
Abstract
Malignant melanoma reveals rapidly increasing incidence and mortality rates worldwide. By now, BRAF inhibition is the standard therapy for advanced melanoma in patients carrying BRAF mutations. However, only approximately 50% of melanoma patients harbor therapeutically attackable BRAF mutations, and overall survival after treatment with BRAF inhibitors is modest. KRAS (Kirsten Rat sarcoma) proteins are acting upstream of BRAF and have a major role in human cancer. Recent approaches awaken the hope to use KRAS inhibition (KRASi) as a clinical tool. In this study, we identified wild-type KRAS as a novel therapeutic target in melanoma. KRASi functions synergistically with BRAF inhibition to reduce melanoma proliferation and to induce apoptosis independently of BRAF mutational status. Moreover, acquired resistance to BRAF inhibitors in melanoma is dependent on dynamic regulation of KRAS expression with subsequent AKT and extracellular-signal regulated kinase activation and can be overcome by KRASi. This suggests KRASi as novel approach in melanoma—alone or in combination with other therapeutic regimes.
Similar content being viewed by others


Introduction
Malignant melanoma is one of the most deadly and most common types of cancer worldwide.1 Until 2011, disseminated melanoma was treated inefficiently with cytotoxic agents that failed to improve overall survival.2, 3 Today, targeted therapies with BRAF (v-Raf murine sarcoma viral oncogene homolog B) inhibitors (BRAFis), MEK (mitogen-activated protein kinase kinase) inhibitors (MEKis) or immunotherapeutic approaches such as programmed death 1 blockade improve survival of melanoma patients.4, 5, 6
BRAF inhibition (BRAFi) is the standard therapy for advanced melanoma in patients carrying BRAF mutations. However, only approximately 50% of melanoma patients harbor therapeutically attackable BRAF mutations, and the increase in progression-free and overall survival after BRAFi is modest.7 Several mechanisms of resistance to BRAFi were proposed.8 One prediction for resistance to BRAFis is that mechanisms enhancing RAF dimerization result in drug resistance. These include alterations that induce RAS activity as the canonical mechanism of RAF dimerization is RAS dependent.9, 10, 11
RAS isoforms have a major role in human cancer, and new technologies have resulted in the development of promising ways to inhibit RAS signaling.12, 13, 14, 15, 16 Encouraging results were also achieved by administration of specific siRNA targeting KRAS (Kirsten Rat sarcoma).12, 17, 18
However, unlike neuroblastoma RAS viral oncogene homolog (NRAS) which is mutated in melanoma in approximately 20%,19 only minor attention has been paid to KRASG12V,20 which is only rarely mutated in melanoma. In contrast, the role of wild-type KRAS in melanoma progression and drug resistance is completely unexplored and therefore was addressed in this study.
Results
Increased KRAS expression correlates with melanoma progression
First, we aimed to determine KRAS expression in melanoma in vitro and in vivo. KRAS mRNA and protein levels were upregulated in both primary and metastatic human melanoma cell lines as compared with normal human epidermal melanocytes (Figures 1a and b and Supplementary Table S1). In contrast to KRAS, NRAS mRNA levels were upregulated in only a few melanoma cell lines and HRAS mRNA levels were not upregulated or even significantly downregulated, respectively, in melanoma cell lines as compared with normal human epidermal melanocytes (Supplementary Figures S1B and C). Moreover, we performed KRAS immunohistological analysis of a tissue microarray comprising human tissue specimens of melanocyte-derived benign nevi and primary and metastatic human melanoma. Both KRAS staining intensity and KRAS membrane localization were elevated in melanoma tissues as compared with nevi. In melanoma metastasis, KRAS membrane localization was even further enhanced as compared with primary melanoma (P=0.017) (Figures 1c and d, Supplementary Figure S2 and Supplementary Table S2). Ki67 staining revealed increased mitotic activity in melanoma tissues with high KRAS expression (P=0.037) and strong KRAS membrane localization (P=0.022) (Figures 1e and f and Supplementary Figure S3). Accordingly, we found significant correlation between KRAS and CyclinD1 mRNA expression in tissue samples derived from melanoma patients (r=0.73, P<0.0001, Figure 1g and Supplementary Table S3). In silico analysis of a melanoma The Cancer Genome Atlas data set with stratification according to Clark Levels revealed that KRAS expression was significantly enhanced in the ‘high risk’ group as compared with the ‘low risk’ group (P=0.0376) (Supplementary Figure S4A). Additional data showed reduced survival in melanoma patients with high as compared with low KRAS expression (P=0.05) (Supplementary Figure S4B). Together, these findings point to relevance of increased KRAS expression in melanoma growth and progression.

KRAS expression in melanoma. (a) KRAS mRNA and (b) KRAS protein levels (lower panel: exemplary western blotting using actin as loading CTR; upper panel: densitometric analysis) in normal human epidermal melanocytes (NHEM) compared with primary melanoma (PT) cell lines (Mel Juso, Mel Ei, Mel Ho) and metastatic (MET) melanoma cell lines (SkMel3, SkMel28, Mel Im, 451Lu, Mel Ju, Hmb2). Data are represented as mean±s.e.m. (*P<0.05 vs NHEM; the sample number was n>6 for each cell line). (c) KRAS immunohistochemical analysis of a tissue microarray comprising human melanocyte-derived benign nevi (Nevus), primary melanoma (PT) and melanoma metastases (MET). (d) Quantification of KRAS staining intensity and KRAS membrane localization. KRAS staining was analyzed qualitatively differentiating (i) no (‘−’), (ii) low/moderate (‘+’) or (iii) high (‘++’) staining intensity. KRAS membrane localization was described qualitatively differentiating (i) negative (‘−’; only cytoplasmic/endomembranous staining), (ii) low/moderate (‘+’; <50% of cells show positive plasma membrane staining) and (iii) high (‘++’; >50% of cells show positive plasma membrane staining). (*P<0.05 (Fisher’s exact test)). (e) Examples of paired Ki67 and KRAS staining in melanoma tissues with weak (left side) and strong (right side) KRAS expression. (f) Quantification of Ki67 staining (‘−’: <5%; ‘+’: 5–20%; ‘++’ >20% positive cells) in melanoma tissues with low (‘+’) and high (‘++’) KRAS staining intensity/ membrane localization. (*P<0.05 (Fisher’s exact test)). (g) Correlation of KRAS mRNA and CyclinD1 mRNA expression in human melanoma tissue samples (n=27, r=0.73, P<0.0001).
KRAS knockdown reduces tumorigenicity of melanoma cells
To get further insights into the functional role of KRAS in melanoma, we analyzed the effects of KRAS suppression in primary (Mel Juso) as well as metastatic (Mel Im) melanoma cells. Both cell lines carry no mutations in the KRAS, HRAS or NRAS gene locus (Supplementary Table S1). Additional experiments were performed using the metastatic melanoma cell line Htz19 and are shown in Supplementary Materials and Methods. KRAS was suppressed by RNA-interference (RNAi) using two different approaches (single small interfering RNAs (si-RNAs) and si-RNA-pools, referred to as KRAS repression ‘KR’, Supplementary Figure S5). RNAi-mediated KRAS knockdown was confirmed by quantitative reverse transcriptase–PCR to be specific and not to suppress other RAS isoforms (Supplementary Figure S6). In addition to KRAS knockdown, we functionally analyzed melanoma cells after NRAS or HRAS knockdown, respectively. Proliferation was not altered after HRAS or NRAS knockdown (Supplementary Figure S6). In contrast, KRAS knockdown significantly reduced anchorage-independent and -dependent tumor colony formation and colony size as compared with mock-transfected control (CTR) cells (Figures 2a–d and Supplementary Figure S5). Moreover, KRAS depletion caused sustained reduction of tumor cell proliferation (Figure 2e). As shown above, Ki67 staining and CyclinD1 expression correlated with KRAS expression in patient samples in vivo (Figures 1e–g). Accordingly, double immunostaining was performed and showed that KRAS expression correlates with Ki67 staining in melanoma cells (Figures 2f and g and Supplementary Figure S7). Moreover, in both Mel Juso (heterozygous BRAFV600E mutation) and Mel Im (homozygous BRAFV600E mutation) cells, KRAS knockdown significantly attenuated signaling of its downstream mediator activated v-akt murine thymoma viral oncogene (phopsho-AKT). KRAS knockdown also significantly suppressed extracellular-signal regulated kinase (ERK) activity in Mel Juso but not in Mel Im cells. This is consistent with the finding that homozygous BRAFV600E mutations (Mel Im) lead to RAS-independent ERK activation (Figures 2i and j). Tissue microarray analysis revealed stronger phospho-ERK staining in human melanomas with high KRAS membrane localization (P=0.002) and KRAS staining (P=0.007) (Figure 2k and Supplementary Figure S7). In summary, KRAS knockdown reduces tumorigenicity and affects oncogenic mitogen-activated protein kinase (MAPK) and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/AKT signaling in melanoma cells in vitro.


Functional effects of KRAS knockdown in melanoma cells in vitro and comparison of melanoma tissues with low and high KRAS expression. Analyses of melanoma cell lines (Mel Im, Mel Juso) with si-RNA-Pool-mediated KRAS repression (KR) and CTR cells transfected with CTR siRNA. (a, c) Colony number and (b, d) colony diameter in three-dimensional, anchorage-independent colony-formation assays (a, b) and in two-dimensional, anchorage dependent clonogenic assays (c, d) (*P<0.05 vs CTR; n=4). (e) Real-time analysis of cell proliferation. The ‘slope’ of the curves depicts the increasing cell index and ‘doubling times’ summarize the proliferative capacity of melanoma cells (*P<0.05; n=3). (f, g) Double-immunofluorescence staining of KRAS (red) and Ki67 (green) in CTR and si-KRAS (KR) transfected Mel Im melanoma cells and according correlation and quantification (*P<0.05; n=4 replicates). (h, i) (Densitometric) western blotting analysis of pERK/ERK and pAKT/AKT (*P<0.05 vs CTR, n=3) and summary and hypothesis of the effects of RNAi-mediated KRAS knockdown on AKT and ERK signaling in Mel Juso cells carrying heterozygous BRAFV600E mutation and Mel Im cells with homozygous BRAFV600E mutation. (j) KRAS and pERK immunohistochemical analysis of a tissue microarray comprising human primary melanoma (PT) and melanoma metastases (MET). Examples are shown for paired pERK and KRAS staining in melanoma tissues with weak (top panels) and strong (bottom panels) KRAS expression. Diagrams on the right side depict quantification of pERK staining (‘−’: <5 %; ‘+’: 5–20%; ‘++’: >20% positive cells) in melanoma tissues with low (‘+’) and high (‘++’) KRAS staining intensity/membrane localization (*P<0.05 (Fishers exact test)).
KRAS knockdown inhibits in vivo tumorigenicity
To investigate KRAS knockdown effects on melanoma cells in vivo, Mel Im cells with si-RNA-suppressed KRAS and CTR cells were subcutaneously injected into nu/nu mice to induce formation of xenograft tumors. The volume of xenograft tumors was reduced in the KRAS knockdown group (Figures 3a and b and Supplementary Figure S8). Moreover, tumor onset was reduced (Figures 3c and d) in the KRAS knockdown group compared with CTRs. Immunofluorescence analysis showed that the smaller tumors grown in the KRAS-repression group re-expressed KRAS (Supplementary Figure S8). Quantification of KRAS mRNA and protein levels revealed that KRAS was still suppressed (however not significantly) in the KR group (Supplementary Figure S8). Immunohistochemistry showed reduced Ki67 staining in tumors formed by KRAS-suppressed melanoma cells (Figure 3e and Supplementary Figure S8). Furthermore, staining of the endothelial cell marker CD31 was less intense in the KRAS knockdown group (Figure 3f and Supplementary Figure S8). Histological Ki67 and CD31 expression are known independent prognostic markers for melanoma patients.21, 22 Together, these data confirm the antitumorigenic in vitro effects of KRAS inhibition (KRASi) and indicate KRAS as an oncogenic target for melanoma.

KRAS knockdown reduces tumorigenicity of melanoma cells in vivo. Analyses of xenograft tumors formed in nude mice after subcutaneous injection of Mel Im melanoma cells with si-RNA-Pool mediated KRAS repression (KR) or CTR cells transfected with CTR si-RNA (800 000 cells/mouse; n=5 animals per group). (a) Tumor volume at different time points after inoculation (p.i.; upper panel) and images of tumor-bearing mice at day 49 p.i. (lower panel). (b) Tumor volume at the time point of resection (ex vivo, day 56 p.i.), and representative images of explanted xenograft tumors. (c) Kaplan–Meier curve for tumor-free survival (HR=hazard ratio (Mantel–Haenszel)). (d) Tumor onset (mean of tumor-bearing mice day 21 until day 56 p.i.). (e) (Quantification of) Ki67 staining (Ki67-positive cells per field; left panel) and (f) CD31 immunostaining and quantification (number of CD-31-positive vessels per field) of xenograft tumors (*P<0.05 vs CTR).
Small-molecule KRASi reduces proliferation and induces apoptosis in melanoma
The marked effects of KRAS knockdown raised the question whether pharmacological inhibition of KRAS could be an effective approach for the treatment of melanoma. By now, attempts to target KRAS failed in clinical studies.12 Recently, a small-molecule inhibitor—‘Deltarasin’ (DR)—was identified (Supplementary Figure S9). DR binds to the delta subunit of rod-specific photoreceptor phosphodiesterase (PDE6δ), a protein that regulates the trafficking of KRAS between membranes. DR has been reported to reduce proliferation of pancreatic adenocarcinoma cells.16 Here we found that DR caused dose-dependent toxicity in Mel Juso (Figure 4a) and Mel Im cells (Figure 4b). Additionally, we explored the function of DR in NRAS-mutated WM1366 melanoma cells (NRASQ61L) and in BRAF-NRAS wild-type WM3211 melanoma cells (Supplementary Figure S10). As anticipated, DR mediated a dose-dependent inhibition of proliferation. However, DR revealed stronger effects in BRAF-NRAS wild-type than in NRAS-mutated cells, pointing to selective KRASi mediated by this compound (Supplementary Figure S10). Quantification of lactate dehydrogenase release into the supernatant (Figure 4c) and microscopical analysis (Figure 4d and Supplementary Figure S9) showed cell toxicity upon treatment with DR doses as low as 2 or 5 μM, respectively. In contrast, doses up to 20 μM DR caused no toxic effects in human fibroblasts or normal human epidermal melanocytes (Figures 4e and f). Fluorescence-activated cell sorting analysis revealed marked time-dependent induction of apoptosis in response to DR treatment (Figure 4g and Supplementary Figure S9). According to RNAi-mediated KRAS suppression (Figures 2h and i), DR exerted significant reduction of AKT and/or ERK activation in Mel Juso and Mel Im cells, respectively (Figure 4h). Next, we evaluated the possible emergence of surviving tumor cells and drug resistance in response to DR. To generate resistance as described for other drugs,23, 24 we performed long-term treatment of Mel Juso and Mel Im cells using increasing DR doses. After 28 days, no surviving tumor cells were detected (Figure 4j). Together, these results demonstrate strong antitumorigenic effects of the small-molecule KRASi DR on melanoma cells.

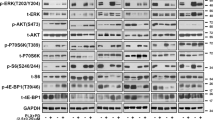
KRASi with the small-molecule DR reduces proliferation and induces apoptosis in melanoma cells in vitro. Melanoma cell lines (Mel Im, Mel Juso) were treated with different concentrations of the KRAS small-molecule inhibitor DR or solvent (DMSO) as CTR. (a, b) Real-time cell proliferation analysis. The cell index as dimensionless parameter for cell proliferation was used to determine the half maximal inhibitory concentration (IC50, small graphs, n=2) of DR. (c) Lactate dehydrogenase (LDH) levels in cell supernatants after treatment with low dose DR (2 μM, 24 h, n=4). (d) Microscopic images of melanoma cell lines treated with 5 μM DR or DMSO for 24 h. (e) Microscopic images of human fibroblast cell lines (derived from two different donors) and normal human epidermal melanocytes (NHEM) treated with a high DR dose (20 μM, 48 h) or DMSO. (f) LDH levels in supernatants of DR or DMSO-treated NHEM (n=4). (g) Fluorescence-activated cell sorting analysis of apoptosis in melanoma cells in response to treatment with DR (5 μM, for 8, 16 or 24 h). Depicted are representative images; experiments were repeated at least three times for each time point and cell line. (h) (Densitometric) western blotting analysis of pERK/ERK and pAKT/AKT (*P<0.05) in Mel Juso cells carrying heterozygous BRAFV600E mutation and Mel Im cells with homozygous BRAFV600E mutation (DR 3 μM, 24 h, n=3). (i) Long-term treatment (28 days) of melanoma cells with increasing doses of DR (up to IC50 or 2 × IC50, respectively) to evaluate the emergence of surviving tumor cells and drug resistance. (*P<0.05 vs DMSO).
KRASi prevents BRAFi-induced paradoxical activation of proliferation and combined KRASi and BRAFi synergistically affects tumor cell apoptosis
BRAFi is one of the few clinical approaches with modest survival benefits for metastasized melanoma patients carrying BRAF mutations.6, 7 Combination regimens have been developed to break acquired resistance. Combined BRAFi and MEK inhibition (MEKi) improves survival as compared with single-agent approaches and has received regulatory approval, giving evidence of the importance of the MAPK pathway in melanoma therapy.25, 26 Here we performed a novel approach combining BRAFi with inhibition of its upstream mediator KRAS. For BRAFi, we used the clinically approved standard drug PLX-4032 (‘Vemurafenib’, referred to as ‘PLX’). We analyzed the effects of DR (5 μM) and PLX (10 μM) on three-dimensional colony formation as well as on proliferation of preformed colonies. In BRAFi primary sensitive Mel Im melanoma cells (Supplementary Figure S11 and Supplementary Table S1), DR was similarly effective as PLX and the DR+PLX combination resulted in almost complete abolishment of colony formation and colony growth (Figure 5a). In BRAFi primary resistant Mel Juso cells, PLX enhanced ERK activation, colony formation and colony growth (Figure 5b and Supplementary Figure S11). Comparable effects have been reported before in BRAFi-resistant melanoma cells and have been explained by a ‘paradox’ transactivation of wild-type BRAF proteins.9, 27 In contrast to PLX, DR markedly inhibited colony formation and proliferation also in Mel Juso cells (Figure 5b). Noteworthy, this effect was also present when DR was combined with PLX, thereby completely abolishing the paradoxical growth-enhancing effects of PLX in this primary BRAFi-resistant melanoma cells.

KRASi prevents BRAFi-induced paradoxical activation of proliferation and combined KRAS and BRAFi synergistically affects melanoma apoptosis. Mel Im melanoma cells (primary sensitive for BRAFi) and Mel Juso cells (primary BRAFi resistant) were treated with the KRAS small-molecule inhibitor DR (5 μM), the BRAFi PLX (10 μM) or the combination of DR and PLX. Solvent (DMSO) served as CTR. (a, b) Time-dependent measurement of colony diameters (upper panel), microscopic images (median panels) and quantification of colony diameters (day 19 after seeding; lower panels) in anchorage-independent colony-formation assays (n=4). (c) Fluorescence-activated cell sorting (FACS) analysis of apoptosis (left panel: representative images of Annexin and propidium iodide (PI) staining; right panel: quantification (%) of apoptotic cells (*P<0.05; n=3). (d) Mel Im and Mel Juso were transfected with a si-RNA-Pool against KRAS (si-KR) or CTR si-RNA (si-CTR) and subsequently treated with PLX (10 μM) or solvent (DMSO) for 24 h. Depicted is FACS analysis of apoptosis according to c) (*P<0.05; n=3).
To further analyze the combined KRASi and BRAFi effects on cell viability, we used short-term treatment intervals. After 5 h, DR or PLX alone exhibited no or only slight effects on apoptosis of melanoma cells. However, the combination of DR and PLX significantly enhanced apoptosis (Figure 5c and Supplementary Figure S11). Accordingly, PLX treatment caused a marked induction of apoptosis in melanoma cells with RNAi-mediated KRAS knockdown but exhibited no (Mel Juso) or only slight (Mel Im) effects on apoptosis of CTR-transfected cells (Figure 5d and Supplementary Figure S11). Together, these data reveal synergistic effects of combined BRAFi and KRASi on melanoma cell viability and apoptosis.
KRASi prevents emergence of BRAFi resistance and KRAS is dynamically regulated in BRAFi-resistant melanoma cells
Next, we wanted to analyze whether KRASi affects the emergence of acquired drug resistance to BRAFi. After treatment with increasing doses of PLX, we observed survival of a moderate proportion of BRAFi-sensitive Mel Im cells. This confirms the common finding of rapid development of acquired resistance to BRAFi as reported in numerous studies. However, the combination of DR and PLX was sufficient to completely abolish the emergence of surviving tumor cells (Figure 6a). Therefore, we explored the function of KRAS in acquired resistance to BRAFi in more detail, using two pairs of BRAFi-sensitive (wild-type, ‘wt’)/BRAFi-resistant (‘R’) BRAFV600E melanoma cell lines (451Lu and SkMel28).23 The resistant cell lines reveal acquired drug resistance and show cross-resistance to different BRAFis, including PLX. The cell lines do not have secondary BRAF mutations beyond V600E or de novo mutations or copy number alterations in NRAS, C-KIT or PTEN (Supplementary Table S1).23 As expected, PLX inhibited proliferation in both BRAFi-sensitive (‘wt’) cell lines. In contrast, in the presence of PLX, proliferation was even enhanced in both cell lines with acquired BRAFi resistance (Figure 6b). Interestingly, we found that KRAS mRNA and protein expressions were elevated in resistant cell lines as compared with non-resistant cell lines (Figure 6c). In contrast, other RAS-isoforms (that is, HRAS and NRAS) were not regulated or even downregulated in resistant as compared with non-resistant cell clones (Supplementary Figure S12). It had to be noted that the resistant cell clones were continuously incubated with PLX to remain drug resistance and to proliferate properly, pointing to a possible effect of PLX treatment on KRAS expression. Indeed, quantitative reverse transcriptase–PCR analysis revealed a dose-dependent and dynamic alteration of KRAS mRNA expression in resistant (Figure 6d) and also in some non-resistant (Supplementary Figure S13) melanoma cell clones in response to PLX treatment. Removal of PLX led to decrease of PLX-induced elevation of KRAS mRNA levels in SkMel28-R cells (Supplementary Figure S13). Western blotting analysis confirmed that PLX treatment dose-dependently enhanced KRAS protein expression in BRAFi-resistant cell lines (Figures 6e and f and Supplementary Figure S13). These findings indicate that dynamic KRAS expression is a critical mediator of melanoma drug resistance to BRAFi.

KRASi prevents emergence of BRAFi resistance and KRAS expression is regulated dynamically in acquired resistance to BRAFi. (a) Images of Mel Im melanoma cells (primary sensitive for BRAFi) and Mel Juso cells (primary BRAFi resistant) treated for 21 days with the BRAFi PLX (up to 20 μM) or a combination of 10 μM PLX and the KRASi DR (5 μM). Treatment with solvent (DMSO) served as CTR. (b) Analysis of cell proliferation (slope of cell index) of SkMel28 and 451Lu melanoma cell lines with acquired PLX-resistance (‘R’) compared with PLX-sensitive wild-type (‘wt’) cells after treatment with PLX (10 μM) or DMSO. (c) KRAS mRNA and protein expression in PLX-resistant (R) and PLX-sensitive (wt) SkMel28 and 451Lu cells. (d) KRAS mRNA expression of SkMel28-R and 451Lu-R cells after 24 and 48 h treatment with different PLX concentrations (*P<0.05 compared with CTR (0 μM PLX)). (e) Western blotting analysis of pAKT/AKT, pERK/ERK and KRAS in 451Lu-R and SkMel28-R cells after treatment with 0, 5 or 10 μM PLX for 8 h. Numbers represent densitometric analysis as indicated. (f) Quantification of KRAS protein in relation to AKT protein expression in SkMel28-R and 451Lu-R cells treated with different concentrations of PLX (0, 5 or 10 μM). (g) Densitometric quantification of activated AKT (pAKT/AKT) protein expression (y axis) in dependence of PLX doses (x axis) in SkMel28-R and 451Lu-R cells. (h) Western blotting analysis of pAKT/AKT (left panel) and pERK/ERK (right panel) levels in 451Lu-R and SkMel28-R cells transfected with si-KRAS (KR) or CTR si-RNA (CTR) and subsequently treated with different concentrations of PLX (0, 5 or 10 μM). (*P<0.05; n=3 for all panels).
KRAS expression enhances MAPK and PI3K signaling in acquired resistance to BRAFi
Epidermal growth factor receptor-induced activation of the PI3K/AKT pathway was reported to be crucial for BRAFi resistance.28, 29 Furthermore, a dynamic RAF kinase switch induces BRAFi resistance that can be overcome by co-targeting MEK and insulin-like growth factor 1 receptor-dependent PI3K signaling.23 Next to epidermal growth factor receptor and insulin-like growth factor 1 receptor, KRAS is known to activate the PI3K/AKT pathway. Here we observed that treatment of BRAFi-resistant cells with PLX dose-dependently increased activation of AKT (Figures 6e and g). Notably, KRAS knockdown was sufficient to inhibit PLX-induced increase of AKT activation in both BRAFi-resistant cell lines (Figure 6h and Supplementary Figure S13). Furthermore, BRAFi-resistant cell lines showed dose-dependent ‘paradoxically’ increased ERK activation upon treatment with PLX (Figure 6e and Supplementary Figure S13). PLX-induced ERK activation was significantly reduced after KRAS knockdown in SkMel28-R cells (Figure 6h). Also, in 451Lu-R cells, KRAS knockdown caused a slight but not significant inhibition of PLX-induced ERK activation (Figure 6h and Supplementary Figure S13). In summary, our data point to an intrinsic mechanism of KRAS-mediated MAPK and PI3K activation that has not been described before in BRAFi-resistant melanoma cells.
KRASi re-sensitizes BRAFi-resistant cells to overcome chemotherapy resistance
In the following experiments, we aimed at revealing the functional role of KRASi in acquired resistance to BRAFi. KRAS knockdown markedly reduced proliferation in both PLX-treated BRAFi-resistant cell lines (Figures 7a and b and Supplementary Figure S14). KRAS knockdown also restored PLX-induced dose-dependent inhibition of proliferation in SkMel28-R (Supplementary Figure S14). According to the described KRAS mRNA and protein alterations in response to PLX, this indicates that resistant cell clones can dynamically react within KRAS signaling, making them more dependent on KRAS in the presence of higher PLX doses. Also KRASi with DR led to dose-dependent inhibition of proliferation of SkMel28-R and 451Lu-R cells (Figures 7c and d and Supplementary Figure S14). Analysis of apoptosis revealed that BRAFi-resistant cell lines showed cross-resistance to MEK and even combined BRAFi/MEKi. However, combining DR and PLX exerted synergistic apoptotic effects in BRAFi-resistant cells (Figures 7e and f and Supplementary Figure S14). RNAi-mediated KRAS knockdown confirmed increased apoptosis in response to BRAFi compared with CTR cells (Supplementary Figure S14). In addition, KRAS knockdown reduced mRNA expression of the antiapoptotic genes BCL-XL and/or BCL2 in BRAFi-resistant SkMel28-R and 451Lu-R cells as well as in Mel Im and Mel Juso cells (Figure 7g and Supplementary Figure S14). Both ERK and AKT signaling are known to promote transcription of apoptosis-inhibiting genes, such as BCL2 and BCL-XL.30, 31, 32, 33 Thus, AKT- and ERK-mediated transcriptional regulation of apoptosis-related genes supports the apoptosis ‘sensitizing’ effect of KRASi. Finally, we examined possible cross-resistance to DR in acquired resistance to BRAFi and found that 8 μM DR was sufficient to completely abolish the emergence of surviving BRAFi-resistant melanoma cells over a sustained period (Figure 7h).

KRASi re-sensitizes BRAFi-resistant melanoma cells to overcome chemotherapy resistance.(a, b) Real-time cell proliferation analysis of SkMel28-R and 451Lu-R melanoma cells (both with acquired resistance against BRAFi PLX) transfected with si-KRAS (KR) or CTR si-RNA (CTR) and subsequently treated with 5 or 10 μM PLX. (c, d) Real-time cell proliferation analysis of SkMel28-R and 451Lu-R cells treated with 5 or 10 μM PLX in combination with KRASi DR (5 μM)) or solvent (DMSO). (a, c: representative proliferation curves; b, d: Summarized slopes of proliferation curves). (e, f) Fluorescence-activated cell sorting analysis (Annexin and propidium iodide (PI) staining) of apoptotic SkMel28-R and 451Lu-R melanoma cells after treatment with PLX (10 μM), DR (5 μM), the MEK1/2-inhibitor U0126 (U0;10 μM) or combinations. (e: representative data sets for SkMel28-R; percentage of values depict apoptotic cells. f: Quantification for both resistant cell lines). (g) BCL2- and BCL-XL mRNA expression SkMel28-R cells transfected with si-KRAS (KR) or CTR si-RNA (CTR) transfected cells. (h) Images of 451Lu-R and SkMel28-R cells during long-term treatment (28 days) with DR as performed to evaluate the emergence of surviving tumor cells and drug resistance. (i) Summary and hypothesis of signaling effects of KRAS knockdown/inhibition in acquired PLX resistance. Top: ERK-induced negative feedback mechanisms on RAS signaling are attenuated and BRAFis cause paradox transactivation of other RAF proteins, resulting in ongoing RAF and ERK activation (which is at least partly RAS dependent). Apart from ERK signaling, KRAS-dependent AKT signaling ensures survival of cancer cells during treatment with PLX. Bottom: Inhibition of KRAS reduces KRAS-dependent AKT and RAF signaling, resulting in regain of drug sensitivity. (*P<0.05; n=3 for all panels).
Together, we show that acquired drug resistance to BRAFi can be mediated by dose-dependent dynamic KRAS expression affecting antiapoptotic AKT and mitogenic ERK signaling. Moreover, KRASi by RNAi or DR can re-sensitize BRAFi-resistant melanoma cells for PLX treatment (Figure 7i).
Discussion
Mutations in RAS genes have been associated with ~30% of all human tumors, with KRAS being the most frequently mutated.34, 35, 36 Interestingly, son of sevenless-mediated cross-activation of wild-type RAS by oncogenic RAS was reported to be essential for tumorigenesis underscoring complementary functions of oncogenic and wild-type RAS in tumor cells.37 Recent copy number analysis of single circulating melanoma cells confirm our results by identifying amplifications of KRAS.38 In another study, copy number changes leading to amplification of KRAS were shown to be associated with improved treatment response to sorafenib targeting CRAF in melanoma, giving evidence that wild-type KRAS is crucial in melanoma and acts via RAF proteins other than BRAF.39
In this study, we directly and functionally uncover the importance of wild-type KRAS in melanoma and provide evidence that KRAS is a novel therapeutic target independent of BRAF mutational status. We revealed that increased KRAS expression in human melanoma cells and tissues correlated with mitotic activity, while KRAS knockdown reduced melanoma cell growth and attenuated AKT and/or ERK signaling in melanoma cells. PI3K/AKT signaling has been shown to contribute to melanoma development and inhibitors of PI3K signaling restrain melanoma cell growth.40
Downstream of KRAS, RAF kinases are the primary signaling relays. The catalytic activity of RAF depends on an allosteric mechanism driven by kinase dimerization, and RAF inhibitors can paradoxically induce ERK signaling by stimulating RAF dimerization.9, 41 Accordingly, small-molecule inhibition of ERK dimerization has been shown to prevent tumorigenesis by RAS-ERK pathway oncogenes.42 Given that RAF dimerization canonically depends on RAS activation,23, 43, 44 we asked whether co-targeting the upstream effector of RAF/ERK signaling might be a novel and effective therapeutic approach in melanoma. Indeed, combined KRASi and BRAFi synergistically induced tumor cell apoptosis and prevented emergence of acquired drug resistance. Moreover, KRASi prevented paradoxical BRAFi-induced proliferation. Fitting to this, a recent study identified CRAF as a key mediator of BRAF resistance and showed that combined pan-RAF and MEKi could overcome intrinsic and acquired resistance to single-agent RAF/MEKi.45 Also our findings suggest that KRAS mediates drug resistance via RAF proteins that are not dimerization dependent such as CRAF and not mutated BRAFV600E, because wild-type RAF protein signaling is still dimerization and therefore RAS dependent.
Chemoresistance in melanoma is still poorly characterized,5 and acquired resistance to BRAFi is the main reason for only modest progression-free survival of melanoma patients harboring BRAFV600E mutations that are treated with the current standard therapy.7 Our study supports the hypothesis that acquired resistance to BRAFi can be mediated by dose-dependent upregulation of antiapoptotic AKT and pro-proliferative ERK signaling and that these signaling pathways at least partly depend on KRAS. We found that resistant cell lines upregulated pERK and pAKT in response to BRAFi. It has been shown recently that BRAFi resistance is mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model.46 Here we observed that KRAS protein expression correlated significantly with pAKT/AKT upregulation and KRAS knockdown reduced pERK and/or pAKT levels in resistant cell lines.
Moreover, KRAS repression re-sensitized BRAFi-resistant cells to inhibition of proliferation and induction of apoptosis. Next to mutated KRAS (G12D), overexpressed wild-type KRAS was shown to enhance resistance to apoptosis in melanoma cell line M229.47 Interestingly, a transcriptomic analysis by Hugo et al.48 showing that non-genomic alterations recur highly in acquired BRAFi-resistant melanoma revealed that wild-type KRAS is indeed recurrently overexpressed in melanoma with acquired resistance to BRAF. In our study, we extend these findings and discovered that endogenous KRAS is indeed a therapeutic target contributing to drug resistance in melanoma. These findings further foster the concept that targeting KRAS or co-targeting KRAS and BRAF could be rational approaches for melanoma treatment. Promising results for prolongation of tumor-free survival in melanoma patients were reported for combination of BRAFis and MEKis.23, 43, 44 However, combination regimes with BRAFis and MEKis are recognized to produce double resistance,49 a phenomenon that was also apparent in the resistant cell clones used in this study. Moreover, BRAFi and MEKi in melanoma can lead to the development of KRAS-mutant tumors.50 One can speculate that this side effect could be undermined by co-targeting KRAS.
Pharmacologically, KRAS has been suggested to be ‘undruggable’ for many years. The main reason for this is that RAS proteins do not present suitable pockets for drug binding, except for the GDP/GTP-binding site, which adheres GDP/GTP very tightly in picomolar affinities.12, 13, 14, 15, 16, 51 Recently, a number of new approaches to address RAS activity have led to the revival of KRAS as a molecular target.52, 53 Here we investigated the effect of the recently developed inhibitor of KRAS-trafficking DR.16 We demonstrate strong antitumor effects in melanoma cells, and even after long-term incubation, we did not detect resistant cells. Moreover, we show for the first time that combinatory approaches of KRASi and BRAFi reveal synergistic antitumorigenic effects in melanoma cells.
Interestingly, recent studies reveal therapeutic cooperation between programmed death 1/programmed death ligand 1 immune check point inhibitors and RAS/MAPK pathway inhibitors in breast cancer.54 This indicates that also in melanoma a combination with the newly approved immunotherapies4, 55 could be a further promising therapeutic field of application of inhibitors of KRAS signaling.
In summary, based on our data and previous studies, we hypothesize that combination of inhibition of mutated BRAF (using PLX-4032) and KRAS (using RNAi or DR) leads to apoptosis instead of survival in BRAFi-sensitive melanoma (Figure 8a). In primary resistance, BRAFi is known to induce paradoxic activation of non-mutated RAF proteins, which is RAS dependent and could be overcome by simultaneous KRASi (Figure 8b). Acquired drug resistance to BRAFi can be mediated by attenuation of feedback mechanisms leading to the here shown dose-dependent dynamic KRAS expression, which can also be undermined by KRASi (Figure 8c).

Hypothesis on effects of combined BRAFi and KRASi in BRAFi sensitivity and primary and acquired resistance. (a) In BRAFi-sensitive melanoma, BRAFi leads to ERK suppression resulting in known attenuation of negative feedback effects on RAS expression, including KRAS, leading to pro-survival AKT signaling, which can be undermined by simultaneous KRASi. (b) In primary BRAFi resistance, BRAFi is known to induce paradoxic trans-activation mediated by heterodimerization with non-mutated BRAF wild type and other RAF proteins. Importantly, dimerization of non-mutated RAF is (K)RAS dependent and can be prevented by KRASi. (c) Acquired drug resistance to BRAFi is mediated by known attenuation of feedback mechanisms leading to dose-dependent dynamic KRAS expression as seen in our experiments. The resulting RAF and PI3K activation can be suppressed by KRASi, thereby restoring BRAFi sensitivity.
Materials and methods
Melanoma cells
Human melanoma cell lines were previously described (Supplementary Table S1). Furthermore, BRAFi-sensitive wild-type and BRAFi-resistant BRAFV600E 451Lu and SkMel28 melanoma cell lines were described by Villanueva et al.23
For KRASi, we used a recently developed small-molecule inhibitor that inhibits cGMP phosphodiesterase delta subunit (PDE6δ)-mediated KRAS trafficking to the cell membrane (DR; Cayman Chemicals, Ann Arbor, MI, USA). A complete list of used inhibitors and detailed treatment conditions are described in the figure legends and in Supplementary Material and Methods.
Sampling and handling of patient material were carried out in accordance with the ethical principles of the Declaration of Helsinki. Use of human tissue material had been approved by the local ethics committee of the University of Regensburg (application numbers 09/11 and 03/151).
Analysis of tumorigenicity of melanoma cells in nude mice
Experiments were conducted according to the German Physiological Society principles for the use of laboratory animals (Granted permission number 54-2532.4-5/14, Government of Bavaria). In all, 800 000 cells in 100 μl of phosphate-buffered saline were injected subcutaneously into the right flank of 4–8-week-old male athymic nu/nu mice. Detailed conditions and quantification methods are described in Supplementary Material and Methods.
Analysis of patient material
Human melanoma tissue samples (Supplementary Table S3) were analyzed using quantitative reverse transcriptase–PCR to determine KRAS and CyclinD1 mRNA levels. Furthermore, a tissue microarray containing tissue samples from melanocyte-derived benign nevi, primary melanomas and melanoma metastasis (Supplementary Table S2) was used for immunohistochemical analysis of pERK/ERK, pAKT/AKT, Ki67 and KRAS. Detailed conditions and quantification methods are described in Supplementary Materials and Methods.
Statistical analysis
Results are expressed as mean±s.e.m. (if not depicted otherwise in figure legends). Comparison between groups was made using Student’s t-test or one-way analysis of variance, if appropriate. Tissue microarray analysis was performed using Fisher’s exact test. Spearman and Pearson correlation coefficients were used for correlation analysis. The level of significance was P<0.05 (depicted in figures as ‘NS’, that is, not significant, and ‘*’, that is, P<0.05). If not depicted otherwise in figure legends, the number of independent experiments was n⩾3. Statistical calculations were performed using the GraphPad Prism Software (GraphPad Software, Inc., San Diego, CA, USA) and SPSS (SPSS Statistics 23, IBM Corp., Armonk, NY, USA).
Additional and more detailed methods and protocols can be found in Supplementary Materials and Methods.
Abbreviations
- AKT:
-
v-akt murine thymoma viral oncogene
- BRAF:
-
v-Raf murine sarcoma viral oncogene homolog B
- BRAFi:
-
BRAF inhibition/inhibitor
- CTR:
-
control
- DR:
-
Deltarasin
- ERK:
-
extracellular-signal regulated kinase
- KR:
-
KRAS (Kirsten Rat sarcoma)
- KRASi:
-
KRAS inhibition/inhibitor
- MAPK:
-
mitogen-activated protein kinase
- MEK:
-
mitogen-activated protein kinase kinase
- MEKi:
-
MEK inhibition/inhibitor
- NRAS:
-
neuroblastoma RAS viral oncogene homolog
- PI3K:
-
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PLX:
-
PLX-4032 (Vemurafenib).
References
Leiter U, Eigentler T, Garbe C . Epidemiology of skin cancer. Adv Exp Med Biol 2014; 810: 120–140.
Harries M, Malvehy J, Lebbe C, Heron L, Amelio J, Szabo Z et al. Treatment patterns of advanced malignant melanoma (stage III-IV) - a review of current standards in Europe. Eur J Cancer 2016; 60: 179–189.
Dummer R, Hauschild A, Guggenheim M, Keilholz U, Pentheroudakis G . Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012; 23 (Suppl 7): vii86–vii91.
Chapuis AG, Lee SM, Thompson JA, Roberts IM, Margolin KA, Bhatia S et al. Combined IL-21-primed polyclonal CTL plus CTLA4 blockade controls refractory metastatic melanoma in a patient. J Exp Med 2016; 213: 1133–1139.
Matin RN, Chikh A, Chong SL, Mesher D, Graf M, Sanza P et al. p63 is an alternative p53 repressor in melanoma that confers chemoresistance and a poor prognosis. J Exp Med 2013; 210: 581–603.
Karimkhani C, Gonzalez R, Dellavalle RP . A review of novel therapies for melanoma. Am J Clin Dermatol 2014; 15: 323–337.
Luke JJ, Ott PA . New developments in the treatment of metastatic melanoma - role of dabrafenib-trametinib combination therapy. Drug Healthc Patient Saf 2014; 6: 77–88.
Monsma DJ, Cherba DM, Eugster EE, Dylewski DL, Davidson PT, Peterson CA et al. Melanoma patient derived xenografts acquire distinct Vemurafenib resistance mechanisms. Am J Cancer Res 2015; 5: 1507–1518.
Lito P, Rosen N, Solit DB . Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013; 19: 1401–1409.
Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468: 973–977.
Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov 2013; 3: 338–349.
McCormick F . KRAS as a therapeutic target. Clin Cancer Res 2015; 21: 1797–1801.
Cox AD, Der CJ, Philips MR . Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res 2015; 21: 1819–1827.
Stephen AG, Esposito D, Bagni RK, McCormick F . Dragging Ras back in the ring. Cancer Cell 2014; 25: 272–281.
Schmick M, Vartak N, Papke B, Kovacevic M, Truxius DC, Rossmannek L et al. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 2014; 157: 459–471.
Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013; 497: 638–642.
Yuan TL, Fellmann C, Lee CS, Ritchie CD, Thapar V, Lee LC et al. Development of siRNA payloads to target KRAS-mutant cancer. Cancer Discov 2014; 4: 1182–1197.
Xue W, Dahlman JE, Tammela T, Khan OF, Sood S, Dave A et al. Small RNA combination therapy for lung cancer. Proc Natl Acad Sci USA 2014; 111: E3553–E3561.
Posch C, Cholewa BD, Vujic I, Sanlorenzo M, Ma J, Kim ST et al. Combined inhibition of MEK and Plk1 has synergistic antitumor activity in NRAS mutant melanoma. J Invest Dermatol 2015; 135: 2475–2483.
Milagre C, Dhomen N, Geyer FC, Hayward R, Lambros M, Reis-Filho JS et al. A mouse model of melanoma driven by oncogenic KRAS. Cancer Res 2010; 70: 5549–5557.
Gimotty PA, Van Belle P, Elder DE, Murry T, Montone KT, Xu X et al. Biologic and prognostic significance of dermal Ki67 expression, mitoses, and tumorigenicity in thin invasive cutaneous melanoma. J Clin Oncol 2005; 23: 8048–8056.
Depasquale I, Thompson WD . Microvessel density for melanoma prognosis. Histopathology 2005; 47: 186–194.
Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010; 18: 683–695.
Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep 2013; 4: 1090–1099.
Johnson DB, Sosman JA . Therapeutic advances and treatment options in metastatic melanoma. JAMA Oncol 2015; 1: 380–386.
Burotto M, Chiou VL, Lee JM, Kohn EC . The MAPK pathway across different malignancies: a new perspective. Cancer 2014; 120: 3446–3456.
Sondergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med 2010; 8: 39.
Wang J, Huang SK, Marzese DM, Hsu SC, Kawas NP, Chong KK et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J Invest Dermatol 2015; 135: 532–541.
Gross A, Niemetz-Rahn A, Nonnenmacher A, Tucholski J, Keilholz U, Fusi A . Expression and activity of EGFR in human cutaneous melanoma cell lines and influence of vemurafenib on the EGFR pathway. Target Oncol 2015; 10: 77–84.
Sale MJ, Cook SJ . That which does not kill me makes me stronger; combining ERK1/2 pathway inhibitors and BH3 mimetics to kill tumour cells and prevent acquired resistance. Br J Pharmacol 2013; 169: 1708–1722.
Wilson BE, Mochon E, Boxer LM . Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol 1996; 16: 5546–5556.
Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N . MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem 2000; 79: 355–369.
Yang J, Song Q, Cai Y, Wang P, Wang M, Zhang D . RLIP76-dependent suppression of PI3K/AKT/Bcl-2 pathway by miR-101 induces apoptosis in prostate cancer. Biochem Biophys Res Commun 2015; 463: 900–906.
Grabocka E, Pylayeva-Gupta Y, Jones MJ, Lubkov V, Yemanaberhan E, Taylor L et al. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell 2014; 25: 243–256.
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D . RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011; 11: 761–774.
Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 2012; 483: 613–617.
Jeng HH, Taylor LJ, Bar-Sagi D . Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat Commun 2012; 3: 1168.
Ruiz C, Li J, Luttgen MS, Kolatkar A, Kendall JT, Flores E et al. Limited genomic heterogeneity of circulating melanoma cells in advanced stage patients. Phys Biol 2015; 12: 016008.
Wilson MA, Zhao F, Khare S, D'Andrea K, Wubbenhorst B, Roszik J et al. Copy number changes are associated with response to treatment with carboplatin, paclitaxel, and sorafenib in melanoma. Clin Cancer Res 2015; 22: 374–382.
Sweetlove M, Wrightson E, Kolekar S, Rewcastle GW, Baguley BC, Shepherd PR et al. Inhibitors of pan-PI3K signaling synergize with BRAF or MEK inhibitors to prevent BRAF-mutant melanoma cell growth. Front Oncol 2015; 5: 135.
Lavoie H, Therrien M . Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 2015; 16: 281–298.
Herrero A, Pinto A, Colon-Bolea P, Casar B, Jones M, Agudo-Ibanez L et al. Small molecule inhibition of ERK dimerization prevents tumorigenesis by RAS-ERK pathway oncogenes. Cancer Cell 2015; 28: 170–182.
Richman J, Martin-Liberal J, Diem S, Larkin J . BRAF and MEK inhibition for the treatment of advanced BRAF mutant melanoma. Expert Opin Pharmacother 2015; 16: 1285–1297.
Queirolo P, Picasso V, Spagnolo F . Combined BRAF and MEK inhibition for the treatment of BRAF-mutated metastatic melanoma. Cancer Treat Rev 2015; 41: 519–526.
Whittaker SR, Cowley GS, Wagner S, Luo F, Root DE, Garraway LA . Combined pan-RAF and MEK inhibition overcomes multiple resistance mechanisms to selective RAF inhibitors. Mol Cancer Ther 2015; 14: 2700–2711.
Perna D, Karreth FA, Rust AG, Perez-Mancera PA, Rashid M, Iorio F et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc Natl Acad Sci USA 2015; 112: E536–E545.
Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014; 4: 80–93.
Hugo W, Shi H, Sun L, Piva M, Song C, Kong X et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 2015; 162: 1271–1285.
Smyth T, Paraiso KH, Hearn K, Rodriguez-Lopez AM, Munck JM, Haarberg HE et al. Inhibition of HSP90 by AT13387 delays the emergence of resistance to BRAF inhibitors and overcomes resistance to dual BRAF and MEK inhibition in melanoma models. Mol Cancer Ther 2014; 13: 2793–2804.
Carlino MS, Kwan V, Miller DK, Saunders CA, Yip D, Nagrial AM et al. New RAS-mutant pancreatic adenocarcinoma with combined BRAF and MEK inhibition for metastatic melanoma. J Clin Oncol 2015; 33: e52–e56.
The KRAS-PDEdelta interaction is a therapeutic target. Cancer Discov 2013; 3: OF20.
Milroy LG, Ottmann C . The renaissance of Ras. ACS Chem Biol 2014; 9: 2447–2458.
Ledford H . Cancer: the Ras renaissance. Nature 2015; 520: 278–280.
Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res 2015; 22: 1499–1509.
Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP . Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med 2014; 211: 715–725.
Acknowledgements
This work was supported by the German Research Association (DFG) (Research Training Group ‘RTG 1962/1’, University of Erlangen), the German Cancer Aid (Deutsche Krebshilfe), the Bavarian Research Network for Molecular Biosystems (BioSysNet) and the Interdisciplinary Center for Clinical Research (IZKF) Erlangen (J55, to PD and D24, to AB). We thank Meenhard Herlyn (The Wistar Institute, Philadelphia, USA) for providing the resistant cell lines. Furthermore, we thank Rudolf Jung, Annette Serwotka and Darleen Schönwälder for technical assistance.
Author contributions
PD, CH and AKB conceived the project, analyzed the data and wrote the paper. PD designed and performed most of the experiments. TS helped in planning and performing mouse experiments. SK contributed to data analysis and manuscript creation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Dietrich, P., Kuphal, S., Spruss, T. et al. Wild-type KRAS is a novel therapeutic target for melanoma contributing to primary and acquired resistance to BRAF inhibition. Oncogene 37, 897–911 (2018). https://doi.org/10.1038/onc.2017.391
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2017.391
- Springer Nature Limited
This article is cited by
-
Cooperative genomic lesions in HRAS-mutant cancers predict resistance to farnesyltransferase inhibitors
Oncogene (2024)
-
Characterization of two different alginate-based bioinks and the influence of melanoma growth within
Scientific Reports (2024)
-
C-Jun drives melanoma progression in PTEN wild type melanoma cells
Cell Death & Disease (2019)