
Abstract
Background:
Heterozygous mutations of SFTPC, the gene-encoding surfactant protein C (SP-C), result in interstitial lung disease (ILD). However, characterization of mutations located in the mature domain of precursor SP-C (proSP-C) is limited. This study examined the molecular pathogenesis of such a mutation of ILD.
Methods:
We employed sequencing of SFTPC and established A549 cells stably expressing several proSP-C mutants. Histopathology and transmission electron microscopy (TEM) of lung tissue from a pediatric patient with ILD were assessed. Effects of mutant proSP-C were evaluated by western blotting, immunofluorescence, and TEM.
Results:
Sequencing of SFTPC revealed a novel heterozygous mutation, c.163C>T (L55F). In lung tissue, abnormal localization of proSP-C was observed by immunohistochemistry, and small and dense lamellar bodies (LBs) in type II alveolar epithelial cells (AECs) were detected by TEM. TEM of A549 cells stably expressing proSP-CL55F displayed abnormal cytoplasmic organelles. ProSP-CL55F exhibited a band pattern similar to that of proSP-CWT for processed intermediates. Immunofluorescence studies demonstrated that proSP-CL55F partially colocalized in CD63-positive cytoplasmic vesicles of A549 cells, which was in contrast to proSP-CWT.
Conclusion:
We detected a novel c.163C>T mutation located in the mature domain of SFTPC associated with ILD that altered the subcellular localization of proSP-C in A549 cells.
Similar content being viewed by others


Main
The hydrophobic surfactant protein C (SP-C) is synthesized from the 191 or 197-amino-acid precursor SP-C (proSP-C), which is encoded by SFTPC on human chromosome 8p21.3. ProSP-C is an integral membrane protein that contains four regions: a short cytoplasmic N-terminal domain (residues 1–23), a mature SP-C region (residues 24–58) mainly composed of a transmembrane domain (residues 33–58), a non-BRICHOS region (residues 59–89), and a BRICHOS domain (residues 90–197) (1). ProSP-C undergoes palmitoylation and a series of proteolytic C- and N-terminal cleavages to yield a 3.7-kDa mature peptide within subcellular compartments of the endoplasmic reticulum (ER), multivesicular bodies, lamellar bodies (LBs), and others (2). Mature SP-C is exclusively produced by type II alveolar epithelial cells (AECs), packaged into LBs, which are a subclass of lysosomal-like organelles, and subsequently secreted to the alveolar surface. Meanwhile, phospholipids and proteins are endocytosed from the alveolar surface into early endosomes and routed back either to lysosomes for degradation or LBs for recycling (3).
Interstitial lung disease (ILD) in infants and children represents a heterogeneous group of respiratory disorders that are mostly chronic and associated with higher morbidity and mortality than those reported in adults (4,5,6). SP-C is critical in reducing alveolar surface tension at the air/liquid interface to prevent alveolar collapse at the end of expiration. Since the first report of an infant and mother with ILD due to a mutation in the SFTPC gene (4), several additional mutations have been identified in ILD patients that were associated with abnormal expression of SP-C (7,8). The great majority of SFTPC mutations are located in the BRICHOS domain, and associated with chronic accumulation of misfolded proSP-C (9,10). In contrast, in vitro studies have shown that mutations located in the non-BRICHOS region, such as proSP-CI73T, misdirected proSP-C to the plasma membrane, leading to the accumulation of abnormally processed isoforms (11). Thus, mutations involving different domains of proSP-C may represent different pathogeneses of ILD. Although several mutations located within the mature domain of SFTPC have been recognized (7,8,12), the pathway and mechanism of such mutations remain unclear.
In the present study, we describe a novel heterozygous SFTPC mutation associated with ILD in a young Japanese girl. Lung biopsy samples performed using transmission electron microscopy (TEM) revealed abnormal subcellular organelles. We also investigated whether the subcellular localization of novel proSP-CL55F was altered in A549 cells.
Results
Case Summary
The patient was a young girl who had been delivered at full-term to nonconsanguineous parents without any apparent perinatal diseases. She presented with poor feeding at 2 mo of age and exhibited a failure to thrive. She was hospitalized at the age of 5 mo due to cough and tachypnea. High-resolution computed tomography at the age of 5 mo showed bilateral ground glass opacities and minimal reticulation ( Figure 1a ). Histopathological evaluation of her lung tissue by open-lung biopsy revealed type II AEC hyperplasia and alveolar interstitial fibrosis with obvious inflammation ( Figure 1d ) as compared with a control ( Figure 1c ). According to clinical features, biochemical indicators, and radiographic and pathological findings, she was given a diagnosis of ILD. She was treated with methylprednisolone pulse therapy followed by oral prednisolone in addition to cyclosporine and azathioprine. Due to her persistently elevated supplementary oxygen requirement, the patient required conventional mechanical ventilation and hydroxychloroquine was added to her regimen. These treatments were associated with a slowing of her disease progression, and she was discharged at 2 y of age with home oxygen therapy. At the age of 10 y, her pulmonary function began to deteriorate rapidly. She successfully underwent living-donor right single-lobe lung transplantation from her mother soon afterwards (13). Three months after transplantation, high-resolution computed tomography demonstrated a well-inflated lung allograft without obvious atelectasis ( Figure 1b ).

Characteristics of interstitial lung disease in this patient. (a,b) High-resolution computed tomography (HRCT) images observed before (a) and after (b) right single-lobe lung transplantation. (a) HRCT image at 5 mo of age showed bilateral ground-glass pulmonary opacities, small cysts, and several thickened septae. (b) HRCT revealed normal right lung parenchyma and a round tracheal shape after lung transplantation. (c) Normal lung tissue, hematoxylin and eosin. (d) The patient’s lung tissue showed interstitial thickening, type II alveolar epithelial cell hyperplasia, and alveolar interstitial fibrosis with obvious inflammation consisting of lymphocytes, plasma cells, and macrophages, hematoxylin and eosin (scale bars = 100 μm).
SFTPC Genetic Analysis
Polymerase chain reaction (PCR) followed by direct sequencing of SFTPC, SFTPB, and ATP-binding cassette subfamily A, member 3 (ABCA3) were performed, and three heterozygous substitutions at bases c.163, c.413, and c.557 were detected in SFTPC (Supplementary Figure S1 online). The c.163C>T in exon 2 located at codon 55 in the mature transmembrane domain resulted in a leucine to phenylalanine substitution (p.L55F). Neither 61 healthy Japanese volunteers, nor the patient’s parents, carried this c.163C>T mutation.
The patient’s mother and several volunteers also possessed substitutions of c.413C>A and c.557G>A, which have been described as common coding SNPs in previous studies (14). Paternal gene sequences were identical to those of healthy control volunteers.
Lung Histology
To evaluate whether the proSP-C mutation influenced the expression of surfactant proteins or ABCA3, immunohistochemical assessment of the girl’s lung tissue specimens was performed. In type II AECs, proSP-C exhibited eccentrically located cytoplasmic distribution adjacent to subcellular organelles ( Figure 2c , d ) in comparison with normal controls ( Figure 2a , b ). Immunostaining for SP-B, precursor SP-B (proSP-B), and ABCA3 showed granular cytoplasmic expression in type II AECs of the patient (Supplementary Figure S2a–f online) in amounts comparable to those of controls (data not shown).

Immunohistochemical staining for precursor surfactant protein C (proSP-C) and electron microscopic images of type II alveolar epithelial cells (AECs). (a–d) Immunohistochemical staining of lung tissue samples for proSP-C. Immunostaining for proSP-C was detected in lung tissue specimens from a control (a) and the patient (c) at low-power magnification (scale bars = 100 μm). Solid boxes were magnified on the left for better resolution (right) (scale bars = 50 μm). (b) Normal staining for proSP-C in type II alveolar epithelial cells (AECs) of normal lung tissue. (d) Type II AECs of patient lung tissue showed abnormal focal brown staining of the cytoplasm adjacent to subcellular organelles. (e–h) Ultrastructure of type II AECs. Type II AECs of normal lung tissue (e) and the patient (g) at low-power magnification (scale bars = 2 μm). Solid boxes were magnified on the left for better resolution (right) (scale bars = 500 nm). (f) High-power magnification of normal lung type II AECs showed numerous lamellar bodies (LBs) filled with concentric lamellae having diameters of 1–2 μm. (h) High-power magnification of patient lung type II AEC showed multiple smaller condensed LBs with an electron-dense core (dark arrows).
Congo red is a well-known marker for β-sheet amyloid aggregates. A common feature seen in BRICHOS domain mutations of proSP-C is a high tendency to misfold and form Congo red-positive aggregates (15,16). However, we did not detect any amyloid fibrils as identified by Congo red-positivity as green birefringent materials in the explanted lung tissue of the patient (Supplementary Figure S2g,h online).
TEM of Lung Tissue
Patient lung tissue samples were examined by TEM to characterize the alterations in subcellular organelles related to ILD. Smaller, denser, and abnormally shaped LBs were observed in type II AECs as 0.5 μm organelles ( Figure 2g , h ), which was in contrast to normal LBs having regular concentric “onion skin” lamellae and a diameter of 1–2 μm ( Figure 2e , f ).
Processing of ProSP-CL55F
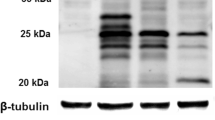
To identify potential processing differences between proSP-CL55F and previously reported mutations, we analyzed proSP-CI73T (17) and proSP-CA116D (18) in the non-BRICHOS and BRICHOS domains of proSP-C, respectively. Semiquantitative PCR showed that A549 cells stably expressing green fluorescent protein (GFP)-tagged proSP-C isoforms expressed comparable levels of proSP-C mRNA (data not shown). Lysates of A549 cells stably expressing GFP-tagged proSP-C isoforms were also analyzed by western blotting using anti-GFP to assess relative protein expression among proSP-CL55F, proSP-CI73T, proSP-CA116D, and proSP-CWT.
As shown in Figure 3 , a 27 kDa product was expressed in cells transfected with the pAcGFP-C1 vector alone. We observed doublet bands for proSP-CWT and proSP-CL55F that likely corresponded to the primary translation product of the fusion protein (48 kDa) and its known palmitoylated isoform (50 kDa) (19).

Processing features of GFP/proSP-CWT GFP/proSP-CL55F, GFP/proSP-CI73T, or GFP/proSP-CA116D. (a) Total protein from A549 cells stably transfected with pAcGFP-C1 vector, GFP/proSP-CWT, GFP/proSP-CL55F, GFP/proSP-CI73T, or GFP/proSP-CA116D was analyzed for expression of proSP-C by western blotting using antibodies directed against GFP or β-actin. (b) Densitometric quantitation of band intensity normalized to β-actin from three separate experiments. Data of proSP-CL55F (black column), proSP-CI73T (gray column), and proSP-CA116D (hatched column) were expressed as fold change over proSP-CWT (white column) values in each experiment. ProSP-CL55F yielded Band 1 similar to those of proSP-CWT. Band 2 of proSP-CL55F was observed at a reduced expression level as compared with proSP-CI73T. The lowest processing intermediate of proSP-CL55F (Band 3) indicated a significant decrease as compared with those of proSP-CWT, proSP-CI73T, and proSP-CA116D. Significant differences were indicated by asterisks. *P < 0.05, **P < 0.001.
ProSP-CL55F yielded bands similar to those of proSP-CWT for the primary translation product of the fusion protein (48 kDa). Two lower molecular weight intermediates were observed at a reduced expression level, and the lowest processing intermediate (Band 3) displayed a significant decrease as compared with those of proSP-CWT, proSP-CI73T, and proSP-CA116D. In proSP-CI73T cells, a band around 37 kDa migrated to a higher molecular weight compared with those in proSP-CWT, proSP-CL55F, and proSP-CA116D.
Abnormal Subcellular Localization of ProSP-CL55F
Immunofluorescence analysis was performed to investigate the subcellular localization of proSP-CL55F in A549 cells. ProSP-CL55F poorly colocalized with CD63/LAMP3 as compared with proSP-CWT ( Figure 4a ). According to quantitative analysis, the correlation of proSP-CL55F was significantly lower than those of proSP-CWT and proSP-CA116D, but was similar to that of proSP-CI73T ( Figure 4b ). Similarly to proSP-CI73T, which was reported to localize at the plasma membrane, proSP-CL55F also demonstrated signs of partially trafficking toward the plasma membrane ( Figure 4a ). Increased plasma membrane localization was observed for proSP-CL55F as compared with proSP-CWT at low magnification (Supplementary Figure S3 online). On the other hand, proSP-CL55F was detected in early endosome antigen (EEA1)-positive vesicles at a level similar to that of proSP-CWT. Statistical analysis using Pearson’s correlation coefficient showed no significant differences among proSP-CL55F, proSP-CWT, proSP-CI73T, and proSP-CA116D (Supplemental Figure S4 online). Likewise, no remarkable difference was seen between proSP-CWT and mutant proSP-C when we analyzed colocalization with ER-Tracker-Red (data not shown).

Intracellular localization of proSP-CWT and mutant proSP-C forms cultured for 12 h in stably expressing A549 cells. (a) Immunofluorescence analysis with a CD63/LAMP3 antibody showed complete colocalization of proSP-CWT and proSP-CA116D and partial colocalization of proSP-CL55F and proSP-CI73T (scale bars = 5 μm). Green channel: proSP-C visualized by GFP fluorescence; Red channel: LAMP-positive structures used to denote lamellar bodies; Merge: overlapped green and red channels for visualization of the colocalization between different SP-C forms and CD63/LAMP3 (yellow). Nuclei were visualized by DAPI (blue). Regions delineated by white boxes were enlarged to illustrate colocalization with the plasma membrane of proSP-CL55F and proSP-CI73T (white arrows). (b) Pearson’s correlation coefficients for proSP-CWT, proSP-CL55F, proSP-CI73T, and proSP-CA116D colocalization with CD63/LAMP3. **P < 0.001.
Altered Subcellular Organelles in ProSP-CL55F
To further characterize the structure of subcellular organelles, A549 cells transfected with either proSP-CWT or mutant proSP-C isoforms were evaluated by TEM.
At the ultrastructural level, nontransfected A549 cells contained various cellular components, such as early endosomes and late endosomes, within the cytoplasm (data not shown). Cells transfected with proSP-CWT showed an increased number of these organelles, which featured regular “onion skin” electron-dense and concentric lamellae with phospholipid membranes ( Figure 5a , b ). Numerous disorganized lysosomal-like organelles with a double membrane (white arrows) and tightly packed electron-dense dark bodies (black arrow) were observed in A549 cells stably expressing proSP-CL55F or proSP-CI73T. A549 cells transfected with proSP-CA116D displayed several lysosomal-like organelles with irregularly arranged and eccentrically packed lamellae ( Figure 5 ).

Characterization of cellular components in A549 cells by transmission electron microscopy (TEM). Representative TEM images of A549 cells stably expressing proSP-CWT, proSP-CL55F, proSP-CI73T, or proSP-CA116D (left) (scale bars = 5 μm). Solid boxes were magnified on the left for better resolution (right) (scale bars = 500 nm). (a,b) In A549 cells transfected with proSP-CWT, normal lysosomal-like organelles were observed. (c,d) Tightly packed electron-dense dark granules (dark arrow) and numerous disorganized lysosomal-like organelles with a double membrane (white arrow) were evident in mutant proSP-CL55F cells. Abnormal lysosomal-like organelles (white arrow) with a double membrane were detected for proSP-CI73T (e,f) and proSP-CA116D (g,h).
Discussion
In the present study, we described the radiological findings and histopathological and ultrastructural characteristics of a child associated with ILD at initial diagnosis. A novel mutation in one SFTPC allele (c.163C>T) in the region encoding the mature peptide of proSP-C was identified in the infant, who eventually required lung transplantation due to chronic respiratory symptoms. This mutation was not detected in her parents, healthy control volunteers, or the literature, nor has it been listed in large publicly available databases, such as the 1000 Genomes Project (http://browser.1000genomes.org/index.html), or Exome Variant Server (http://evs.gs.washington.edu/EVS/), suggesting that it was a de novo variant.
The clinical presentations of ILD depending on patient age and outcome in infants and children have been associated with disorders of SP-C, SP-B, and ABCA3 expression. According to previous reports, recessive SP-B gene mutations were often found as a cause of lethal respiratory failure in the newborn period; most infants with an SP-B deficiency presented within hours of birth with respiratory failure that required mechanical ventilation (20). ABCA3 gene mutations have also been described in fatal respiratory diseases in newborns and severe chronic lung disease in infancy (21,22). In contrast, SFTPC mutations found in autosomal dominant diseases were often associated with ILD severities ranging from mild to severe and affected patients from the neonatal period (4) to adulthood (6). In the current study, a child presented with the symptoms, signs, and radiographic findings typical of ILD at the age of 5 mo and exhibited a history of persistent respiratory insufficiency for 9 y, indicating that SFTPC mutations were a possible contributor to her condition.
Immunohistochemical staining for proSP-C displayed abnormal distribution within secretory compartments in type II AECs of patient lung tissue specimens as compared with a control, indicating a potential association between proSP-CL55F and the alteration of subcellular proSP-C localization. This was supported by morphological alteration of type II AECs detected by TEM evaluation of lung tissue sections, which provided distinctive characterization by small and densely packed LBs with electron dense inclusions in type II AECs.
To investigate the pathogenesis of proSP-CL55F associated with ILD, we constructed a model of A549 cells stably transfected with proSP-CWT, proSP-CL55F, proSP-CI73T, or proSP-CA116D. TEM examination of A549 cells stably transfected with proSP-CL55F uncovered abnormal lysosomal-like organelles and increased dense core bodies of lysosomal-like organelles, suggesting that proSP-CL55F might have disrupted normal intracellular storage and secretion of SP-C in lysosomal-like organelles.
Western blotting analysis showed similar patterns of intermediate products among proSP-CWT, proSP-CL55F, and proSP-CA116D, suggesting that proSP-CL55F and proSP-CA116D did not interfere with the cleavage step of proSP-C at the C-terminus. An aberrant 37 kDa band was identified for proSP-CI73T and supported the findings of abnormal protein processing. ProSP-CL55F showed a significant reduction in an intermediate processing band as compared with proSP-CWT, proSP-CI73T, and proSP-CA116D, which indicated a loss in proSP-CL55F expression in the processing pathway of A549 cells.
Immunofluorescence analysis of proSP-CWT, proSP-CL55F, proSP-CI73T, and proSP-CA116D in transfected A549 cells was performed to observe subcellular localization. The A549 cell line has been widely studied as a model for type II AECs to characterize the early steps of proSP-C maturation in vitro. Although A549 is a nonlamellar body cell line, when the wild-type isoform of proSP-C is transfected into A549 cells, proSP-C localizes to acidic, lysosomal-like organelles (23). CD63/LAMP3 is used to identify LBs, late endosomes, and lysosomes in type II AECs. Similarly to proSP-CI73T, proSP-CL55F also trafficked partially to CD63/LAMP3-positive vesicles, presumably for routing to the plasma membrane. Mutations located in the non-BRICHOS domain of proSP-C have been described as mistargeting proteins. Previous studies showed that proSP-CI73T was directed to plasma membrane and resulted in abnormal accumulation of the protein in endosome/lysosome compartments in vitro (11). These data suggested that proSP-CL55F might be related to some extent to alterations in the subcellular localization of proSP-CI73T in A549 cells, although we could not confirm this result by western blotting. Ultrastructural images of proSP-CI73T with increased numbers of large, electron-dense vesicles containing organellar debris have demonstrated the disruption of degradation pathways through autophagy (24). Therefore, the western blotting results, alterations in subcellular localization, and abnormally large electron-dense bodies in proSP-CL55F in our system might have been due in part to degradation pathway disruption by autophagic processes.
Consisting of a transmembrane and short extramembrane domain, the mature peptide of proSP-C is known to form a very rigid and stable α-helix (25). Valine is typically over-represented (>50% of amino acids) in the transmembrane domain of proSP-C. The hydrophobic peptides of SP-C and amyloid β-peptide are derived from the transmembrane region of their precursor protein with higher propensities for the β strand than helix conformation (26). However, Congo red staining in this patient revealed no formation of amyloid-like deposits, which indicated that proSP-CL55F might not induce proSP-C misfolding and aggregation.
The main limitation of this study was that our A549 system corresponded to a homozygous rather than a heterozygous SFTPC mutation. To date, all known ILD-related SP-C mutations have been heterozygous, and thus a normal allele is also expressed in vivo situation (27). Therefore, in previous multiple heterozygous expressions of mutant proteins associated with autosomal dominant diseases performed in vitro, cells coexpressing combinations of wild-type and mutant proteins were adopted to analyze the mechanism of genetic dominance of interfering mutations (9,28). Additional evaluations are required to establish the precise in vivo pathogenic mechanism of proSP-CL55F.
In summary, we uncovered a new pathogenic mutation in SFTPC located in the mature domain of proSP-C in a Japanese girl with ILD. Functional analyses of transfected A549 cells expressing this mutant proSP-CL55F protein revealed that it may have caused altered subcellular localization. Further studies are needed for a more detailed understanding of the pathogenic mechanisms of genetic onset of this mutation.
Methods
This study was approved by the institutional review board of the Shinshu University School of Medicine. Written informed consent was obtained from the patient’s guardians and Japanese healthy volunteers.
Genetic Analyses
Genetic analyses were conducted on the patient from the age of 12 mo. Genomic DNA was extracted from peripheral blood samples of the patient, her parents, and 61 healthy control subjects using the QIAamp DNA blood mini kit (Qiagen GmbH, Hilden, Germany). The coding exons of SFTPC were amplified by PCR using previously described primers (29). PCR products were confirmed by sequencing.
Immunohistochemistry of Lung Tissue
Lung tissue sections from the patient obtained by open lung biopsy at 5 mo of age were prepared from formalin-fixed paraffin-embedded samples. Control lung tissue was resected for malignancy. Immunohistochemistry was performed with antibodies against proSP-C, SP-B, ABCA3 (Abcam, Cambridge, UK), or proSP-B (Novocastra, Newcastle, UK), according to standard protocols (30).
Congo Red Staining of Lung Tissue
Formalin-fixed paraffin-embedded lung tissue sections obtained by open chest biopsy at 5 mo of age were treated by Congo red solution and dehydrated using increasing concentrations of ethanol. The fluorescence signals were observed by polarization microscopy, and images were collected.
GFP/SP-C cDNA Constructs
To obtain wild-type human proSP-C (Met1 to Ile197), mRNA was extracted from sputum samples of healthy volunteers with the QIAamp RNA blood mini kit (Qiagen GmbH). cDNA fragments of SFTPC were amplified by the primer pairs A and D ( Table 1 ) at the enzyme sites of BglII and EcoRI, respectively.
For mutant GFP/proSP-CL55F, the cDNA of wild-type human proSP-C served as the template using the primers by overlap extension PCR with a two-round, four-primer technique ( Table 1 ). For construction of GFP/proSP-CI73T and GFP/proSP-CA116D, mutagenesis was also performed by overlap extension PCR, as described previously (9,18,31). Following digestion with BglII at the 5’ end and EcoRI at the 3’ end, PCR products were purified for cloning into pAcGFP-C1 vectors (Clontech, Mountain View, CA). These constructs were confirmed by sequencing.
Generation of Stably Transfected A549 Cells
A549 lung epithelial cells were nucleofected with GFP/proSP-CWT, GFP/proSP-CL55F, GFP/proSP-CI73T, or GFP/proSP-CA116D using the Amaxa SF Cell line 4D-Nucleofector X kit (Lonza, Cologne, Germany).
Transfected A549 cells were treated with 600 μg/ml G418 (Roche, Indianapolis, IN) as a neomycin selection marker in a 5% CO2 incubator at 37 °C. Control cells not carrying the resistance marker were used to verify cell death by G418. After 4 wk, several neomycin-resistant clones were selected. Neomycin-resistant cells were assessed and sorted by a BD FACS Aria II cell sorter (Becton-Dickinson, Mountain View, CA) using a 488-nm laser. A549 cells stably transfected with GFP/proSP-CWT, GFP/proSP-CL55F, GFP/proSP-CI73T, or GFP/proSP-CA116D were generated for further analysis.
RNA Extraction and PCR of Stably Transfected A549 Cells
Total RNA was extracted from stably transfected A549 cells and reverse transcription to cDNA was performed using the PrimeScript RT reagent kit (TaKaRa Bio, Shiga, Japan). For semiquantitative RT-PCR, four 10-fold serial dilutions of template cDNAs from GFP/proSP-CWT, GFP/proSP-CL55F, GFP/proSP-CI73T, or GFP/proSP-CA116D were amplified using primer pairs A and D ( Table 1 ) and confirmed by sequencing. The amplification conditions of SFTPC and GAPDH were 1 cycle at 94 °C for 1 min, followed by 35 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min.
Western Blotting
Stably transfected A549 cells were cultured for 12 h, harvested, and subjected to protein extraction. Western blotting was performed using primary antibodies, as follows: GFP (Clontech) at a 1:5,000 dilution and β-actin (MBL, Nagoya, Japan) at a 1:1,000 dilution. Peroxidase-labeled antibodies to mouse IgG and rabbit IgG (H+L; KPL, Gaithersburg, MD), respectively, were used as secondary antibodies. β-actin was used as the standard for normalizing protein samples. Detection was performed using an ECL system and target proteins were quantified using a densitometric image analyzer with NIH Image version 1.61 software (Bethesda, MD).
Fluorescence Microscopy
Stably transfectedA549 cells were cultured for 12 h on Lab-Tek II CC2 chamber slides (Thermo Fisher Scientific, Rochester, NY). The cells were fixed and incubated with primary antibodies for CD63/LAMP3 (BioLegend, San Diego, CA) or EEA1 (Cell Signaling Technology, Danvers, MA) at 1:100 dilutions. Species-specific Alexa Fluor 555- and 568-conjugated secondary antibodies (Invitrogen, Eugene, OR), respectively, were used at 1:500 dilutions. Samples were mounted using ProLong Gold Antifade Reagent with DAPI and examined using an LSM 5 EXCITER confocal laser scanning microscope (Carl Zeiss, Jena, Germany). Cells were also stained with ER-Tracker-Red (BODIPY TR, Glibenclamide, Invitrogen, Eugene, OR).
Images of single costained cells were captured for colocalization analysis. Colocalizations between GFP/proSP-C and CD63/LAMP3 or EEA1 were quantified using ZEN2009 software (Carl Zeiss) based on Pearson’s correlation coefficient.
TEM Examination
Small lung tissue specimens obtained from the patient at 5 mo of age and stably transfected A549 cells were cultured for 12 h and fixed. After dehydration through a series of ethanol gradients, the samples were embedded in EPON resin. Ultrathin sections (70–80 nm) were prepared and double-stained with uranyl acetate and lead citrate. Ultrastructural investigation was carried out using a JEOL JEM-1230 TEM device.
Statistical Analysis
Statistical analyses were performed by one-way ANOVA using SPSS version 17.0 software (SPSS, Chicago, IL). All values are expressed as the mean ± SE. The level of significance was defined as a P value of <0.05.
Statement of Financial Support
No financial assistance was received to support this study.
Disclosure
The authors have no financial disclosures indicating any financial ties to products in the study or potential/perceived conflicts of interest.
References
Beers MF, Mulugeta S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol 2005;67:663–96.
Nogee LM. Alterations in SP-B and SP-C expression in neonatal lung disease. Annu Rev Physiol 2004;66:601–23.
Mulugeta S, Beers MF. Surfactant protein C: its unique properties and emerging immunomodulatory role in the lung. Microbes Infect 2006;8:2317–23.
Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 2001;344:573–9.
Turcu S, Ashton E, Jenkins L, Gupta A, Mok Q. Genetic testing in children with surfactant dysfunction. Arch Dis Child 2013;98:490–5.
Lawson WE, Grant SW, Ambrosini V, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 2004;59:977–80.
Nogee LM, Dunbar AE 3rd, Wert S, Askin F, Hamvas A, Whitsett JA. Mutations in the surfactant protein C gene associated with interstitial lung disease. Chest 2002;121(3 Suppl):20S–1S.
Baekvad-Hansen M, Nordestgaard BG, Tybjaerg-Hansen A, Dahl M. Two novel mutations in surfactant protein-C, lung function and obstructive lung disease. Respir Med 2010;104:418–25.
Wang WJ, Mulugeta S, Russo SJ, Beers MF. Deletion of exon 4 from human surfactant protein C results in aggresome formation and generation of a dominant negative. J Cell Sci 2003;116(Pt 4):683–92.
Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol 2005;32:521–30.
Beers MF, Hawkins A, Maguire JA, et al. A nonaggregating surfactant protein C mutant is misdirected to early endosomes and disrupts phospholipid recycling. Traffic 2011;12:1196–210.
Willander H, Askarieh G, Landreh M, et al. High-resolution structure of a BRICHOS domain and its implications for anti-amyloid chaperone activity on lung surfactant protein C. Proc Natl Acad Sci USA 2012;109:2325–9.
Chen F, Matsukawa S, Ishii H, et al. Delayed chest closure assessed by transesophageal echocardiogram in single-lobe lung transplantation. Ann Thorac Surg 2011;92:2254–7.
Markart P, Ruppert C, Wygrecka M, et al. Surfactant protein C mutations in sporadic forms of idiopathic interstitial pneumonias. Eur Respir J 2007;29:134–7.
Thurm T, Kaltenborn E, Kern S, Griese M, Zarbock R. SFTPC mutations cause SP-C degradation and aggregate formation without increasing ER stress. Eur J Clin Invest 2013;43:791–800.
Nerelius C, Martin E, Peng S, et al. Mutations linked to interstitial lung disease can abrogate anti-amyloid function of prosurfactant protein C. Biochem J 2008;416:201–9.
Abou Taam R, Jaubert F, Emond S, et al. Familial interstitial disease with I73T mutation: A mid- and long-term study. Pediatr Pulmonol 2009;44:167–75.
Zarbock R, Woischnik M, Sparr C, et al. The surfactant protein C mutation A116D alters cellular processing, stress tolerance, surfactant lipid composition, and immune cell activation. BMC Pulm Med 2012;12:15.
Vorbroker DK, Dey C, Weaver TE, Whitsett JA. Surfactant protein C precursor is palmitoylated and associates with subcellular membranes. Biochim Biophys Acta 1992;1105:161–9.
Wilder MA. Surfactant protein B deficiency in infants with respiratory failure. J Perinat Neonatal Nurs 2004;18:61–7.
Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med 2004;350:1296–303.
Bullard JE, Wert SE, Whitsett JA, Dean M, Nogee LM. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med 2005;172:1026–31.
Maguire JA, Mulugeta S, Beers MF. Multiple ways to die: delineation of the unfolded protein response and apoptosis induced by Surfactant Protein C BRICHOS mutants. Int J Biochem Cell Biol 2012;44:101–12.
Hawkins A, Guttentag SH, Deterding R, et al. A non-BRICHOS SFTPC mutant (SP-CI73T) linked to interstitial lung disease promotes a late block in macroautophagy disrupting cellular proteostasis and mitophagy. Am J Physiol Lung Cell Mol Physiol 2015;308:L33–47.
ten Brinke A, Posthuma G, Batenburg JJ, et al. The transmembrane domain of surfactant protein C precursor determines the morphology of the induced membrane compartment in CHO cells. Eur J Cell Biol 2003;82:285–94.
Gustafsson M, Thyberg J, Näslund J, Eliasson E, Johansson J. Amyloid fibril formation by pulmonary surfactant protein C. FEBS Lett 1999;464:138–42.
Thomas AQ, Lane K, Phillips J 3rd, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002;165:1322–8.
Calebiro D, de Filippis T, Lucchi S, et al. Intracellular entrapment of wild-type TSH receptor by oligomerization with mutants linked to dominant TSH resistance. Hum Mol Genet 2005;14:2991–3002.
Guillot L, Epaud R, Thouvenin G, et al. New surfactant protein C gene mutations associated with diffuse lung disease. J Med Genet 2009;46:490–4.
Amin RS, Wert SE, Baughman RP, et al. Surfactant protein deficiency in familial interstitial lung disease. J Pediatr 2001;139:85–92.
Woischnik M, Sparr C, Kern S, et al. A non-BRICHOS surfactant protein c mutation disrupts epithelial cell function and intercellular signaling. BMC Cell Biol 2010;11:88.
Acknowledgements
We would like to acknowledge the participants in this research study. We also appreciate the assistance of doctors Lika’a Fasih Y. Al-Kzayer, and Le Thanh N. Uyen of Shinshu University School of Medicine.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figure S1
(TIFF 10079 kb)
Supplementary Figure S2
(TIFF 14066 kb)
Supplementary Figure S3
(TIFF 17874 kb)
Rights and permissions
About this article

Cite this article
Liu, T., Sano, K., Ogiwara, N. et al. A novel surfactant protein C L55F mutation associated with interstitial lung disease alters subcellular localization of proSP-C in A549 cells. Pediatr Res 79, 27–33 (2016). https://doi.org/10.1038/pr.2015.178
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.178
- Springer Nature America, Inc.