
Abstract
Background
Visceral fat accumulation increases the risk of developing type 2 diabetes and metabolic syndrome, and is associated with excessive glucocorticoids (GCs). Fat depot-specific GC action is tightly controlled by 11ß-hydroxysteroid dehydrogenase (11ß-HSD1) coupled with the enzyme hexose-6-phosphate dehydrogenase (H6PDH). Mice with inactivation or activation of H6PDH genes show altered adipose 11ß-HSD1 activity and lipid storage. We hypothesized that adipose tissue H6PDH activation is a leading cause for the visceral obesity and insulin resistance. Here, we explored the role and possible mechanism of enhancing adipose H6PDH in the development of visceral adiposity in vivo.
Methods
We investigated the potential contribution of adipose H6PDH activation to the accumulation of visceral fat by characterization of visceral fat obese gene expression profiles, fat distribution, adipocyte metabolic molecules, and abdominal fat-specific GC signaling mechanisms underlying the diet-induced visceral obesity and insulin resistance in H6PDH transgenic mice fed a standard of high-fat diet (HFD).
Results
Transgenic H6PDH mice display increased abdominal fat accumulation, which is paralleled by elevated lipid synthesis associated with induction of lipogenic transcriptor C/EBPα and PPARγ mRNA levels within adipose tissue. Transgenic H6PDH mice fed a high-fat diet (HFD) gained more abdominal visceral fat mass coupled with activation of GSK3β and induction of XBP1/IRE1α, but reduced pThr308 Akt/PKB content and browning gene CD137 and GLUT4 mRNA levels within the visceral adipose tissue than WT controls. HFD-fed H6PDH transgenic mice also had impaired insulin sensitivity and exhibited elevated levels of intra-adipose GCs with induction of adipose 11ß-HSD1.
Conclusion
These data provide the first in vivo mechanistic evidence for the adverse metabolic effects of adipose H6PDH activation on visceral fat distribution, fat metabolism, and adipocyte function through enhancing 11ß-HSD1-driven intra-adipose GC action.
Similar content being viewed by others


Introduction
Obesity is a growing global public health problem that affects billion people worldwide. Overweight, especially visceral obesity has been recognized as the most dangerous risk factor for developing type 2 diabetes mellitus (T2DM), metabolic syndrome, and cardiovascular diseases [1, 2]. Hormone glucocorticoids (GCs) regulate adipose tissue differentiation, function, distribution, insulin sensitivity, and in excess, will lead to the pathological state. Patients with GC excess (Cushing’s syndrome) develop pronounced abdominal visceral fat deposition associated with the progress of metabolic syndrome with dyslipidemia, insulin resistance, T2DM, and hypertension [3]. In target cells, GC action is regulated by 11ß-hydroxysteroid dehydrogenase type 1 (11ß-HSD1) at pre-receptor levels [4]. 11ß-HSD1 is an intracellular endoplasmic reticulum (ER) lumen-resident enzyme, and its major function in vivo is the NADPH-dependent conversion of inert cortisone (11-dehydrocorticosterone in rodents) to active cortisol (corticosterone) and therefore amplifies tissue GC production. Increasing GC production in adipose tissue caused by 11ß-HSD1 induces pronounced visceral fat accumulation and insulin resistance in mice [5]. Clinical observations also have shown that adipose 11ß-HSD1 activity is increased in typical obese humans and patients with metabolic syndrome [6, 7]. In contrast, mice lacking 11β-HSD1 are resistant to diet-induced obesity and metabolic syndrome [8]. These results implicate the importance of adipose 11ß-HSD1-driven local GC production in the progression of visceral obesity and T2DM.
11β-HSD1 amplification of intracellular active GC production requires a high [NADPH]/[NADP+] ratio, which can be crucially provided by the enzyme hexose-6-phosphate dehydrogenase (H6PDH) [9, 10]. H6PDH is an enzyme localized to the ER lumen that converts glucose-6-phosphate (G6P) and NADP to produce NADPH, thereby driving 11β-HSD1 reductase activity and consequently intracellular GC production [11, 12]. Patients with gene mutations in H6PDH show defects in 11ß-HSD1 activity and GC production [13]. Consistent with the physiological relevance of H6PDH for 11ß-HSD1, H6PDH knockout mice are unable to reproduce active cortisol from cortisone by 11ß-HSD1, and have improved lipogenesis and insulin sensitivity [14]. These results demonstrate that H6PDH is required for 11β-HSD1 amplifying local GC signaling related to the progression of obesity and insulin resistance.
In contrast, subcutaneous H6PDH mRNA levels are elevated in the worsening metabolic disorders associated with increased local cortisol production in obese patients with the metabolic syndrome [15]. Similarly, there is increased expression of H6PDH in abdominal visceral and/subcutaneous fat in patients with T2DM as well as in obese diabetic rodents compared to normal controls [16,17,18]. These observations imply that fat-specific elevation of H6PDH expression may represent an emerging mechanism in the pathogenesis of metabolic syndrome and thus agents that reduce endogenous 11β-HSD1 and consequently can improve or delay the progress of T2DM and obesity. We recently reported that transgenic mice overexpressing H6PDH in adipose tissue (aP2-H6PDH Tg mice) exhibit feature of metabolic syndrome with abdominal region fat accumulation, fasting hyperglycemia, dyslipidemia with lipolysis, and elevated intra-adipose GC levels [19], implicating that adipose tissue H6PDH is able to couple the regulation of cellular GC metabolism and fat mass linked to the pathogenesis of obesity and insulin resistance. Thus, the aP2-H6PDH transgenic mouse may serve as a good model to further explore the potential impact of adipose H6PDH activation in the etiology of visceral adiposity and the adverse metabolic consequence relative to visceral obesity/metabolic syndrome in humans. However, it is unclear whether enhanced adipose tissue H6PDH expression would render the transgenic mice even more susceptible to obesity and insulin resistance when fed a Western-type diet.
In order to gain the new insight of these important issues, we conducted in vivo studies to investigate the key molecular mechanism underlying the adverse effects of enhanced adipose H6PDH on abdominal fat deposition by analysis of lipogenic and adipogenic metabolism profile of visceral adipose tissue in transgenic aP2-H6PDH mice. We also investigated at the molecular levels whether adipose tissue H6PDH activation would exacerbate visceral adiposity and insulin resistance in obesity by characterization of obese gene expression profiles, adiposity signaling molecules, visceral fat distribution, browning maker gene expression, adipose metabolic parameters, and abdominal visceral fat-specific GC action mechanisms underlying the diet-induced obese syndrome in HFD-fed transgenic aP2-H6PDH mice. Our data provide the first in vivo mechanistic evidence for the adverse metabolic effects of enhanced adipose H6PDH on fat distribution, adipose function, and homeostasis through enhancing 11ß-HSD1-driven intra-adipose GC action.
Materials and methods
Animals and experimental models
Adult male transgenic mice overexpressing H6PDH under the adipocyte fatty acid-binding protein (aP2) gene (aP2/H6PDH Tg) and their age-matched (10 weeks) wild-type (WT) C57blk/6 control mice were bred and genotyped, as previously described [19]. Eight animals have been used for each experimental group, the numbers of animal used were determined to achieve desired power of 0.8. Animals were housed in standard SPF conditions under a 12:12-h light–dark cycle with water ad libitum. Mice were maintained on either a control diet (Research Diets D12328) or a HFD (45 % energy from Fat, Research Diets D12451; Research Diets Inc., New Brunswick, NJ, USA) for 4 or 14 weeks. For each genotype, mice are divided randomly into the control diet or HFD group. Food intake and body weights were recorded weekly. At the end of the experiments, blood samples were collected, and adipose fat depots were weighed, dissected, and frozen rapidly in liquid nitrogen. Animal protocols were approved by the Institutional Animal Care and Use Committee of the Charles R. Drew University (Los Angeles, California, USA). Blood samples were analyzed for insulin and FFA levels using ELISA kit (Abcam, Cambridge, MA, USA), as previously described [18].
Glucose and insulin tolerance tests
After 20 weeks on a control or a HFD, aP2/H6PDH transgenic and wild-type mice were fasted overnight, and blood samples were taken at the indicated time points following the intraperitoneal injection (i.p) with glucose (1.0 g/kg d-glucose). For the insulin tolerance test, mice were injected with insulin after fasting for 6 h (i.p, 0.75 units/kg; Novolin R; Eli Lilly, Indianapolis, IN), and the glucose levels were measured at the indicated time points.
Glucose uptake in adipose tissue
Glucose uptake was measured as previously reported [20]. Briefly, fresh epididymal fat pads were cut into less than 5-mg pieces and then incubated with 0.5% BSA at 37 °C for 30 min in DMEM. Tissues were washed with 1% BSA in Ringer bicarbonate-HEPES (KRBH) buffer (Sigma K4002) and incubated in KRBH solution with 0.1% BSA in the absence or presence of 100 nM insulin for 5 min at 37 °C. Tissues were further incubated for 30 min in the same solution with adding 10 mmol/L glucose and 2.0 μCi 2-[3H]DG (NEN Life Science, Boston, MA). The uptake reaction was terminated, and radioactivity was measured by using a liquid scintillation counter.
RNA extraction and quantitative real-time RT-PCR
Total RNA was isolated from tissues using the TRIzol reagent (Invitrogen), and the first-strand cDNA was synthesized from 2.0 μg mRNA using a commercially available kit (Thermo Fisher Scientific). The primers were designed with Primer express software 2.0 and were as follows: mouse 11ß-HSD1 (F: 5′-CCTTGGCCTCATAGACACAGAAAC-3′; R: 5′-GGAGTCAAAGGCGATTTT CA-3′), H6PDH (F: 5′-TGGCTACGGGTTGTTTTTGAA-3′; R: 5′-TA TACAGGTAC ATCTCCTCTTCCT-3′), ACC (F: 5′-TGTAAATCTGGCTGCATCCATTAT-3′; R: 5′-TGGTAGACTGCCCGTGTGA-3′), ACL (F: 5′ ATGCCAAGACCA TCCTCTCACT-3′; R: 5′-TCTCACAATGCCCTTGAAGGT-3′), GR (F: 5′ -TGCTATGCTTTGCTCCTGAT CTG-3′;R: 5′-TGTCAGTTGATAAAACCGCTGC-3′), FOXO1 (F: 5′-GGACAGCCGC GCAAGACCAG-3′; R: 5′-TTGAATTCTTCCAGCCCGCCGA-3′), SIRT1 (F: 5′-AATC CCGGACTTCAGATCCC C-3′;R: 5′-CAACATGAAAAAGGGCTTGGG-3′), TXB 1(F: 5′-CGCTGTGGGACGAGTTCAAT-3′; R:5′-CGGCCATGGGATCCATT-3′), CD137 (F: 5′-TTGGGAACATTTAATGACCAGA-3′; R: 5-TCCCGGTCTTAAGCACAGAC-3′), IREα (F: 5′-CCTA CAAGAGTATGTGGAGC-3′; R: 5′- GGTCTCTGTGAACAATGTT GAGAG-3′), XBP1 (F: 5′-GGATTTGGAAGAAGAGAACCACAA-3′; R: 5′-CCGTGAG TTTT CTCCCGTAAAA-3′) and G6PT (F: 5′-AGGCCTTGTAGGAAGCATTGC-3′; R: F: 5′ TCACCGTTACTCGGAAGAGGAA-3′). RT-PCR was performed using SYBR kits in the ABI Prism 7700 System (Thermo Fisher Scientific). Relative mRNA expression levels were calculated using the comparative CT method and were expressed as a relative value after normalization to 18S.
Oil red staining with mounting medium DAPI
Fat tissue was fixed in freshly prepared 4% paraformaldehyde, then imbedded in O.C.T., and frozen sections were cut and stained with Oil Red O (Sigma, St. Louis, MO). Cells were mounted with Vestashield mounting solution with 4′, 6′-diamidino-2-phenylindole (Vector Laboratories) and visualized under a fluorescence microscope.
Adipose tissue corticosterone assays
Corticosterone levels were measured in adipose tissue as previously described [21,22,23]. Briefly, epididymal and mesenteric fat depots were isolated, weighed and homogenized in dPBS buffer (PH 7.4) by a tissue solubilizer. The adipose tissue steroids were extracted twice with ethyl acetate (5 mL/g adipose tissue). The extractions were evaporated and the residues were dissolved in sample analysis buffer. Adipose corticosterone levels were measured using a corticosterone EIA kit (Oxford Biomedical Research). 11ß-HSD1 activity was evaluated by the addition of 1 mM NADPH and 250 nM 11-DHC with [3H]11-DHC as the tracer to microsomes in KRB solution at 37 °C for 1–2 h. Steroids were separated and the conversion of [3H]11-DHC to [3H]CORT was determined by scintillation counting of radioactivity, as previously described [19].
Protein extraction and Western blot analysis
Adipose tissues were homogenized on ice and protein concentrations were determined by Bradford assay as previously described [18]. Adipose proteins were fractionated on 4–15% acrylamide SDS-PAGE gels (BioRad, Hercules, CA, USA) and transferred onto nitrocellulose membranes for the Western analysis of pSer79 ACC, pSer455ACL, p-threonine308 kinase Akt/PKB, IR, IRS-1, GLUT4, pSer9 GSK3β, pSer724 IREα, ACC, ACL, XBP1, p-IREα, IREα, and GAPDH (Cell Signaling, Danvers MA). Proteins signals were quantified by Eagle Eye II Quantitation System (Stratagene, CA, USA).
Statistical analyses
All data are expressed as the mean ± SEM. Data were compared using an unpaired Student’s t-test. To compare multiple groups, one-way ANOVA was used. When ANOVA revealed significant differences, then group comparisons were performed using the Newman–Keul’s post hoc test. A P < 0.05 was considered significant.
Results
Transgenic aP2-H6PDH mice have altered lipogenic gene expression in adipose tissue
Adipose H6PDH expression is associated with abdominal fat storage [14, 19]. To ascertain whether elevated adipose H6PDH expression influences lipogenic profile, we assessed the expression of lipid synthase acetyl-CoA carboxylase (ACC) and ATP-citrate lyase (ACL), the major markers of lipid content in adipose tissue of aP2/H6PDH Tg mice. As shown in Fig. 1, epididymal fat mRNA levels encoding ACC and ACL in aP2-H6PDH Tg mice were increased compared with that of the WT controls (P < 0.01; Fig. 1a). Furthermore, epididymal fat total ACC and ACL protein levels were increased with elevated (1.9-fold and 2.8-fold respectively) phosphorylated levels of Ser79ACC and Ser455 ACL in aP2-H6PDH mice compared with WT controls (Fig. 1b, c). Similarly, higher ACC and ACL mRNA levels (Fig. 1d) with elevated ACL, pSer79ACC, and pSer455 ACL contents were observed in mesenteric visceral fat from the aP2-H6PDH Tg mice, but with unchanged total ACC protein levels compared to that of WT controls (Fig. 1e, f). In addition, subcutaneous adipose ACC and ACL mRNA levels were higher in aP2-H6PDH Tg mice compared with WT controls (Fig. 1g). Moreover, ACC and ACL protein expression levels were markedly elevated with increased levels of pSer79ACC and pSer455 ACL in aP2-H6PDH mice compared with WT controls (Fig. 1h, i). Consistent with the increased lipid biosynthesis in adipose tissue, fatty acid synthase (FAS) mRNA levels were increased by 2-fold and 2.8-fold, respectively, in epididymal and mesenteric visceral fat in aP2-H6PDH Tg mice compared with WT controls (Fig. 2a, c). Real-time RT-PCR analysis also showed that FFA re-esterification enzyme PEPCK mRNA for TG synthesis, were markedly elevated in total adipose tissue from aP2-H6PDH Tg (Fig. 2a, c, e; P < 0.01). These data indicate that aP2-H6PDH Tg mice had increased adipose lipid content.

Lipogenic gene ACC and ACL mRNA and protein expression in epididymal fat (Ep fat), mesenteric fat (Mes fat) and subcutaneous fat (Sub fat) of aP2-H6PDH (■) or WT (□) mice. a, d, g Relative expression of ACC and ACL mRNA levels were measured by real-time RT-PCR and normalized to 18S (n = 8). b, c, e, f, h, i Relative expression levels of t-ACC, t-ACL, pSer79 ACC, and pSer455 ACL proteins were normalized to GAPDH. Data are means ± SE of 7–8 mice/group. *P < 0.01 vs. WT controls; **P < 0.001 vs. WT controls

Alterations of obese gene expression in epididymal fat (Ep fat), mesenteric fat (Mes fat) and subcutaneous fat (Sub fat) between aP2-H6PDH (■) or WT (□) mice. a, c, e Relative expression levels of lipid metabolic gene FAS and PEPCK, and lipogenic gene transcriptor C/EBPα, PPARγ, and SREBP mRNA levels in epididymal fat (a), mesenteric fat (c), and subcutaneous fat (e) were normalized to 18 S. b, d, f Relative expression of leptin, FOXO1, and SIRT1 mRNA levels in epididymal fat (b), mesenteric fat (d) and subcutaneous fat (f) of aP2-H6PDH Tg (■) or WT (□) mice. Data are mean ± SE from six to eight mice per group. *P < 0.01 vs. WT controls; **P < 0.005 vs. WT controls
The effects of enhanced adipose tissue H6PDH expression on obese gene expression in fat tissue
To investigate the mechanism underlying the differential changes in lipogenesis in aP2-H6PDH Tg and WT mice, the mRNA encoding lipogenic gene transcriptors C/EBPα, PPARγ, and SREBP were analyzed. C/EBPα mRNA levels were increased by 4-fold in epididymal fat, 3.6-fold in mesenteric fat (Fig. 2a, c), and 3-fold in subcutaneous fat (Fig. 2e) in aP2-H6PDH mice compared with controls. Adipose PPARγ, a key factor regulating fat distribution mRNA expression were increased by 2.5-fold in epididymal fat, 2-fold in mesentiric fat with a more than 6-fold in subcutaneous fat of the aP2-H6PDH Tg mice compared with WT controls (P < 0.01; Fig. 2a, c, e). Moreover, SREBPα mRNA expression was increased by 1.9-fold in epididymal fat, 1.7-fold in mesenteric visceral fat, and 5-fold in subcutaneous fat (P < 0.01; Fig. 2a, c, e) in aP2-H6PDH Tg mice, respectively. Consistent with the phenotype of fat accumulation, aP2-H6PDH Tg mice also had elevated fat mRNA levels encoding leptin, FOXO1 and SIRT1 in adipose tissue (Fig. 2b, d, c). These results support that aP2-H6PDH Tg mice had upregulation of obese genes associated with fat accumulation.
Transgenic aP2-H6PDH mice exhibit increased ER stress marker expression in fat tissue
Considering that the ER stress proteins have been shown to be crucial for adiposity in fat tissue through mechanism integrating various pathways leading to obesity [24,25,26], we analyzed the expression levels and the phosphorylation state of XBP1 and IREα, two key transcriptional factors of ER stress in adipose tissue. Real-time RT-PCR analysis revealed that epididymal fat IREα and XBP1 mRNA levels were increased by 1.5-fold and 1.45-fold in aP2-H6PDH Tg mice compared with their WT controls (Fig. 3a). Parallel to these observations, IREα protein level was increased with elevated pSer298 IREα content by 2.7-fold in epididymal fat of aP2-H6PDH mice (P < 0.01; Fig. 3c, e). Consistent with these results, aP2-H6PDH Tg mice also had elevated XBP1 protein expression in epididymal fat compared with WT controls (Fig. 3c, e). These data indicate that activation of IREα/XBP1 pathway is involved in adipose adiposity of aP2/H6PDH Tg mice.

ER stress response and the relative alterations of Akt/PKB and GSKβ gene expression in epididymal fat (Ep fat) of aP2-H6PDH Tg (■) and their WT controls (□). a, b Relative expression of IREα, XBP1, AKT, and GSK3β mRNA levels measured by real-time RT-PCR and normalized to 18S. c, e Western blotting analyses were performed to compare the expression levels of t-REα, XBP1, and pSer724 IRE. d, f Relative adipose p-Th308 Akt and pSer9 GSK3β protein was standardized to GAPDH. *P < 0.01 vs. WT controls; **P < 0.02 vs. WT controls
Transgenic aP2-H6PDH mice activated GSK3ß with suppressed Akt signaling in fat tissue
Since Akt can regulate adipogenesis via inhibition of GSK3ß [27, 28], a master activator of adipogenesis, we next examined if the molecular basis for the induction of visceral fat adiposity in aP2-H6PDH Tg mice was associated with altered Akt and GSK3ß signaling. Consistent with visceral fat obesity, epididymal fat GSK3ß mRNA levels (Fig. 3b; P < 0.01) were higher with elevated pSer9GSK3ß content by 1.7-fold in aP2-H6PDH Tg mice (Fig. 3d, f). Reciprocally, epididymal fat Thr308Akt/PKB phosphorylation in aP2-H6PDH Tg mice was lower, with no effect of Akt mRNA and total Akt protein levels compared to their respective WT controls (Fig. 3d, f). These data indicate that activation of GSK3ß with reduction of Akt signaling may be a potential mechanism for adipogenesis in adipose tissue of aP2-H6PDH Tg mice.
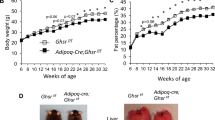
Transgenic aP2-H6PDH mice exhibits exacerbation of HFD-induced accumulation of visceral fat
To further investigate the impact of enhanced adipose H6PDH in the development of visceral obesity, we assessed the effect of high-fat feeding on energy homeostasis, body weight, food intake, and abdominal visceral fat pad mass in transgenic aP2-H6PDH mice. The mean body weight gain of mice fed HFD for 14 weeks were significantly increased when compared with regular chow diet feeding (Fig. 4a). However, there were no significant differences in body weight (Fig. 4a) and food intake (data not shown) between aP2/H6PDH mice and WT controls on control diet or HFD. In contrast, HFD-fed aP2/H6PDH mice gained more visceral fat mass (increased by 27% in epididymal fat and 23% in mesenteric visceral fat (Fig. 4b) than that of WT controls fed HFD, reflecting altered fat distribution with increased visceral fat mass. Oil red O staining with mounting media DAPI showed increased lipid droplet content in visceral fat from aP2-H6PDH Tg mice fed on both control diet and HFD when compared with their respective WT controls (Fig. 4c, d). Consistent with adipose redistribution, mesenteric visceral fat PPARγ, SREBP, and C/EBPα expression were higher with elevated mRNA levels of IRE and XBP1 (Fig. 4d, e), which paralleled to the elevated IREα and XBP1 protein with activation of pSer298 IREα in mesenteric fat of aP2/H6PDH mice (Fig. 4f). Likewise, HFD-fed aP2-H6PDH Tg mice also had increased mesenteric fat mRNA levels encoding leptin (Fig. 4d) with upregulation of Ser9 GSK3ß phosphorylation, but reduced p-Thr308Akt/PKB level (Fig. 4f). These data show that elevation of H6PDH in adipose tissue promotes the HFD-induced phenotype of abdominal fat accumulation.
In contrast, we found that HFD-fed aP2-H6PDH Tg mice had decreased browning marker gene CD137 and browning-related gene GLUT4 mRNA levels in mesenteric fat, but did not significantly change TBX mRNA levels (Fig. 4e) in visceral fat, reflecting decreased the browning of white adipose tissue in aP2-H6PDH Tg mice may be involved in the development of HFD-induced abdominal visceral obesity.

Body weight and visceral fat mass of aP2-H6PDH transgenic and WT mice. a Body weight gain of aP2-H6PDH and WT mice fed chow diet (C) or high-fat diet (HFD) (○, non-Tg mice fed chow diet; ■, Tg mice fed chow diet; △, non-Tg mice fed HFD; ● Tg mice fed HFD, n = 8–10 mice/group). b Ep fat and Mes fat mass of H6PDH mice fed control chow diet or HFD. c Lipid droplet accumulation was monitored by fluorescent microscopy using Oil Red O staining with DAPI. d, e Relative mesenteric fat obese gene mRNA levels in aP2-H6PDH Tg (▨) and their WT controls (■) and WT mice fed HFD. f Relative alterations of Akt, p-Th308Akt and pSer9 GSK3β content in Mes fat of mice fed HFD. *P < 0.01 and #P < 0.02 vs. WT controls; **P < 0.02 vs. HFD-fed controls; †P < 0.02 and ‡P < 0.01 vs. HFD-fed WT controls. g Schematic diagram representing the consequences of enhancing adipose H6PDH expression. H6PDH activation provides the fuel for the production of 11β-HSD1-driven intra-adipose CORT and results in fat accumulation through: (1) CORT stimulation of IREα/XBP1 pathway activates C/EBPα and PPARγ expression, facilitating lipogenesis and TG synthesis, (2) C/EBPα can activate 11β-HSD1, further enhancing CORT availability and (3) CORT-mediated suppression of p-Akt induces insulin resistance and activation of GSK3β signaling that is required for adipogenesis. Additionally, GSK3β activation can promote CORT to bind/activate GR and further amplifying GC action in adipose tissue
Transgenic aP2-H6PDH mice have impaired glucose tolerance and insulin sensitivity
We then determined if elevated adipose H6PDH would render the transgenic mice more susceptible to diet-induced insulin resistance. We measured the rate of glucose disposal and insulin sensitivity by the glucose and insulin tolerance tests and analyzed adipose metabolic function. The fasting blood glucose levels of aP2-H6PDH Tg and WT mice fed HFD were higher than those of their respective controls fed chow diet (WT control diet: 158 ± 12.6 mg/dL; WT high-fat diet: 274 ± 17 mg/dL; H6PDH Tg control diet: 198 ± 11 mg/dL; H6PDH Tg high-fat diet: 337 ± 14 mg/dL) (Fig. 5a). Furthermore, higher plasma glucose levels during GTT (Fig. 5a) were observed in HFD-fed aP2-H6PDH Tg mice compared to that of WT mice fed HFD. During the ITT, aP2-H6PDH Tg mice on HFD also had decreased glucose disposal in response to insulin compared with WT mice fed HFD (Fig. 5b). Moreover, plasma FFA, which is stimulated by GCs [30] and can cause adipose insulin resistance[31], was elevated in HFD-fed aP2-H6PDH Tg mice (Fig. 5c). Additionally, basal and insulin-stimulated glucose uptake in adipose tissue were lower in aP2-H6PDH mice (Fig. 5d), reflecting the cause of adipose insulin resistance.

Glucose and insulin tolerance tests in aP2-H6PDH Tg mice and WT controls. a Glucose tolerance test (GTT; □, non-Tg mice fed chow diet (C); ○, non-Tg mice fed high-fat diet (HFD); ■, Tg mice fed chow diet; ● Tg mice fed HFD). b Insulin tolerance test (ITT; △, non-Tg mice fed chow diet; ○, Tg mice fed chow diet; ▲, non-Tg mice fed HFD; ● Tg mice fed HFD). c Serum FFA levels. d Adipose glucose uptake in visceral fat tissue from aP2-H6PDH mice and WT control. Data are means ± SE of 7–8 mice/group. †P < 0.02 vs. HFD-fed WT controls; *P < 0.001 and #P < 0.01 vs. WT controls; **P < 0.01 vs. HFD-fed WT controls
Transgenic aP2-H6PDH mice exhibit enhanced 11β-HSD1 expression and corticosterone level in fat tissues
Adipose H6PDH expression have shown to be associated with corticosterone production in adipose tissue [14, 17]. We next determined if the adverse metabolic phenotypes observed in aP2-H6PDH Tg mice fed on HFD are partly due to changes in local GC metabolism in this tissue. We found that HFD-fed aP2-H6PDH Tg mice exhibited elevated H6PDH and 11β-HSD1 mRNA expression in epididymal fat and mesenteric visceral fat (Fig. 6a, b). Moreover, we observed that epididymal fat and mesenteric fat G6PT mRNA, the key driver supplying G6P for H6PDH-mediated NADPH generation within the ER lumen for 11β-HSD1 [11], were higher in HFD-fed aP2-H6PDH Tg mice (P < 0.01; Fig. 6b). Consistent with these observations, epididymal and mesenteric fat 11β-HSD1 protein expression and activity were higher in HFD-fed aP2-H6PDH mice than their respective HFD-fed controls (Fig. 6c, d). In parallel with the increase in 11β-HSD1 activity, adipose corticosterone levels were increased by 38% in epididymal fat and by 35% in mesenteric fat in aP2-H6PDH Tg mice compared with WT controls fed HFD (Fig. 6c), indicating increased intra-adipose GC production. Furthermore, adipose GC receptor (GRα) mRNA that is activated by GCs [30], were also elevated in epididymal and mesenteric fat of aP2-H6PDH Tg mice fed on HFD (Fig. 6a, b), reflecting increased GR-mediated local GC action in abdominal adipose tissue. However, there were no significant differences in plasma corticosterone levels between aP2-H6PDH Tg and WT mice fed on HFD, although HFD-fed aP2-H6PDH Tg mice had slightly elevated plasma corticosterone levels (Fig. 6d). These results suggest that elevated intra-adipose GC levels may account, at least in part, for the adverse metabolic phenotype observed in aP2-H6PDH Tg mice fed on HFD, despite normal circulating GC levels.

Adipose target gene expression and corticosterone levels in epididymal fat (Ep fat) and mesenteric fat (Mes fat) of aP2-H6PDH (white bar) and WT mice fed HFD (black bar). a, b Quantitative real-time RT-PCR analysis demonstrating the relative alterations of H6PDH, G6PT, 11β-HSD1, and GR mRNA levels in Ep fat (a) and Mes fat (b) of aP2-H6PDH and WT mice fed HFD. c Western blot analysis of expression of 11β-HSD1 in epididymal fat (Ep fat) and mesenteric fat (Mes fat) of aP2-H6PDH and WT mice fed HFD. d Adipose 11β-HSD1 reductase activity was measured in adipose microsomes using 11-dehydrocorticosterone (DHC) as the substrate in the presence of NADPH. Enzyme activity was expressed as % DHC converted to corticosterone (B). e Tissue corticosterone concentrations in epididymal fat and mesenteric visceral fat of aP2-H6PDH and WT mice fed HFD. f Plasma corticosterone levels in aP2-H6PDH and WT mice fed HFD. Data are means ± SE of 7–8 mice/group. *P < 0.001 vs. HFD-fed WT controls; †P < 0.01 vs. HFD-fed WT controls
Discussion
The data presented here suggest that a new fat depot-specific mechanism driving the adverse fat distribution and visceral fat accumulation is the result of enhanced adipose H6PDH-mediated induction of intra-adipose GC production with upregulation of 11ß-HSD1. We found that H6PDH transgenic mice induced visceral fat adiposity and the adverse metabolic phenotypes are correlated with elevated 11ß-HSD1-driven local corticosterone level in visceral adipose tissue. We observed that aP2-H6PDH Tg mice exhibit elevated 11ß-HSD1 and increased epididyminal and mesenteric visceral fat mass, hyperglycemia, insulin resistance, hyperlipidemia along with elevated GC-inducible lipogenic enzyme ACC and ACL expression. Increased adipose tissue H6PDH could elevate ER luminal NADPH ability to drive 11ß-HSD1 amplification of local GC action and consequently leads to lipogenesis and increased adipose mass linked to visceral fat deposition. This interpretation is further supported by recent report revealing that patients with obesity and metabolic syndrome have increased H6PDH mRNA levels and elevated cortisol production with increased 11ß-HSD1 in visceral fat and subcutaneous fat [15,16,17,18]. In addition, adipose H6PDH activation was correlated with elevated G6PT mRNA level in visceral fat of aP2-H6PDH Tg mice. Elevated G6PT expression ensures ER luminal G6P availability to increase H6PDH and may thus provide an additional mechanism to maintain adipose H6PDH activation in the aP2-H6PDH Tg mice.
Interestingly, we found that the visceral adiposity and its adverse metabolic phenotype were tightly associated with elevated intra-adipose corticosterone levels in aP2-H6PDH mice, but circulating corticosterone levels were elevated only slightly in response to HFD. Endorsing this hypothesis, is the demonstration of increased adipose GC production leading to visceral obesity and metabolic syndrome, despite the normal plasma GC levels in aP2-11ß-HSD1 mice [5]. Our data suggest that activated H6PDH coupled with elevation of 11ß-HSD1-driven intracellular GC production in adipose tissue may be an emerging mechanism to account for the accumulation of visceral fat by increased lipogenesis in aP2-H6PDH mice.
It is well known that tissue-specific action of GCs is mediated by activation of intracellular GR [31]. We found that aP2-H6PDH mice have enhanced 11ß-HSD1 and simultaneously induced fat GR expression with concomitant elevation of adipose GSK3ß phosphorylation that is activated by GCs [31]. Indeed, GSK3ß has been identified as the key ligand-dependent transcription activator and its functional signaling is required for GC-dependent transcriptional activation of GR [32, 33], a ligand-activated transcription factor that is crucially required for GC action. These findings support a concept that activation of GSK3ß signaling itself may be an additional mechanism to ensure adipose H6PDH-coupled 11ß-HSD1-driven local GC action. Induction of GSK3ß signaling could allow GCs to bind/activate intracellular GR and thus promotes GR-mediated intra-adipose corticosterone action leading to visceral fat accumulation and the adverse metabolic phenotype observed in aP2-H6PDH mice. This is consistent with the previous data showing that GSK3ß is required for adipose adipogenesis in insulin-resistant animals [34, 35]. Additionally, we also found that the pThr308Akt content, a marker of adipose insulin sensitivity that is suppressed by GCs [23], were reduced coupled with decreased glucose uptake in response to elevated local GC levels with concomitant induction of GSK3ß in adipose tissue of aP2-H6PDH Tg mice. Reduction in phosphorylated AKT may indicate failures in insulin signaling and, therefore, a lower GLUT4 translocation to the cell membrane. This could explain in part the insulin resistance observed in this animal model.
Given that Akt is also a negative regulator of obesity and insulin resistance through inhibition of GSK3ß [35, 36], reduction of p-Thr308Akt could also promote GSK3ß activation and, in turn, visceral fat adiposity and local insulin resistance. These findings support our hypothesis that visceral fat lipid accumulation of aP2-H6PDH mice may be mediated, at least, in part by activation of GSK3β with suppression of Akt.
Consistent with increased lipogenic gene expression prolife of adipose tissue, SREBP expression is elevated in response to increased H6PDH coupled with 11ß-HSD1 in visceral fat in aP2-H6PDH mice. SREBP is stimulated by insulin [37] and aP2-H6PDH mice had elevated insulin levels, which is associated with high-SREBP levels and visceral fat accumulation observed in aP2/H6PDH Tg mice. Importantly, we also observed that another two key lipogenic transcriptional activator mRNA encoding PPARγ and C/EBPα that are positively regulated by GCs [38, 39], were also elevated in response to activation of obese gene mRNA levels encoding SIRT1 and FOXO1 with induction of 11ß-HSD1 in adipose tissue of aP2-H6PDH mice. Moreover, subcutaneous fat PPARγ and C/EBPα mRNA levels were also increased in aP2-H6PDH mice. Elevated adipose PPARγ and C/EBPα could thus promote the progress of lipogenesis and adipogenesis linked the development of adipose adiposity in aP2-H6PDH mice. This interpretation is supported by studies reporting that activation of C/EBPα and PPARγ are necessary and sufficient to initiate adipogenesis and obesity in insulin-resistant rodents [40, 41]. In addition, aP2-H6PDH mice also exhibit reduced expression of visceral fat CD137 [42, 43], the key gene for begin and browning of white adipocytes that is decreased in adipose tissue in obese animals [42, 43]. Moreover, we also observed that brown fat-related gene encoding GLUT4 expression that is high in browning of white adipocytes [44], was decreased in fat tissue of aP2-H6PDH mice. These findings suggest that reduced adipose CD137 and GLUT4 may also contribute to adipose adiposity in aP2-H6PDH mice.
In addition, the current study also observed that induction of adipose C/EBPα and PPARγ expression paralleled the elevated expression of adipose IREα and XBP1, two key ER stress transcription factors activated by GCs [45] in response to elevated adipose GC levels in aP2-H6PDH mice. Indeed, IREα/XBP1 are required for the transcriptional activator of C/EBPα during adipogenesis [24] and aP2-H6PDH mice show elevated IREα expression and its phosphorylation in visceral fat tissues. Consistent with this concept, we also found that the adipose XBP1 levels, a specific substrate for IRE1α [46, 47], were increased in aP2-H6PDH mice, suggesting that activation of IREα/XBP1 itself could promote the activation of C/EBPα and lipogenesis in adipose tissue. This is consistent with the role of IREα/XBP1 in inducing lipogenesis and adipogenesis [24]. Moreover, IREα/XBP1 is upregulated by insulin [48] and elevated plasma insulin levels may contribute to higher IREα and XBP1 in visceral fat from aP2-H6PDH mice. Consistent with the earlier finding that PPARγ is activated by XBP1 [33], here, we observed elevated XBP1 and increased PPARγ expression in fat tissue of aP2-H6PDH mice. These findings imply that elevation of XBP1/IREα could contribute to high C/EBPα and PPARγ levels, which is sufficient to trigger adipogenesis and the progression of fat accumulation in aP2-H6PDH mice. This interpretation is supported by earlier observation that adipose IREα and XBP1 are increased with obesity and insulin resistance in obese mice [25, 26]. Adipose H6PDH activation-mediated induction of IREα/XBP1 may thus provide an additional mechanism to maintain C/EBPα and PPARγ linked to increased lipogenesis and fat accumulation in aP2-H6PDH mice.
In summary, our current findings provide the first in vivo mechanistic evidence for the contribution of increased adipose H6PDH expression to fat accumulation, insulin resistance, and its relative metabolic disorders. We further demonstrated that the adverse effects of H6PDH activation on adipose function and insulin sensitivity may be largely driven by elevated 11ß-HSD1 amplifying local GC action relative to changes in obese gene expression profile and reduction of glucose uptake associated with reduction of insulin signaling molecules. These results imply that fat-specific manipulation of H6PDH may have potential as a new therapeutic strategy, and we may expect that selective inhibition of adipose H6PDH may represent novel and potential pharmaco-therapeutic approaches to target fat 11ß-HSD1 and treat T2DM and visceral obesity.
References
Després JP, Lemieux I, Bergeron J, Pibarot P, Mathieu P, Larose E, et al. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol. 2008;28:1039–49.
Himabukuro M. Cardiac adiposity and global cardiometabolic risk: new concept and clinical implication. Circ J. 2009;73:27–34.
Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17:245–61.
Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect “Cushing’s disease of the omentum. Lancet. 1997;349:1210–3.
Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–70.
Tomlinson JW, Stewart PM. Mechanisms of disease: selective inhibition of 11beta-hydroxysteroid dehydrogenase type 1 as a novel treatment for the metabolic syndrome. Nat Clin Pract Endocrinol Metab. 2005;1:92–9.
Morton NM, Seckl JR. 11beta-hydroxysteroid dehydrogenase type 1 and obesity. Front Horm Res. 2008;36:146–64.
Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, et al. 11β-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. PNAS. 1997;94:14924–9.
Mziaut H, Korza G, Hand AR, Gerard C, Ozols J. Targeting proteins to the lumen of endoplasmic reticulum using N-terminal domains of 11beta- hydroxysteroid dehydrogenase and the 50-kDa esterase. J Biol Chem. 1999;274:14122–9.
Odermatt A, Arnold P, Stauffer A, Frey BM, Frey FJ. The N-terminal anchor sequences of 11beta-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. J Biol Chem. 1999;274:28762–70.
Hewitt KN, Walker EA, Stewart PM. Minireview: hexose-6-phosphate dehydrogenase and redox control of 11 beta-hydroxysteroid dehydrogenase type 1 activity. Endocrinology. 2005;146:2539–43.
Chou JY, Matern D, Mansfield BC, Chen YT. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr Mol Med. 2002;2:121–43.
Draper N, Walker EA, Bujalska IJ, Tomlinson JW, Chalder SM, Arlt W, et al. Mutations in the genes encoding 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6- phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nat Genet. 2003;34:434–9.
Bujalska IJ, Hewitt KN, Hauton D, Lavery GG, Tomlinson JW, Walker EA, et al. Lack of hexose-6-phosphate dehydrogenase impairs lipid mobilization from mouse adipose tissue. Endocrinology. 2008;149:2584–91.
Alberti L, Girola A, Gilardini L, Conti A, Cattaldo S, Micheletto G, et al. Type 2 diabetes and metabolic syndrome are associated with increased expression of 11beta-hydroxysteroid dehydrogenase 1 in obese subjects. Int J Obes. 2007;31:1826–31.
Uçkaya G, Karadurmus N, Kutlu O, Corakçi A, Kizildağ S, Ural AU, et al. Adipose tissue 11 beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase gene expressions are increased in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2008;82:S135–40.
Wang Y, Nakagawa Y, Liu L, Wang W, Ren X, Anghel A, et al. Tissue-specific dysregulation of hexose-6-phosphate dehydrogenase and glucose-6-phosphate transporter production in db/db mice as a model of type 2 diabetes. Diabetologia. 2011;54:440–50.
Yan C, Yang H, Wang Y, Dong Y, Yu F, Wu Y, et al. Increased glycogen synthase kinase-3β and hexose-6-phosphate dehydrogenase expression in adipose tissue may contribute to glucocorticoid-induced mouse visceral adiposity. Int J Obes. 2016;40:1233–41.
Wang Y, Liu L, Du H, Nagaoka Y, Fan W, Lutfy K, et al. Transgenic overexpression of hexose-6-phosphate dehydrogenase in adipose tissue causes local glucocorticoid amplification and lipolysis in male mice. Am J Physiol Endocrinol Metab. 2014;306:E543–51.
Stolic M, Russell A, Hutley L, Fielding G, Hay J, MacDonald G, et al. Glucose uptake and insulin action in human adipose tissue—influence of BMI, anatomical depot and body fat distribution. Int J Obes Relat Metab Disord. 2002;26:17–23.
Rönquist-Nii Y, Edlund PO. Determination of corticosteroids in tissue samples byliquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2005;37:341–50.
Szymczak J, Milewicz A, Thijssen JH, Blankenstein MA, Daroszewski J. Concentration of sex steroids in adipose tissue after menopause. Steroids. 1998;63:319–21.
Wang Y, Yan C, Liu L, Wang W, Du H, Fan W, et al. 11β-hydroxysteroid dehydrogenase type 1 shRNA ameliorates glucocorticoid-induced insulin resistance and lipolysis in mouse abdominal adipose tissue. Am J Physiol Endocrinol Metab. 2015;308:E84–95.
Sha H, He Y, Chen H, Wang C, Zenno A, Shi H, et al. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009;9:556–64.
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40.
Ozcan U, Cao Q, Yilmaz E, Lee AH, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61.
Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, et al. Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008;7:159–72.
Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–3.
Morgan SA, McCabe EL, Gathercole LL, Hassan-Smith ZK, Larner DP, Bujalska IJ, et al. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc Natl Acad Sci USA. 2014;111:E2482–91.
Kalderon B, Mayorek N, Berry E, Zevit N, Bar-Tana J. Fatty acid cycling in the fasting rat. Am J Physiol. 2000;279:E221–7.
Liu Y, Nakagawa Y, Wang Y, Sakurai R, Tripathi PV, Lutfy K, et al. Increased glucocorticoid receptor and 11β-hydroxysteroid dehydrogenase type 1 expression in hepatocytes may contribute to he phenotype of type 2 diabetes in db/db mice. Diabetes. 2005;54:32–40.
Yun SI, Yoon HY, Jeong SY, Chung YS. Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3β. J Bone Miner Metab. 2009;27:140–8.
Rubio-Patiño C, Palmeri CM, Pérez-Perarnau A, et al. Glycogen synthase kinase-3β is involved in liganddependent activation of transcription and cellular localization of the glucocorticoid receptor. Mol Endocrinol. 2012;26:1508–20.
Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3β-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. 2008;28:7309–22.
Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell. 2010;143:897–910.
Moon MH, Jeong JK, Lee JH, Park YG, Lee YJ, Seol JW, et al. Antiobesity activity of a sphingosine 1-phosphate analogue FTY720 observed in adipocytes and obese mouse model. Exp Mol Med. 2012;44:603–14.
Lin J, Yang R, Tarr PT, Wu PH, Handschin C, Li S, et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1β coactivation of SREBP. Cell. 2005;120:261–73.
Williams LJ, Lyons V, MacLeod I, Rajan V, Darlington GJ, Poli V, et al. C/EBP regulates hepatic transcription of 11beta-hydroxysteroid dehydrogenase type 1. A novel mechanism for cross-talk between the C/EBP and glucocorticoid signaling pathways. J Biol Chem. 2000;275:30232–9.
Galitzky J, Bouloumié A. Human visceral-fat-specific glucocorticoid tuning of adipogenesis. Cell Metab. 2013;18:3–5.
Yeh WC, Cao Z, Classon M, McKnight SL. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995;9:168–81.
Vidal-Puig A, Jimenez-Linan M, Lowell BB, Hamann A, Hu E, Spiegelman B, et al. Regulation of PPARγ gene expression by nutrition and obesity in rodents. J Clin Invest. 1996;97:2553–61.
Carey AL, Vorlander C, Reddy-Luthmoodoo M, Natoli AK, Formosa MF, Bertovic DA, et al. Reduced UCP-1 content in in vitro differentiated beige/brite adipocyte derived from preadipocytes of human subcutaneous white adipose tissues in obesity. PLoS ONE. 2014;9:e91997.
Fu T, Seok S, Choi S, Huang Z, Suino-Powell K, Xu HE, et al. MicroRNA 34a inhibits beige and brown fat formation in obesity in part by suppressing adipocyt fibroblast growth factor 21 signaling and SIRT1 function. Mol Cell Biol. 2014;34:4130–42.
Singh R, Braga M, Reddy ST, Lee SJ, Parveen M, Grilalva L, et al. Follistatin targets distinct pathways to promote brown adipocyte characteristics in brown and white adipose tissues. Endocrinology. 2017;158:1217–30.
Zhou JY, Zhong HJ, Yang C, Yan J, Wang HY, Jiang JX. Corticosterone exerts immunostimulatory effects on macrophages via endoplasmic reticulum stress. Br J Surg. 2010;97:281–93.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29.
Nekrutenko A, He J. Functionality of unspliced XBP1 is required to explain evolution of overlapping reading frames. Trends Genet. 2006;22:645–8.
Park SW, Zhou Y, Lee J, Lu A, Sun C, Chung J, et al. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat Med. 2010;16:429–37.
Author contributions
LL, YW, and JW contributed to the design and performance of experiments. LL, YW, JW, YD, SC, XL, and YL acquired and analyzed the data. MJ generated aP2- H6PDH transgenic mouse line. YL drafted and writing of the manuscript and supervised this project. MJ, KL, TCF, and CH reviewed this manuscript. All authors discussed and agreed on the results, and gave final approval.
Funding
YL is supported by NIH grant SC1DK104821 and LSI grant F00333003007. TCF is supported by NIH grant 2R24DA017298. LL is supported by grants from the Project of National Natural Science Foundation of China (8147102 and 8127086) and the Shanghai Leading Talent (SLJ15055). YD is supported by AHA grant 12SDG8760-002.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
These author contributed equally: L. Liu, Y.Wang and J. Wang
Rights and permissions
About this article

Cite this article
Liu, L., Wang, Y., Wang, J. et al. Enhanced hexose-6-phosphate dehydrogenase expression in adipose tissue may contribute to diet-induced visceral adiposity. Int J Obes 42, 1999–2011 (2018). https://doi.org/10.1038/s41366-018-0041-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-018-0041-1
- Springer Nature Limited