, β21, β12, β22, ···, β1n, β2n)‘2n×1
\({\mathbf{X}} = \left[ {\begin{array}{*{20}{c}} {1\;\;0} \\ {0\;\;1} \\ \ldots \\ {1\;\;0} \\ {0\;\;1} \end{array}} \right]_{2n \times 2},{\boldsymbol{V = }}\left[ \begin{array}{l}V_1\\ \,\,\,\,\,\,V_2\\ \,\,\,\,\,\,\,\,\,\,\, \ldots \\ \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,V_n\end{array} \right]_{2n \times 2n}\)
where B is the vector of regression coefficient, X is the design matrix, and V is the variance-covariance matrix for all studies.
The generalized least-squared estimator \({\hat{\mathbf \beta }}\) of overall regression coefficients is given by
which has a normal distribution with mean β and covariance matrix Σ given by
Under the null hypothesis of no association to either phenotype, that is, β = 0 (for both traits), the score statistic
will asymptotically follow a chi-squared distribution with 2 degrees of freedom.
Two univariate test-statistics are constructed similarly. Specifically,
where \({\hat{\mathbf \beta }}_1\) and \({\hat{\mathbf \beta }}_2\) are two elements in \({\hat{\mathbf \beta }}\), and Σ11 and Σ22 are two diagonal elements in Σ. Under the null hypothesis of no univariate association, that is, β1 = 0 or β2 = 0, T1 or T2 will follow a chi-squared distribution with 1 degree of freedom.
In silico replication
Pleiotropic SNPs identified in the discovery meta-analysis were replicated by the UKB sample. Briefly, the UKB sample involves ~500,000 participants (aged between 40–69) from across the United Kingdom, a large prospective cohort study. The ethical approval for the UKB study came from the North West Centre for Research Ethics Committee (11/NW/0382) and informed consent was obtained from all participants. This study used the data under the UKB application number 41 542, which was included in the general ethical approval of the UKB study.
All subjects had genome-wide genotypes at 784 256 autosome markers and were imputed into the UK10K haplotype, 1000 Genomes phase 3, and Haplotype Reference Consortium reference panels. All the included subjects are those who self-reported as white (data field 21000). Subjects whose self-reported gender is inconsistent with the genetic gender, who had been genotyped but not imputed or who had withdrawn their consents were removed. Based on the genome-wide genotyped variants, a set of unrelated subjects were sampled and subsequently analyzed with KING [27].
Body composition was quantified by bioelectrical impedance approach. LFM was quantified as the sum of fat mass at legs (data fields 23112 and 23116), and LLM was quantified as the sum of fat-free mass at legs (data fields 23113 and 23117). Phenotype modeling of both LFM and LLM were similar to that in the discovery samples. The difference is that both phenotypes were mandatorily adjusted by the top 10 principal components to adjust the potential population structure. Association was again examined by the univariate/bivariate linear regression model.
Age-dependent effect
The identified SNPs were evaluated for their age-dependent effects to both traits in the replication UKB sample. Specifically, the UKB sample was divided into the following 6 age strata, 38–45 years (N = 41765), 46–50 years (N = 47453), 51–55 years (N = 57674), 56–60 years (N = 74275), 61–65 years (N = 92510) and 66–73 years (N = 56414). The association of each trait was examined in each age stratum by a linear regression model. Regression coefficients from all age stratum were then meta-regressed on median age of each stratum. Statistical evidence was corrected for multiple testing by Bonferroni correction.
Functional annotation
We annotated the functional relevance of the identified SNPs by a series of tools and resources, including HaploReg [28], Genotype-Tissue Expression (GTEx) project data [29] (GTEx Analysis Release v.7) and 3DIV [30].
HaploReg is an information database that annotates non-coding SNPs into different functional categories, with information from multiple large experimental projects. Functional categories include promoter, enhancer, DNase hypersensitivity region, transcription factor binding sites and others.
GTEx project, providing a cross-organ eQTL research platform, is a large database to collect mRNA sequencing of multiple human organs and currently collects genotypes and gene expression from 544 donors throughout the body, and can study eQTL in various tissues.
3DIV (3D-genome Interaction Viewer and database) is a database to collect publicly available human Hi-C data, which provides 66 billion uniformly processed raw Hi-C read pairs obtained from 80 different human cell/tissue types. Hi-C (high-throughput chromatin conformation capture) [31] is a method to detect long-range chromatin interactions, allowing extensive genome-wide investigation of 3D chromatin structure. 3DIV extracted the annotation of promoters and gene bodies, chromatin interaction frequencies, histone ChIP-seq signals and coordinates of disease-associated SNPs, enhancer/super-enhancers annotation from gene annotations, Hi-C experiments, ChIP-seq experiments and GWAS studies.
Software availability
All in-house software implementing our proposed methods were available in the Supplementary File 1.
Results
GWAS meta-analysis
Table 1 summarizes the basic characteristics of the samples. A total of 12,517 subjects from six samples are included in the meta-analysis. Principle components analysis was applied to each individual sample and no population outliers are observed. The 1000 genomes sequencing project generated 12 403 269 bi-allelic variants. After removing variants either of low-frequency or of poor imputation accuracy, 6,879,267 variants are qualified for analysis. 88% (6,035,487) of them are SNPs, and the remaining 12% (843,780) are INDEL variants.
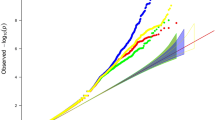
A logarithmic quantile–quantile plot of meta-analysis test statistics for univariate analyses and bivariate analysis showed a marked deviation in the tail of the distribution, implying the possible existence of true associations (Fig. 1).

Results were plotted for univariate GWAS of LFM (blue), univariate GWAS of LLM (green), and bivariate GWAS of both traits (red).
Univariate analysis of (lean-adjusted) LFM identified 43 variants at the GWS, mapping to 9 distinct loci (Supplementary Table S1): 2q24.2 (lead SNP rs16846291, p = 1.77 × 10−8), 2q24.3 (rs10170665, p = 1.96 × 10−8), 2q36.3 (rs2943648, p = 1.97 × 10−9), 3p14.1 (rs9860730, p = 3.05 × 10−8), 6p21.1 (rs11967262, p = 1.25 × 10−8), 8p23.2 (rs200550880, p = 8.92 × 10−9), 10q22.3 (rs3895291, p = 1.29 × 10−8), 12q24.31 (rs4765528, p = 7.68 × 10−13) and 21q21.1 (rs12627504, p = 8.92 × 10−9). Here, an independent locus was defined as a genomic region of 500 kb length from either side of the lead SNP. rs4765528 (p = 7.68 × 10−13) is the most significant SNP associated with LFM, which is located into 12q24.31. 12q24.31 was reported to be associated with BMI (lead SNP rs7133378, p = 1.00 × 10−8) [32], while rs4765528 and rs7133378 are in strong LD pattern (LD r2 = 0.80). 2q24.3 was previously reported to be associated with BFM with the same lead SNP rs10170665 [33]. None of the remaining 7 loci was previously reported to be associated with BFM, but 4 have been reported for BMI.
Univariate GWAS meta-analysis of (fat-adjusted) LLM identified 89 variants at the GWS level mapping to 14 distinct loci (Supplementary Table S2): 3q22.1 (rs1389271, p = 2.14 × 10−8), 3q24 (rs4296557, p = 7.56 × 10−10), 3q27.1 (rs16857317, p = 1.25 × 10−8), 5p15.31 (rs143036230, p = 4.22 × 10−8), 6p21.1 (rs513688, p = 5.03 × 10−9), 9q21.13 (rs12351661, p = 1.96 × 10−8), 9q34.3 (rs112034122, p = 3.14 × 10−8), 10p12.1 (rs4749219, p = 2.53 × 10−8), 10q26.2 (rs557974347, p = 2.73 × 10−8), 11q12.3-q13.1 (rs532653744, p = 4.14 × 10−8), 12p13.32 (rs149168716, p = 3.97 × 10−8), 15q14 (rs8041675, p = 3.80 × 10−8), 17q21.31 (rs12150327, p = 5.76 × 10−9) and 19p13.3 (rs10414354, p = 2.52 × 10−9). rs4296557 (p = 7.56 × 10−10) at 3q24 is the most significant SNP associated with LLM, which is a novel locus. Of the remaining 13 loci, 6 (9q34.3, 10p12.1, 12p13.32, 15q14, 17q21.31 and 19p13.3) were reported to be associated with LBM [34, 35].
In bivariate GWAS analysis, 13 of the above 23 (9 + 14) loci remain significant at the GWS level (Supplementary Table S3). All the above lead SNPs in bivariate GWAS are nominally significant (p < 0.05) in both univariate analyses.
In addition, the bivariate analysis identified 5 loci at the GWS level that were not identified in univariate analysis of either phenotype: 4q28.2 (rs12512163, p = 3.95 × 10−8), 5q13.1 (rs4976033, p = 1.93 × 10−9), 5q31.2 (rs11371364, p = 6.01 × 10−9), 8q24.11 (rs111960203, p = 4.34 × 10−8) and 18q21.32 (rs371326986, p = 9.04 × 10−9). Manhattan plot of the univariate analyses and bivariate analysis are displayed in Fig. 2.

Results were plotted for univariate GWAS of LFM (blue), univariate GWAS of LLM (green), and bivariate GWAS of both traits (red).
In silico replication in the UKB sample
We replicated the 18 bivariate lead SNPs in the UKB sample. The lead SNP rs11371364 at 5q31.2 and rs548671603 at 10q22.3 were not found in the imputed UKB genotypes and were replaced by the second lead SNPs rs6895170 (bivariate pdiscovery = 6.37 × 10−9, LD r2 = 0.87) and rs375369367 (bivariate pdiscovery = 5.86 × 10−9, LD r2 = 0.70), respectively.
Among the 18 bivariate associated variants, four SNPs were significant in both univariate analyses as well as in bivariate analysis (Supplementary Table S4). These significant SNPs include rs1024137 (pLFM = 9.30 × 10−15, pLLM = 1.20 × 10−12, pbivariate = 4.07 × 10−13), rs4976033 (pLFM=5.40 × 10−7, pLLM = 2.30 × 10−4, pbivariate = 6.35 × 10−7), rs4765528 (pLFM=5.80 × 10−16, pLLM = 2.30 × 10−7, pbivariate = 1.88 × 10−11) and rs371326986 (pLFM = 4.40 × 10−2, pLLM = 5.50 × 10−46, pbivariate = 2.35 × 10−95). Among the 4 replicated loci, 2q36.3 was previously reported to be associated with LBM (lead SNPs rs2943656, p = 2 × 10−11, r2 = 0.88) [36] and BMI (rs2176040, p = 1.00 × 10−8, r2 = 0.98) [37]. 5q13.1 (lead SNP rs4976033) has been reported to be associated with LBM (rs34287, p = 6.00 × 10−10) [35], but the reported lead SNP is in weak LD with the one in the present study (r2 < 0.1). 12q24.31 was reported to be associated with BMI (lead SNP rs7133378, p = 1.00 × 10−8, r2 = 0.80) [32] and 18q21.32 was reported to be associated with BFM and LBM (lead SNP rs6567160, p = 3.00 × 10−11 and p = 2.00 × 10−62, respectively, r2 = 0.96) [35, 38]. At last, the left locus at 5q13.1 is a novel locus (Table 2).
Interestingly, among the four replicated SNPs, three (rs1024137, rs4765528 and rs4976033) were oppositely associated with LFM versus LLM, suggesting an opposite regulatory mechanism towards fat mass and lean mass development.
Validation of the FHS sample by MERLIN
Univariate association signals of the above four lead SNPs in the FHS sample were also analyzed by MERLIN [25] for validation. The results, as listed in Supplementary Table S5, showed very comparable signals. The difference was probably due to that MERLIN analyzed hard-called genotypes while the method used in the present study analyzed imputed dosage.
Age-dependent effect
The age-dependent effects at the four lead SNPs were listed in Fig. 3. No significant result was identified after multiple testing adjustment (α = 0.05/8 = 6.25 × 10−3), but the effect of rs4765528 on leg fat mass was nominally significant (p = 0.02), implying a possible age-dependent effect of rs4765528. Specifically, the effect of rs4765528 tends to get smaller as mean age increased.

The UKB sample was divided into the following 6 age strata, 38–45 years (N = 41765), 46–50 years (N = 47453), 51–55 years (N = 57674), 56–60 years (N = 74275), 61–65 years (N = 92510) and 66–73 years (N = 56414). The association of each trait was examined in each age stratum by a linear regression model. Regression coefficients from all age stratum were then meta-regressed on median age of each stratum. No significant result was identified after multiple testing adjustment (α = 0.05/8 = 6.25 × 10−3), but the effect of rs4765528 on leg fat mass was nominally significant (p = 0.02), implying a potential age-dependent effect at this SNP.
Functional annotation
We annotated each of the four replicated variants and their neighboring SNPs with strong LD (r2 > 0.8) with HaploReg, GTEx portal and 3DIV. rs1024137, the lead SNP in 2q36.3, is an expression quantitative trait loci (eQTL) and was associated with the expression of insulin receptor substrate 1 (IRS1) gene in subcutaneous adipose (p = 9.58 × 10−9). rs1024137 has 81 neighbor variants with strong LD structure, and some of them have promoter and enhancer activity marked by different histone marks. In the GTEx portal, rs1024137 was associated with the expression of IRS1 gene in subcutaneous adipose (p = 3.80 × 10−16) and visceral omentum adipose (p = 1.90 × 10−10). Moreover, rs1024137 and IRS1 have chromatin interaction in GM12878 cells through the analysis of 3DIV. GM12878, transformed with the Epstein–Barr virus, is a lymphoblastoid cell line and has more stable chromosome karyotypes compared with commonly used human cell lines [39].
rs4976033, the lead SNP at the 5q13.1 region, has no neighbor variant with strong LD structure. Its surrounding region has promoter and enhancer activity marked by H3K4me1 histone mark in skeletal muscle. In the study of 3DIV, rs4976033 has chromatin interaction with enhancer activity region of PIK3R1 in GM12878 cells.
The lead SNP rs4765528 in 12q24.31 is an intron variant in ZNF664 gene. It is an eQTL and is associated with the expression of CCDC92 gene in subcutaneous adipose (p = 5.71 × 10−9) and visceral omentum adipose (p = 5.01 × 10−9). It is also associated with the expression of DNAH10OS and RP11-380L11.4 genes in skeletal muscle (p = 1.50 × 10−13 and 1.10 × 10−12, respectively).
rs371326986 in 18q21.32 is annotated to be associated with the expression of MC4R gene in testis (p = 6.70 × 10−5). rs371326986 and MC4R have chromatin interaction in GM12878 cells through the study of 3DIV.
New locus/gene associated with LFM and LLM
5q13.1 is a newly identified locus that was not associated with either LFM or LLM related traits previously. Allele G at this SNP tended to decrease LFM (β = −0.072) and increase LLM (β = 0.07). A regional plot of rs4976033 plotted by LocusZoom [40] was displayed in Fig. 4. It is located at the 3′ of phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1, 116.6 kb) gene. Compared with lean, non-diabetics, obese and type 2 diabetic humans have 2–3-fold higher abundance of all PIK3R1 protein isoforms in skeletal muscle [41].

X-axis represents the region of 500 kb on either side of rs4976033 and the genes exist in this region. Y-axis represents −log10(P value). The r2 values are from the European samples. The European recombination rates are indicated with a blue line on the right-hand y-axis.
We constructed a protein-protein interaction network of PIK3R1 (Fig. 5) with STRING [42]. Ten proteins are predicted functional partner of PIK3R1. Among them, IRS1, PTEN, RAC-alpha serine/threonine-protein kinase (AKT1) and epidermal growth factor receptor were reported to be associated with adipocytes and skeletal muscle development.

Ten genes are predicted functional partners of PIK3R1, which may have possible mechanism contribute to LFM and/or LLM.
Discussion
In the present study, we performed a bivariate genome-wide association meta-analysis for (lean-adjusted) LFM and (fat-adjusted) LLM in 12,517 subjects from six samples and further replicated the significant association signals in the UKB sample. We identified four genomic loci, 2q36.3, 5q13.1, 12q24.31 and 18q21.32, which may play pleiotropic effects for both LFM and LLM.
2q36.3, 5q13.1, 12q24.31 and 18q21.32 are four important chromosome regions. Previous studies have demonstrated the association between these regions and different diseases, such as cardiovascular disease [32], coronary artery disease [43] and metabolic syndrome [44], suggesting the complex etiology of these regions. Variants in these regions were reported to be associated with lean mass or obesity-related traits in previous reports [36, 45, 46].
At 2q36.3, the lead SNP rs1024137 is located at the 3′ of IRS1 (500.2 kb) gene. IRS1 is a modulator of insulin signaling pathway, which can mediate the control of various cellular processes through insulin and participates in growth hormone and adipocytokine signaling pathways [36]. Studies have shown that genetic disruption of MG53 increases IRS1 levels, improves insulin signaling in skeletal muscle of mouse and insulin resistance in diet-induced obese (DIO) mice [47].
At 5q13.1, the lead SNP rs4976033 is located at the 3′ of PIK3R1 (116.6 kb) gene. Motohashi et al. [48] have shown that miR-128a regulate PIK3R1, IRS1 and other target genes involved in insulin signaling at mRNA and protein level. miR-128a is highly expressed in skeletal muscle and increases during myoblast differentiation. PTEN is a negative regulator of AKT1 and deletions in muscle or adipose tissue all lead to improved glucose tolerance in healthy or obese/diabetic mice [49]. AKT1 is the member of the AKT kinase family, which is dispensable for the differentiation of Myf5-lineage adipocytes and skeletal muscle development [50].
rs4765528 at 12q24.31 is an intron variant in zinc finger protein 664 (ZNF664) gene. ZNF664 is a krueppel C2H2-type zinc-finger protein of unknown function, which may be involved in transcriptional regulation. SNPs near ZNF664 are highly correlated with adiponectin signaling in Asians [51]. Population-level genetic analyses combined with experiments in cellular models implicate CCDC92 and DNAH10 as previously unrecognized molecules influencing adipocyte differentiation [52].
At 18q21.32, the lead SNP rs371326986 is located at the 3′ of melanocortin 4 receptor (MC4R, 187.1 kb). MC4R is crucial in the regulation of body weight and monogenic forms of obesity commonly result from mutations in MC4R gene [53]. SNPs present in the intergenic region upstream of MC4R were associated with BMI [54].
Early GWAS and their meta-analysis focused on the analysis of single traits, which may lose some valuable information, such as pleiotropic genetic loci. Here, we performed a large scale bivariate GWAS meta-analysis that simultaneously analyzed two related traits fat mass and lean mass. Joint consideration of fat mass and lean mass can provide additional information compared to information contained in individual traits. In addition, weight is made up of various body components, including fat mass, lean mass, bone mass, and other soft tissues. Fat mass is the only component that reflects obesity. We used LFM as a phenotypic trait for obesity rather than BMI, which can strongly explain the association between probable locus and obesity, and provide greater power to detect pleiotropic genes that are important to the pathogenesis of metabolic diseases.
In this study, both studied traits are highly polygenic, that is, hundreds to thousands responsible loci may exist. Our previous theoretical analyses have shown that the identification of one locus from all the responsible loci in a GWAS scan has considerable power even under a small sample size [55]. In contrast, SNPs identified in the discovery sample were very difficult to replicate because it was those particular SNPs that were to be replicated. Therefore, a large replication sample was warranted for successful replication, as the case of UKB sample used in the present study, where the power to replicate the SNPs identified in the discovery sample are over 90%. While we acknowledged that our small-sample GWAS analysis may not be comprehensive in identifying the majority of all associated loci, we were confident that the identified and replicated ones were very likely to be truly associated loci. If using UKB as discovery sample, then the findings were very unlikely to be replicated by our relatively small GWAS.
In conclusion, statistical evidence in our study combined with previously reported biological functions suggest that loci 2q36.3, 5q13.1 12q24.31 and 18q21.32 may play roles in the development of both fat mass and lean mass. Our findings can further enhance our understanding of genetic association between fat mass and lean mass, and provide a rationale for subsequent studies of these implicated genes in the pathophysiology of diseases related to fat mass and lean mass.
References
Penatti M, Lira FSD, Katashima C, Rosa JC, Pimentel GJNH. Sugar intake is correlated with adiposity and obesity indicators and sedentary lifestyle in Brazilian individuals with morbid obesity. Nutr Hosp. 2012;27:1547–53.
Collaborators GBDO, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377:13–27.
Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–81.
Srikanthan P, Horwich TB, Tseng CH. Relation of muscle mass and fat mass to cardiovascular disease mortality. Am J Cardiol. 2016;117:1355–60.
Hunter GR, Weinsier RL, Gower BA, Wetzstein C. Age-related decrease in resting energy expenditure in sedentary white women: effects of regional differences in lean and fat mass. Am J Clin Nutr. 2001;73:333–7.
Yim JE, Heshka S, Albu JB, Heymsfield S, Gallagher D. Femoral-gluteal subcutaneous and intermuscular adipose tissues have independent and opposing relationships with CVD risk. J Appl Physiol. 2008;104:700–7.
Goodpaster BH, Krishnaswami S, Harris TB, Katsiaras A, Kritchevsky SB, Simonsick EM, et al. Obesity, regional body fat distribution, and the metabolic syndrome in older men and women. Arch Intern Med. 2005;14:11–12.
Van Pelt RE, Jankowski CM, Gozansky WS, Wolfe P, Schwartz RS, Kohrt WM. Sex differences in the association of thigh fat and metabolic risk in older adults. Obesity. 2011;19:422–8.
Snijder MB, Visser M, Dekker JM, Goodpaster BH, Harris TB, Kritchevsky SB, et al. Low subcutaneous thigh fat is a risk factor for unfavourable glucose and lipid levels, independently of high abdominal fat. The Health ABC Study. Diabetologia. 2005;48:301–8.
Carey DG, Pliego GJ, Raymond RL, Skau KB. Body composition and metabolic changes following bariatric surgery: effects on fat mass, lean mass and basal metabolic rate. Obesity Surg. 2006;16:469–77.
Arden NK, Spector TD. Genetic influences on muscle strength, lean body mass, and bone mineral density: a twin study. J Bone Miner Res. 1997;12:2076–81.
Thomis MAJCJAP. Determinants and upper-limit heritabilities of skeletal muscle mass and strength. Can J Appl Physiol. 2004;29:186–200.
Karasik D, Zhou Y, Cupples LA, Hannan MT, Kiel DP, Demissie S. Bivariate genome-wide linkage analysis of femoral bone traits and leg lean mass: Framingham study. J Bone Miner Res. 2009;24:710–8.
Visser M, Kritchevsky SB, Goodpaster BH, Newman AB, Nevitt M, Stamm E, et al. Leg muscle mass and composition in relation to lower extremity performance in men and women aged 70 to 79: the health, aging and body composition study. J Am Geriatr Soc. 2002;50:897–904.
Venturelli M, Morgan GR, Donato AJ, Reese V, Bottura R, Tarperi C, et al. Cellular aging of skeletal muscle: telomeric and free radical evidence that physical inactivity is responsible and not age. Clin Sci. 2014;127:415–21.
Ivey FM, Hafer-Macko CE, Macko RF. Exercise training for cardiometabolic adaptation after stroke. J Cardiopulm Rehabil Prev. 2008;28:2–11.
Baumgartner RN, Koehler KM, Dympna G, Linda R, Heymsfield SB, Ross RR, et al. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol. 1998;8:8.
Welch GW, Sowers MR. The interrelationship between body topology and body composition varies with age among women. J Nutr. 2000;130:2371.
Zhang L, Choi HJ, Estrada K, Leo PJ, Li J, Pei YF, et al. Multistage genome-wide association meta-analyses identified two new loci for bone mineral density. Hum Mol Genet. 2014;23:1923–33.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73.
Zhang L, Pei YF, Fu X, Lin Y, Wang YP, Deng HW. FISH: fast and accurate diploid genotype imputation via segmental hidden Markov model. Bioinformatics. 2014;30:1876–83.
Zhang L, Li J, Pei YF, Liu Y, Deng HW. Tests of association for quantitative traits in nuclear families using principal components to correct for population stratification. Ann Hum Genet. 2009;73(Pt 6):601–13.
Zhang L, Bonham AJ, Li J, Pei YF, Chen J, Papasian CJ, et al. Family-based bivariate association tests for quantitative traits. Plos One. 2009;4:e8133.
Chen WM, Abecasis GR. Family-based association tests for genomewide association scans. Am J Hum Genet. 2007;81:913–26.
Konstantopoulos S. Fixed and mixed effects models in meta-analysis. In: IZA Discussion Paper 2198, 2006.
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–73.
Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(D1):D930–D934.
Carithers LJ, Moore HM. The genotype-tissue expression (GTEx) project. Biopreserv Biobank. 2015;13:307–8.
Yang D, Jang I, Choi J, Kim MS, Lee AJ, Kim H, et al. 3DIV: A 3D-genome Interaction Viewer and database. Nucleic Acids Res. 2018;46(D1):D52–D57.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93.
Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet. 2019;104:65–75.
Pei YF, Ren HG, Liu L, Li X, Fang C, Huang Y, et al. Genomic variants at 20p11 associated with body fat mass in the European population. Obesity. 2017;25:757–64.
Tachmazidou I, Suveges D, Min JL, Ritchie GRS, Steinberg J, Walter K, et al. Whole-genome sequencing coupled to imputation discovers genetic signals for anthropometric traits. Am J Hum Genet. 2017;100:865–84.
Hubel C, Gaspar HA, Coleman JRI, Finucane H, Purves KL, Hanscombe KB, et al. Genomics of body fat percentage may contribute to sex bias in anorexia nervosa. Am J Med Genet B. 2019;180:428–38.
Zillikens MC, Demissie S, Hsu YH, Yerges-Armstrong LM, Chou WC, Stolk L, et al. Large meta-analysis of genome-wide association studies identifies five loci for lean body mass. Nat Commun. 2017;8:80.
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206.
Pei YF, Zhang L, Liu Y, Li J, Shen H, Liu YZ, et al. Meta-analysis of genome-wide association data identifies novel susceptibility loci for obesity. Hum Mol Genet. 2014;23:820–30.
Muller RY, Ming C, Hammond, Rio DC, Lee YJ, Biomol Tech JJ. An efficient method for electroporation of small interfering RNAs into ENCODE project tier 1 GM12878 and K562 cell lines. J Biomol Tech. 2015;26:142–9.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7.
Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes. 2005;54:2351–9.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52.
van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122:433–43.
Zabaneh D, Balding DJ. A genome-wide association study of the metabolic syndrome in Indian Asian men. Plos One. 2010;5:e11961.
Kilpelainen TO, Zillikens MC, Stancakova A, Finucane FM, Ried JS, Langenberg C, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet. 2011;43:753–60.
Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–69.
Song R, Peng W, Zhang Y, Lv F, Wu HK, Guo J, et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature. 2013;494:375–9.
Motohashi N, Alexander MS, Shimizu-Motohashi Y, Myers JA, Kawahara G, Kunkel LM. Regulation of IRS1/Akt insulin signaling by microRNA-128a during myogenesis. J Cell Sci. 2013;126(Pt 12):2678–91.
Wong JT, Kim PT, Peacock JW, Yau TY, Mui AL, Chung SW, et al. Pten (phosphatase and tensin homologue gene) haploinsufficiency promotes insulin hypersensitivity. Diabetologia. 2007;50:395–403.
Sanchez-Gurmaches J, Martinez Calejman C, Jung SM, Li H, Guertin DA. Brown fat organogenesis and maintenance requires AKT1 and AKT2. Mol Metab. 2019;23:60–74.
Wu Y, Gao H, Li H, Tabara Y, Nakatochi M, Chiu YF, et al. A meta-analysis of genome-wide association studies for adiponectin levels in East Asians identifies a novel locus near WDR11-FGFR2. Hum Mol Genet. 2014;23:1108–19.
Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49:17–26.
Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34.
Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–75.
Pei YF, Zhang L, Papasian CJ, Wang YP, Deng HW. On individual genome-wide association studies and their meta-analysis. Hum Genet. 2014;133:265–79.
Acknowledgements
We appreciate all the volunteers who participated into this study. The Framingham Heart Study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01-HC-25195). This manuscript was not prepared in collaboration with investigators of the Framingham Heart Study and does not necessarily reflect the opinions or views of the Framingham Heart Study, Boston University, or NHLBI. Funding for SHARe Affymetrix genotyping was provided by NHLBI Contract N02-HL-64278. SHARe Illumina genotyping was provided under an agreement between Illumina and Boston University. Funding support for the Framingham Whole Body and Regional Dual X-ray Absorptiometry (DXA) dataset was provided by NIH grants R01 AR/AG 41398. The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accession phs000342.v14.p 10. The WHI program is funded by the National Heart, Lung, and Blood Institute, National 20 Institutes of Health, U.S. Department of Health and Human Services through contracts N01WH22110, 24152, 32100-2, 32105-6, 32108-9, 32111-13, 32115, 32118-32119, 32122, 42107-26, 42129-32, and 44221. This manuscript was not prepared in collaboration with investigators of the WHI, has not been reviewed and/or approved by the Women’s Health Initiative (WHI), and does not necessarily reflect the opinions of the WHI investigators or the NHLBI. Funding for WHI SHARe genotyping was provided by NHLBI Contract N02-HL-64278. The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accession phs000200.v10.p 3.
Funding
YFP and LZ were partially supported by the funding from National Natural Science Foundation of China (31771417 and 31571291). HWD was partially supported by the National Institutes of Health (R01 AR069055, U19 AG055373, R01 MH104680, R01 AR059781 and P20 GM109036), the Franklin D. Dickson/Missouri Endowment and the Edward G. Schlieder Endowment. This study was also benefited from a project funded by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions. The numerical calculations in this paper have been done on the supercomputing system of the National Supercomputing Center in Changsha. The funders had no role in study design, data collection and analysis, results interpretation, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, Y., Ran, S., Lin, Y. et al. Four pleiotropic loci associated with fat mass and lean mass. Int J Obes 44, 2113–2123 (2020). https://doi.org/10.1038/s41366-020-0645-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-020-0645-0
- Springer Nature Limited