
Abstract
Background:
Diagnosis of adrenocortical neoplasms (ACN), in pediatric age, is based on Wieneke criteria. The p57, a cyclin-dependent kinase inhibitor, acts to negatively regulate cell proliferation and is frequently found dysregulated in cancer. The identification of loss of heterozygosity (LOH) of 11p15, containing the p57 gene, could be a tool for differential diagnosis of benign and malignant ACN.
Methods:
Immunohistochemistry with anti-p57 and microsatellite markers analysis of 11p15 region to value LOH were made in both ACN and surrounded normal adrenal cortex.
Results:
Nine ACN, two clinically benign, two uncertain, and five malignant, were diagnosed. Positive p57 cells were evident in normal adrenal cortex and in one histologically benign ACN. A low/absent expression of p57 was documented in eight ACN independently from the classification on the basis of pathological and clinical criteria. Microsatellite marker analysis confirmed the LOH of 11p15 region in the same ACN.
Conclusion:
LOH of 11p15 has a high prognostic value suggesting the p57 gene is important in ACN pathogenesis. Immunohistochemistry for p57 is a simple and cheap tool that can be used to quickly identify LOH of 11p15 in ACN.
Similar content being viewed by others


Main
Adrenocortical neoplasms (ACN) are rare in pediatric age. Approximately 60% of children with malignant ACN are diagnosed in infancy (1,2). In the neonatal period they are diagnosed either for early virilizing symptoms or discovered incidentally. Although ACN in children can manifest in the context of a familial cancer syndrome (such as Li–Fraumeni or Beckwith–Wiedemann syndromes), the majority of pediatric ACNs arise sporadically (3). Rearrangements, loss of heterozygosity (LOH, loss of one of two alleles of a gene), and abnormal imprinting of the 11p15.5 locus, resulting in low p57 and H19 with elevated insulin growth factor 2 (IGF2) mRNA expression levels, have been reported in sporadic ACNs (4). p57, 700 kb centromeric to the H19/IGF2 locus, is paternally imprinted and expressed from the maternal allele. It encodes a cyclin-dependent kinase (CDK) inhibitor that acts to negatively regulate cell proliferation and to actively direct differentiation (5,6). This gene is particularly expressed in adrenal tissues. In normal adrenal gland, by immunohistochemistry (IHC), cells expressing p57 are confined to the cortical zone, with absent labeling in the medulla (7).
In this study, we have used IHC for p57 to identify ACN associated with LOH of 11p15 in nine children. Since 11p15 LOH has a high prognostic value, being a tumor suppressor and a regulator of embryonic growth, the identification of imprinting errors of the 11p15 region could be another criterion to be considered as a tool in the diagnosis of malignancy in ACN (8).
Patients and Methods
Reviewing our database from 1998 to 2015, a total of nine children with adrenal tumors were recorded. The study was approved by the Bambino Gesù Children’s Hospital Review Board. Clinical and pathologic data are summarized in Table 1 . Patients underwent clinical, radiological, and hormonal evaluation. Histologic diagnosis of benign, uncertain, and malignant adrenocortical tumors was based on widely accepted criteria, including tumor mass, presence of metastasis or recurrence, mitotic rate, nuclear pleomorphism, necrosis and capsular and/or vascular invasion (9). In all cases some normal adrenal tissue was present at the periphery of the neoplastic nodules. Tissue specimens, removed at surgery, were formalin-fixed and paraffin embedded and a small sample of both neoplastic and normal adrenal tissue were snap-frozen in liquid nitrogen and stored at −80°C until required for DNA extraction. To avoid contamination with surrounding tissue, non-neoplastic areas were separated macroscopically. Blood samples were available for all patients except two. Blood and tissues were collected after informed consent given by the patients and with the approval of the local ethic committee.
One child presented neonatal hypoglycemia and during surgical removal of AC mass, a small nodule in body pancreas was observed and took away.
Immunohistochemistry
Nine ACNs were analyzed using formalin-fixed and paraffin embedded tissue. Immunohistochemistry was done with anti-p57 and anti-IGF2 antibody as previously described (10). The following primary antibodies were used: a goat affinity-purified polyclonal antibody raised against a peptide corresponding to amino acids 286 - 305 mapping at the carboxy terminus of human p57 (C-20 cat. no. sc-1040 Santa Cruz Biotechnology, Santa Cruz, CA) and a rabbit affinity-purified polyclonal antibody IGF2 (1:500, Abcam, Cambridge, MA). The p57 immunostaining was strictly nuclear: 10 - 20% cells expressing p57 was in the zona reticularis, 5 - 10% in the zona glomerulosa, while the zona fasciculata was devoid of positive cells. The IGF2 immunostaining was definitely perinuclear with or without significant cytoplasmic staining: the medulla was diffusely and strongly positive, the zona fasciculata was negative whereas the zona reticularis and glomerulosa showed speckled cytoplasmic positivity without perinuclear accumulation.
To confirm β-cell hyperplasia anti-insulin (1:80 mouse monoclonal, Novocastra, Newcastle upon Tyne, UK) was used.
Microsatellite Analysis
All ACNs was analyzed for LOH of the 11p15 region. To evaluate the deletion size, 13 different microsatellite markers covering 20 Mb from 11p15.5 region (containing imprinted genes H19, IGF2, and p57) to 11p15.1 region were studied by PCR on frozen material, blood or formalin-fixed specimens. Selected markers for the 11p15 region were from telomere to centromere (NCBI database): D11S2071, D11S4177, D11S922, D11S4046, THO, D11S1318, D11S4088, D11S4181, D11S1331, D11S875, D11S921, D11S902, and D11S899. DNA was extracted from blood, normal adrenal tissue, and ACN following the QIAMP DNA minikit protocol (QIAGEN, Hilden, DE). Fluorescent markers were amplified using Kapa 2G Fast HS (Kapa Biosystems, Woburn, MA). PCR products were analyzed by an ABI 3130xl sequencer and ABI Genemapper v 3.0 software (Applied Biosystems, Carlsbad, CA).
Results
Histology
All nine tumors were circumscribed and encapsulated. The tumors showed mild to moderate nuclear pleomorphism with prominent nucleoli. Five cases displayed capsular invasion and four cases had evidence of venous invasion. The majority of cases had a diffuse or solid growth pattern in >30% of the tumor. Cellularity varied from medium to high. The mean number of mitotic figures was low (<15/20 HPF, with a range of 2–9 mitoses) in seven, and high (≥15/20 HPF) in two. Rare atypical mitotic figures were observed. Necrosis was present in many cases (six out of nine) and microcalcifications were recognized in five cases.
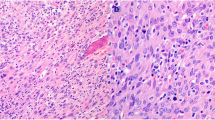
On the base of the microscopic features, according to the Wieneke et al. criteria (9), in our cohort we could recognize: two histologically benign ACN ( Figure 1a ), two ACN with uncertain biological behavior and five histologically malignant ACN ( Figure 1b ).

Benign and malignant ACN immunohistochemistry. (a) Benign adrenal cortical neoplasia: a thick capsul compresses the adrenal cortex (H&E, 10×). (b) A malignant cortical adrenal neoplasia. Highly cellular tumor: cells with compact eosinophilic cytoplasm and nuclear pleomorfism (H&E, 20×). Inset: neoplastic emboli within vascular lumen (10×). (c) p57 immunohistochemistry in benign adrenocortical neoplasms (ACN) with diffuse nuclear staining (10×) and (d) in malignant ACN with sporadic positive neclei (20×). Inset positive nuclei in normal adrenal gland (10×). (e) Insulin Growth Factor 2 (IGF2) immunohistochemistry in benign ACN labels sporadic neoplastic cells (10×) and (f) in malignant ACN: cytoplasm is strongly and diffusely stained (20×). Inset: negative cytoplasm in normal adrenal gland (10×).
On histology the pancreatic nodule was characterized by focal β-cell adenomatous hyperplasia consistent with congenital focal hyperinsulinism.
Immunohistochemistry
A preserved nuclear expression of p57 was observed in the two cases histologically classified as benign ACN ( Figure 1c ). A moderate reduced expression (only multifocal positive nuclei) was evident in two ACN characterized by atypical morphology, but lacking consistent pathological features of malignancy (clinical uncertain ACN) (9). Marked decrease of p57 expression (~80%) was documented in all five malignant ACNs ( Figure 1d ). IGF2 was expressed in all ACN ( Figure 1f ) except that in one benign ACN ( Figure 1e ) and one malignant ACN.
Within focal β-cell adenomatous hyperplasia, anti-insulin positive ( Figure 2a ), was observed absence of p57 expression, while positive nuclei were evident in normal β-cell in Langerhans inlets ( Figure 2b ).

Focal adenomatous hyperplasia of β-cell. (a) Insulin immunohistochemistry showed focal adenomatous hyperplasia of β-cells (4×). (b) p57 was positive in occasional normal β-cells in Langerhans inlets inside adenomatous hyperplasia (20×).
LOH in Adrenal Tumor
Table 2 shows the genetic map of the tested microsatellites. For informative cases, allelic loss is documented if one allele is significantly decreased (<50%) in cells from the ACN compared with the same allele coming from normal tissue and blood. All tested cases of ACN showed extensive 11p15 LOH involving imprinted genes (IGF2, H19, and p57). In one benign ACN microsatellites analysis displayed a preserved alleles asset till p57 (comprehensive of IGF2 and H19), while a LOH was observed in the region below.
Discussion
The prognosis for adrenocortical adenomas and carcinomas is vastly different so it is important to accurately differentiate the two tumor types. The prognosis for malignant ACN is largely dependent on grading and staging of the tumor, but not on histology. Since children with ACN appear to have a more favorable outcome than adults and Weiss score (11) is not a good predictor of prognosis based on histopathological features (12), it is preferable to classify pediatric ACN as clinically benign or clinical malignant tumors, as suggested by Wieneke et al. (9), classification that over time has been validated by other authors (13,14).
Recently several potential biomarkers have been studied to predict the biological behavior and a therapeutic target in childhood ACN. High IGF1R expression (15), low expression of HLA class II antigens (HLA-DRA, HLA-DPA1, and HLA-DPB1) (16) and high CD105 and low CD34 intratumoral microvessel density and VEGF expression above 4.8% (17) on IHC were associated with more aggressive pediatric ACN.
ACNs in children seem to be linked to the physiologic transition from fetus to newborn. After birth, there is a rapid structural and functional remodeling of the fetal zone of the adrenal cortex, a process that provides a balance between proliferation, apoptosis, and differentiation. Adrenal cellular proliferation is regulated by factors that activate or inhibit the cell cycle.
The p57, a check-point of cell cycle, is frequently found dysregulated in cancer. The p57, normally expressed from the maternal allele, is located within an imprinted region on chromosome 11 along with IGF2 and H19. Genetic and epigenetic alterations (LOH or loss of imprinting) in that chromosomal region are associated with sporadic ACN as well as with Beckwith–Wiedemann syndrome. Gene profiling studies have shown that adrenocortical tumors contain only 20% of adult adrenal p57 mRNA levels (18).
The p57 IHC is a helpful tool for the identification of LOH in ACN. In our series all ACN with LOH displayed on IHC a decrease in the p57 staining, while the benign ACN with the p57 biallelic expression preserved, showed a widespread label. Since 11p15 abnormality was found more frequently in clinical malignant ACN than in clinical benign neoplasm (19), it could be considered as further criteria for the differentiation of ACN. Likewise IGF2 overexpression could be evaluated by immunohistochemistry, taking into consideration that IGF2 is expressed at high levels in the fetal adrenal where it plays an important role in regulating proliferation and steroid production (20).
Four patients presented the association with a TP53 mutation. TP53 mutations have a fundamental role in AC carcinoma in the context of Li–Fraumeni syndrome. TP53 has a wide spectrum of functional activity. Mutations that produce near-total loss of tumor-suppressor activity and perturbations in proapoptotic signaling may concur to uncontrolled cell proliferation in adrenal cortex.
Conclusion
LOH of 11p15 is more frequently associated with malignant than with benign adrenocortical neoplasia (78.5 vs. 9.5%) and therefore, it is linked with a higher risk of tumor recurrence. For this reason 11p15 alterations could be used as a biological marker for predicting the clinical behavior in AC neoplasm regardless of the pathologic features.
Immunohistochemical expression of p57 is sharply decreased in malignant ACN in contrast to a preserved staining in benign ACN. Immunohistochemistry with anti-p57 (and anti-IGF2) is a simple and cheap tool that can be used to identified LOH in these neoplasms and may be useful in predicting the outcome and allow the patient to take advantage to the maximum of a close clinical follow-up, further surgery, or the application of adjuvant therapies.
Due to the small number of cases, validation in a larger number of cases as well as in an independent tumor cohort is necessary to confirm these results.
Statement of Financial Support
No financial assistance was received to support this study.
Disclosure
There is no conflict of interest to disclose.
References
Faria AM, Almeida MQ. Differences in the molecular mechanisms of adrenocortical tumorigenesis between children and adults. Mol Cell Endocrinol 2012;351:52–7.
Michalkiewicz E, Sandrini R, Figueiredo B, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol 2004;22:838–45.
Barlaskar FM, Hammer GD. The molecular genetics of adrenocortical carcinoma. Rev Endocr Metab Disord 2007;8:343–8.
Sidhu S, Gicquel C, Bambach CP, et al. Clinical and molecular aspects of adrenocortical tumourigenesis. ANZ J Surg 2003;73:727–38.
Bourcigaux N, Gaston V, Logié A, Bertagna X, Le Bouc Y, Gicquel C. High expression of cyclin E and G1 CDK and loss of function of p57KIP2 are involved in proliferation of malignant sporadic adrenocortical tumors. J Clin Endocrinol Metab 2000;85:322–30.
Matsuoka S, Thompson JS, Edwards MC, et al. Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15. Proc Natl Acad Sci USA 1996;93:3026–30.
Barzon L, Chilosi M, Fallo F, et al. Molecular analysis of CDKN1C and TP53 in sporadic adrenal tumors. Eur J Endocrinol 2001;145:207–12.
Gicquel C, Bertagna X, Gaston V, et al. Molecular markers and long-term recurrences in a large cohort of patients with sporadic adrenocortical tumors. Cancer Res 2001;61:6762–7.
Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol 2003;27:867–81.
Kassem SA, Ariel I, Thornton PS, et al. p57(KIP2) expression in normal islet cells and in hyperinsulinism of infancy. Diabetes 2001;50:2763–9.
Weiss LM, Medeiros LJ, Vickery AL Jr . Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol 1989;13:202–6.
Dehner LP, Hill DA. Adrenal cortical neoplasms in children: why so many carcinomas and yet so many survivors? Pediatr Dev Pathol 2009;12:284–91.
Dall’Igna P, Virgone C, De Salvo GL, et al. Adrenocortical tumors in Italian children: analysis of clinical characteristics and P53 status. Data from the national registries. J Pediatr Surg 2014;49:1367–71.
Magro G, Esposito G, Cecchetto G, et al. Pediatric adrenocortical tumors: morphological diagnostic criteria and immunohistochemical expression of matrix metalloproteinase type 2 and human leucocyte-associated antigen (HLA) class II antigens. Results from the Italian Pediatric Rare Tumor (TREP) Study project. Hum Pathol 2012;43:31–9.
Peixoto Lira RC, Fedatto PF, Marco Antonio DS, et al. IGF2 and IGF1R in pediatric adrenocortical tumors: roles in metastasis and steroidogenesis. Endocr Relat Cancer 2015;23:481–93.
Leite FA, Lira RC, Fedatto PF, et al. Low expression of HLA-DRA, HLA-DPA1, and HLA-DPB1 is associated with poor prognosis in pediatric adrenocortical tumors (ACT). Pediatr Blood Cancer 2014;61:1940–8.
Dias AI, Fachin CG, Avó LR, et al. Correlation between selected angiogenic markers and prognosis in pediatric adrenocortical tumors: Angiogenic markers and prognosis in pediatric ACTs. J Pediatr Surg 2015;50:1323–8.
Rainey WE, Carr BR, Wang ZN, Parker CR Jr . Gene profiling of human fetal and adult adrenals. J Endocrinol 2001;171:209–15.
Leboulleux S, Gaston V, Boulle N, Le Bouc Y, Gicquel C. Loss of heterozygosity at the mannose 6-phosphate/insulin-like growth factor 2 receptor locus: a frequent but late event in adrenocortical tumorigenesis. Eur J Endocrinol 2001;144:163–8.
Lalli E, Figueiredo BC. Pediatric adrenocortical tumors: what they can tell us on adrenal development and comparison with adult adrenal tumors. Front Endocrinol (Lausanne) 2015;6:23.
Acknowledgements
The authors thank Marta Stefanelli for IHC and Gabriele Bacile for his assistance with the figures.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Giovannoni, I., Boldrini, R., Benedetti, M. et al. Pediatric adrenocortical neoplasms: immunohistochemical expression of p57 identifies loss of heterozygosity and abnormal imprinting of the 11p15.5. Pediatr Res 81, 468–472 (2017). https://doi.org/10.1038/pr.2016.239
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.239
- Springer Nature America, Inc.