
Abstract
Background
Reverse electron transport (RET) driven by the oxidation of succinate has been proposed as the mechanism of accelerated production of reactive oxygen species (ROS) in post-ischemic mitochondria. However, it remains unclear whether upon reperfusion, mitochondria preferentially oxidase succinate.
Methods
Neonatal mice were subjected to Rice-Vannucci model of hypoxic-ischemic brain injury (HI) followed by assessment of Krebs cycle metabolites, mitochondrial substrate preference, and H2O2 generation rate in the ischemic brain.
Results
While brain mitochondria from control mice exhibited a rotenone-sensitive complex-I-dependent respiration, HI-brain mitochondria, at the initiation of reperfusion, demonstrated complex-II-dependent respiration, as rotenone minimally affected, but inhibition of complex-II ceased respiration. This was associated with a 30-fold increase of cerebral succinate concentration and significantly elevated H2O2 emission rate in HI-mice compared to controls. At 60 min of reperfusion, cerebral succinate content and the mitochondrial response to rotenone did not differ from that in controls.
Conclusion
These data are the first ex vivo evidence, that at the initiation of reperfusion, brain mitochondria transiently shift their metabolism from complex-I-dependent oxidation of NADH toward complex II-linked oxidation of succinate. Our study provides a critical piece of support for existence of the RET-dependent mechanism of elevated ROS production in reperfusion.
Similar content being viewed by others


Main
Perinatal hypoxic-ischemic (HI) brain injury is one of the major causes of permanent neurological handicap in newborn infants (1). Despite substantial progress in neonatal intensive care and the introduction of neonatal neurocritical care service, our current understanding of this disease does not allow for the development of mechanism-targeted neuroprotective interventions.
HI-brain injury comprises of acute oxygen and substrate deprivation caused by collapse of cerebral circulation. At the cellular level, HI-insult produces a severe bioenergetic crisis which results in cellular demise if nutrient and oxygen supply are not restored in a timely manner. If cerebral circulation is reestablished, then reperfusion initiates full or partial brain recovery. At the same time, reperfusion can also serve as a critical stage of post-ischemic injury. One of the leading mechanisms of reperfusion injury is oxidative stress in which mitochondria are recognized as the primary sites for excessive production of reactive oxygen species (ROS) (2). Robust elevation of ROS production in mitochondria has been associated with oxidation of succinate, which creates reverse electron transport (RET) from complex-II to complex-I (refs 3, 4). In vitro, RET in mitochondria generate ∼100-fold greater ROS compared to conventional, complex-I-dependent forward electron transport (FET, from complex-I to compex-IV) (5). Our previous reports have demonstrated that partial inhibition of the reperfusion-driven recovery of mitochondrial complex-I significantly reduced mitochondrial ROS generation and attenuated HI-brain damage (6, 7). In a model of focal stroke, inhibition of complex-I also afforded neuroprotection which was attributed to attenuation of oxidative stress (8). While these data highlight a pathogenic role of complex-I in post-ischemic oxidative damage, there is no direct evidence that complex-I releases ROS via RET. Recently, Chouchani et al. (9) have shown that in cardiac and brain ischemia, the extent of injury was strongly linked to accumulation of succinate. However, it has never been shown if upon reperfusion mitochondria indeed preferentially oxidize accumulated succinate. In respect to the developing brain, temporal changes in the Krebs cycle metabolites (except succinate) during HI and reperfusion has never been reported. This study addresses this gap in our knowledge and determines whether succinate indeed accumulates in the immature HI-brain and, most importantly, whether upon reperfusion, mitochondria preferentially metabolize succinate.
Methods
Model of neonatal HI-brain injury
All experiments were approved by Institutional Animal Care and Use Committee of Columbia University. HI-brain injury was produced using Rice-Vannucci model adapted to p10-12 neonatal mice (C57Bl6/J) as described previously (6, 7, 10, 11). In brief, the right carotid artery was permanently ligated under isoflurane anesthesia. Cerebral circulation to the hemisphere unilateral to the carotid artery ligation side is provided via Circle of Willis from the contralateral carotid artery. Hypoxic exposure to 8%/92% O2/N2 for 15 min, at an ambient temperature of 37±0.3 °C causes a collapse of systemic and cerebral circulation especially in the ipsilateral hemisphere (12). Reperfusion was achieved by re-oxygenation with room air, which restores cerebral blood flow in the ipsilateral, “ligated” hemisphere (12). Mice were klled at 0 (no reperfusion), 30, and 60 min of reperfusion, and the brain tissue (ischemic hemisphere) was immediately frozen in liquid nitrogen and stored at −80 °C for future measurement of the Krebs cycle metabolites.
Measurement of the Krebs cycle metabolites
Metabolites were analyzed via HPLC as previously described (13). Briefly, brain tissues were deproteinized with perchloric acid and pelleted via centrifugation. Butyrate was spiked into all samples as an internal standard. Metabolites in the supernatant were resolved using two Aminex HPX-87H columns (300 × 7.8 mm; Bio-Rad) in series, at a flow rate of 0.7 ml/min with 10 mM H2SO4 mobile phase, at 35 °C, with absorbance detection at 210 nm. α-keto acids and the Krebs cycle intermediates were quantitated using a 5-point calibration curve constructed with standard solutions.
Measurement of mitochondrial respiration
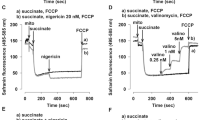
In a separate cohort of HI-mice, brains were obtained at 0, 30 and 60 min of reperfusion and homogenized on ice (five strokes in 7 ml Dounce, A-pestle). Mitochondrial respiration in these homogenates was measured within five minutes following brain isolation, at 32 °C, using a Clark-type electrode (Oxytherm, Hansatech, UK). Brain homogenate (1 mg of protein) was placed in 500 μl of respiration buffer (Tris MOPS 10 mM, KCl 120 mM, KH2PO4 1 mM, EGTA 10 μM, pH 7.4), containing glucose as a substrate (25 mM). Because the respiration buffer contained only glucose as a substrate, we reasoned that mitochondria “selected” substrates for their respiration, depending on their metabolic preference. After recording the initial, basal respiration rates, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 0.3 μM) was added to induce maximal respiration. Once FCCP-accelerated respiration reached a steady state rate, the homogenates were supplemented with complex-I inhibitor, rotenone (0.06 μM). Since mitochondrial respiration on NAD-linked substrates requires an active complex-I, the inhibition of complex-I with rotenone should cease respiration if the mitochondria preferentially oxidize NAD-linked substrates (Figure 1a). In contrast, if mitochondria respire on succinate, a FAD-linked substrate that donates electrons only to complex-II, then the inhibition of complex-I should not affect mitochondrial respiration (Figure 1a). Thus, the response to inhibition of complex-I indicates which substrate, succinate or NAD-linked substrates, are being used in the homogenate. To control for complex-II-dependent oxygen consumption, complex-II was blocked with atpenin (0.2 μM). Finally, non-mitochondrial oxygen consumption was determined by blocking cytochrome c oxidase (cyanide 2 mM). Residual respiration rate following supplementation of cyanide was subtracted from all analyzed rates.

(a) Tracing of brain mitochondrial (0.1 mg of protein/ml) respiration supported with either malate-glutamate (complex-I dependent substrate) or succinate (complex-II dependent substrate). Note, the response to rotenone (Rot) is completely different. (b,c) Cerebral Krebs cycle metabolite content in naïve (n=6), HI-mice at 0 (n=6), 30 (n=5) and 60 min of reperfusion (n=5). *P<0.05 compared to naive. (d) Representative HPLC elution profiles of Krebs cycle metabolites.
Measurement of mitochondrial H2O2 production
Mitochondrial H2O2 production rate was measured in cerebral homogenate using Amplex Ultra-Red assay as described previously (6). Briefly, 0.5 mg of homogenate was suspended in 1 ml of Tris-MOPS buffer supplemented with glucose (25 mM), Amplex Ultra-Red (10 μM), 4 U/ml of horseradish peroxidase (HRP, Invitrogen). H2O2 fluorescence was recorded for 400 s in the absence or in the presence of rotenone (0.06 μM). The calibration curve was obtained by adding several 100 nmol aliquots of freshly made H2O2 to the cuvette containing the respiration buffer, Amplex UltraRed, and HRP. The H2O2 production rate was expressed in pmoles/mg protein/minute. Changes in H2O2 production rate in response to rotenone was expressed in % relative to the basal H2O2 production rate (100%) without rotenone.
Statistical analysis
All data are expressed as mean±SEM. Differences in mitochondrial respiration, H2O2 production rate, and Krebs cycle metabolites between naïve and HI samples at each time-point of reperfusion were compared using Student t-test. Changes of the same variable during reperfusion were analyzed by analysis of variance for repeated measures with Fisher’s post hoc analysis. P value<0.05 was used to determine significance.
Results
The Krebs cycle metabolites
At the end of HI-insult (0 min of reperfusion), all measured NAD-linked substrates, except α-ketoglutarate, were either significantly decreased, or unchanged, compared to naïve controls (Figures 1b–d). With reperfusion, concentrations of NAD-linked substrates either normalized (pyruvate, α-ketoglutarate) or increased, significantly exceeding (malate) the concentrations in naïve samples (Figures 1b–d). The most drastic changes were detected in succinate which increased by ∼30-fold, compared to naïve mice (Figures 1b–d). At 60 min of reperfusion, the succinate level gradually decreased, reaching values similar to those in naïve mice (Figures 1b–d). This reperfusion-driven decline in succinate was associated with gradual elevation of fumarate (Figures 1c). As expected, the level of lactate was significantly elevated at the end of HI-insult and normalized by 60 min of reperfusion (Figures 1c and d).
Mitochondrial respiration
Analysis of mitochondrial respiration in homogenates demonstrated no significant differences in basal (mixed, states 4 and 3) and FCCP-accelerated respiration rates between HI-mice and naïve littermates at all time-points of reperfusion (Figures 2a–c). However, while in naive mice, rotenone almost fully blocked FCCP-accelerated respiration, HI-mice at 0 min of reperfusion exhibited remarkable preservation of their respiration under this condition (Figures 2a and d). At 30 min of reperfusion, the rotenone-insensitive respiration rate remained significantly greater in HI-mice compared to that in naïves. However, compared to the post-rotenone rate at 0 min of reperfusion, it was significantly decreased (Figures 2a and d). At 60 min of reperfusion, HI-mitochondria responded to rotenone in an identical manner to that of controls (Figures 2a and d). This coincided with normalization of succinate level (Figures 1b and d) and inhibition of complex-II suppressed respiration in HI-mice and naïve mice (Figure 2e).

(a) Representative tracings of glucose-supported mitochondrial respiration in brain homogenates obtained from naïve and HI-mice at 0 and 60 min of reperfusion (indicated). Note, a difference in response to inhibition of complex-I with rotenone in HI-samples at 0 min of reperfusion, compared to naïve and HI-samples at 60 min of reperfusion. Compare these data to that in the Figure 1a. (b–e) mitochondrial respiration rates in naive mice (n=11) and HI-mice at 0 (n=6), at 30 (n=6), at 60 min of reperfusion (n=7). *P=0.0001 compared to naives, P<0.0001 compared to HI-0.
Mitochondrial H2O2 production
In the absence of rotenone, basal H2O2 production rate was significantly greater in the homogenates isolated from HI mice at 0 min of reperfusion as compared to naïve mice and HI mice studied at 60 min of reperfusion (Figures 3a and b). Importantly, rotenone significantly (P=0.02) increased H2O2 production rate compared to the basal rate only in naïve mice. In contrast, compared to the basal rate, the same concentration of the rotenone significantly (P=0.0001) decreased H2O2 production rate in the HI-mice studied at 0 min of reperfusion (Figure 3c). In the presence of rotenone, both naïve and HI-mice studied at 60 min of reperfusion exhibited an elevation of the H2O2 production rate, which was significant compared to the rotenone-decreased H2O2 production rate in HI-mice examined at 0 min of reperfusion (Figure 3c).

(a) Basal H2O2 emission rates in naive mice (n=7) and HI-mice at 0 (n=7), at 60 min of reperfusion (n=7). *P=0.0002 compared to naives, P<0.0001 compared to HI-60. (b) Representative tracings of glucose-supported basal H2O2 production in brain homogenates obtained from naïve and HI-mice at 0 and 60 min of reperfusion (indicated). (c) Changes in H2O2 production rate in response to rotenone expressed in % relative to the basal H2O2 production rate without rotenone. *P=0.0002 compared to naives, P=0.002 compared to HI-60.
Discussion
Our study presents three novel findings: (a) in the developing brain HI-insult significantly alters the content of the Krebs cycle metabolites, leading to a dramatic but transient accumulation of succinate and elevation of α-ketoglutarate; (b) in contrast to naïve mice, HI-brain mitochondria at the initiation of reperfusion, markedly preserved their respiration following inhibition of complex-I. In these HI-mitochondria, only the inhibition of complex-II arrested respiration, indicating a shift toward complex-II-dependent mitochondrial metabolism. Finally, (c) the brain homogenate metabolizing glucose demonstrated completely reversed effect of complex-I inhibition on ROS release: rotenone increased production in naïve mice while in HI-mice studied at the initiation of reperfusion, rotenone decreased ROS production.
In the mature brain, ischemia changes concentrations of Krebs cycle metabolites: ∼8–10-fold reduction of all NAD-linked mitochondrial substrates and elevation of succinate content by threefold (14). Our results obtained from the immature HI-brain are in agreement with this report, showing a decrease of malate and pyruvate levels in response to neonatal HI. However, compared to that reported in adult rats, in neonatal rodents, the extent of succinate accumulation was substantially greater (∼30-fold) and concentration of α-ketoglutarate was increased (9, 14). In the model of neonatal asphyxia, significantly elevated levels of plasma-circulating succinate and other Krebs cycle metabolites (fumarate, malate) have been reported in newborn piglets and nonhuman primates (15, 16). By showing changes in the ischemic brain Krebs cycle metabolic profile during HI and reperfusion, our study not only complements these reports, but changes the research focus from the identification of circulating markers of HI-brain injury toward mechanistic significance of succinate accumulation and its preferential oxidation. In line with our data, the measurement of a single Krebs cycle metabolite revealed elevated cerebral succinate level in neonatal rats at the end of HI-insult (17).
In our study, mitochondrial respiration in ex-vivo isolated brain tissue was fueled with glucose. Thus, mitochondria “chose” endogenous substrates depending on their metabolic preferences. Because normoxic brains demonstrated near-full cessation of mitochondrial respiration in response to inhibition of complex-I, one might conclude that in normal brains, bioenergetics are mostly driven by the oxidation of NAD-linked substrates. In contrast, in the HI-brains, the recovery of mitochondrial respiration at the onset of reperfusion mostly depended on the oxidation of succinate. Indeed, the inhibition of complex-I in HI-mitochondria only partially suppressed respiration, but inhibition of complex-II fully ceased mitochondrial respiration. Given that succinate-fueled respiration does not require complex-I activity, we propose that in neonatal HI-brain, mitochondrial substrate utilization is shifted from oxidation of NAD-linked substrates toward the oxidation of succinate. Another important finding is the transient nature of this phenomenon, which is evidenced by gradual normalization of succinate levels, elevation of complex-II product fumarate, and by recovery of NAD-linked substrate concentrations with reperfusion. Most importantly, this was associated with a back-shift of cellular metabolism toward NAD-linked mitochondrial respiration, as at 60 min of reperfusion the response of mitochondrial respiration to inhibition of complex-I was similar to naïve mice. This result is consistent with near-full recovery of NAD-linked mitochondrial respiration with reperfusion (6). In addition, earlier studies have reported a significantly greater tolerance of complex-II to ischemic depression, compared to complex-I (6, 18, 19). This suggests that at the initiation of reperfusion not only is succinate readily available, but also that complex-II is capable of initiating the recovery of mitochondrial respiration.
The mechanistic role in significant increase of mitochondrial ROS production at the onset of reperfusion was assigned to RET. The RET can be supported by oxidation of succinate and generates ROS in complex-I. There are three biological conditions that have to be present for the existence of this mechanism in vivo: (1) relatively active complex-I, (2) availability of succinate and (3) preferential oxidation of succinate in mitochondria. The first condition can be supported by attenuation of oxidative damage to the ischemic brain and heart by partial deactivation of complex-I (6, 7, 8, 20). Attenuation of post-ischemic oxidative stress and suppression of mitochondrial ROS release achieved by the inhibition of succinate accumulation during ischemia (9) strongly argues in favor of the second condition. Our study, by demonstrating preferential succinate oxidation in the mitochondria isolated at the onset of reperfusion adds a critical third piece of evidence in support of a RET-driven mechanism for accelerated mitochondrial ROS release during early reperfusion. Furthermore, here we show that post-HI mitochondria at the initiation of reperfusion generate significantly greater amounts of ROS compared to naïve mice and HI-mice studied at 60 min of reperfusion. Most importantly in these mitochondria which were fueled only with glucose, the response to rotenone differed radically as naïve organelles increased H2O2 emission rate while HI-mitochondria decreased H2O2 emission. This strongly suggests that the elevation in H2O2 production in HI-mitochondria is supported by RET which is sensitive to the rotenone inhibition. It is also well established that mitochondria fueled with NAD-linked substrates increase their H2O2 production in the presence of rotenone (21).
Potential mechanistic significance of this work also relates to succinate-dependent bioenergetics recovery following ischemia. Considering post-ischemic partial inhibition of complex-I activity, preferential mitochondrial oxidation of accumulated succinate is critical for ATP replenishment at the initiation of reperfusion. Earlier we have shown that inhibition of complex-II significantly exacerbates brain injury in this model (6). In cardiac ischemia-reperfusion injury, supplementation of succinate into cardioplegic solution significantly reduced myocardial infarction (22). Neuroprotection associated with ketogenic diet or administration β-hydroxybutyrate in focal stroke was also linked to pre-ischemic accumulation of succinate (23). In neonatal HI-brain injury, the accumulation of succinate was attributed to improved neo-angiogenesis and neuro-recovery governed by G-protein coupled receptor 91 (17). Taken together with evidence for a pathogenic role of succinate accumulation in cardiac and brain ischemia-reperfusion injury (6, 7, 9, 24), these data support an existence of pro-survival and pathogenic mechanisms induced by post-HI accumulation and preferential oxidation of succinate in the developing brain.
In conclusion, our work demonstrates that compared to the normal brains, which mostly oxidize NAD-linked substrates, HI-brains at the initial stage of reperfusion shift their mitochondrial metabolism towards a preferential oxidation of succinate, the event strongly associated with RET-driven elevated release of ROS in mitochondria.
References
Glass HC, Rowitch DH . The role of the neurointensive care nursery for neonatal encephalopathy. Clin Perinatol 2016;43:547–557.
Murphy MP . Understanding and preventing mitochondrial oxidative damage. Biochem Soc Trans 2016;44:1219–1226.
Chance B, Hollunger G . Energy-linked reduction of mitochondrial pyridine nucleotide. Nature 1960;185:666–672.
Kotlyar AB, Vinogradov AD . Slow active/inactive transition of the mitochondrial NADH- ubiquinone reductase. Biochim Biophys Acta 1990;1019:151–158.
Starkov AA . The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci 2008;1147:37–52.
Niatsetskaya ZV, Sosunov SA, Matsiukevich D et al. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. J Neurosci 2012;32:3235–3244.
Sosunov SA, Ameer X, Niatsetskaya ZV et al. Isoflurane anesthesia initiated at the onset of reperfusion attenuates oxidative and hypoxic-ischemic brain injury. PLoS ONE 2015;10:e0120456.
Piantadosi CA, Zhang J . Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke 1996;27:327–331.
Chouchani ET, Pell VR, Gaude E et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014;515:431–435.
Ten VS, Wu EX, Tang H et al. Late measures of brain injury after neonatal hypoxia-ischemia in mice. Stroke 2004;35:2183–2188.
Rice JE 3rd, Vannucci RC, Brierley JB . The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 1981;9:131–134.
Matsiukevich D, Randis TM, Utkina-Sosunova I, Polin RA, Ten VS . The state of systemic circulation, collapsed or preserved defines the need for hyperoxic or normoxic resuscitation in neonatal mice with hypoxia-ischemia. Resuscitation 2010;81:224–229.
Wojtovich AP, Brookes PS . The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim Biophys Acta 2008;1777:882–889.
Folbergrova J, Ljunggren B, Norberg K et al. Influence of complete ischemia on glycolytic metabolites, citric acid cycle intermediates, and associated amino acids in the rat cerebral cortex. Brain Res 1974;80:265–279.
Solberg R, Enot D, Deigner HP et al. Metabolomic analyses of plasma reveals new insights into asphyxia and resuscitation in pigs. PLoS ONE 2010;5:e9606.
Chun PT, McPherson RJ, Marney LC et al. Serial plasma metabolites following hypoxic-ischemic encephalopathy in a nonhuman primate model. Dev Neurosci 2015;37:161–171.
Hamel D, Sanchez M, Duhamel F et al. G-protein-coupled receptor 91 and succinate are key contributors in neonatal postcerebral hypoxia-ischemia recovery. Arterioscler Thromb Vasc Biol 2014;34:285–293.
Sims NR . Selective impairment of respiration in mitochondria isolated from brain subregions following transient forebrain ischemia in the rat. J Neurochem 1991;56:1836–1844.
Almeida A, Allen KL, Bates TE et al. Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J Neurochem 1995;65:698–703.
Ten VS, Starkov A . Hypoxic-ischemic injury in the developing brain: the role of reactive oxygen species originating in mitochondria. Neurol Res Int 2012;2012:542976.
Chen Q, Moghaddas S, Hoppel CL et al. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther 2006;319:1405–1412.
Sakamoto M, Takeshige K, Yasui H et al. Cardio protective effect of succinate against ischemia/reperfusion injury. Surg Today 1998;28:522–528.
Puchowicz MA, Zechel JL, Valerio J et al. Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J Cereb Blood Flow Metab 2008;28:1907–1916.
Pell VR, Chouchani ET, Frezza C et al. Succinate metabolism: a new therapeutic target for myocardial reperfusion injury. Cardiovasc Res 2016;111:134–141.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Statement of Financial Support
This work was supported by NIH grant NS-100850 (V.T., A.S.), MRC grant MR/L007339/1 (A.G.), HL-071158 (P.S.B.), and by an American Heart Association pre-doctoral fellowship 17PRE33410360 (J.Z.)
Rights and permissions
About this article

Cite this article
Sahni, P., Zhang, J., Sosunov, S. et al. Krebs cycle metabolites and preferential succinate oxidation following neonatal hypoxic-ischemic brain injury in mice. Pediatr Res 83, 491–497 (2018). https://doi.org/10.1038/pr.2017.277
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.277
- Springer Nature America, Inc.
This article is cited by
-
Oxygen therapy of the newborn from molecular understanding to clinical practice
Pediatric Research (2019)