
Abstract
Sun-induced skin lesions, in particular actinic keratosis, are generally considered as premalignant skin lesions that can progress into squamous cell carcinoma (SCC) and invasive SCC if left untreated. Therefore, understanding the molecular mechanisms by which the ultraviolet-B (UV-B)-exposed cells are being protected and the signaling pathways that promote the progression of certain premalignant skin lesions to malignant lesions will permit us to prevent or cure skin cancers. In the current study, we found that phospho-p21-activated kinase-1 (Pak1) and Pak1 expression was high in clinical samples of sunlight-induced premalignant skin lesions assessed by immunohistochemistry. Further, we observed that phospho-Pak1 and Pak1 levels are high in UV-B-exposed hairless SKH mouse model skin samples as compared with unexposed skin tissue. Our results from cell line and animal models showed that Pak1 is activated in response to UV-B radiation, and this activated Pak1 translocates from the cytoplasm to the nucleus. Inside the nucleus, Pak1 via C-Fos binds to a specific promoter region of DNA repair kinase ATR (ataxia–telangiectasia and Rad3-related protein) and acts as a transcriptional regulator of ATR. Results from our analysis showed that Pak1 overexpression, knockdown and Pak1 knockout cell line models showed that Pak1 confers protection to keratinocytes from UV-B-induced apoptosis and DNA damage via ATR. To our knowledge, this is the first study that evaluates the functional and clinical significance of a signaling molecule, Pak1, in sun-induced premalignant skin lesions and indicates that increased Pak1 activation and expression could serve as an early warning sign of progression toward non-melanoma skin cancer, if ignored.
Similar content being viewed by others


Introduction
Non-melanoma skin cancer (NMSC)—which includes both basal cell carcinoma (BCC) and squamous cell carcinoma (SCC)—has become a growing health concern over the recent years and is mainly due to overexposure to ultraviolet (UV) radiation.1 The frequency of NMSC is solidly allied to solar UV radiation exposure.2 UV-B is a potent natural carcinogen competent enough to initiate, endorse and facilitate the progression of skin cancer.3 Development of UV-B-induced skin carcinoma is an intricate and multistep process with an intermediate stage of developing skin lesions before the transformation stage. Although not classified as NMSC, these precursor lesions of NMSC-like actinic keratosis and polymorphic light eruptions if left untreated may sometimes transform into SCC.4, 5 Actinic keratosis serves as a precursor for ~60% of SCCs.6 Thus, these lesions can be an early warning sign of skin cancer and also serve as biomarkers in chemopreventive studies.
UV-B induces DNA damage by forming cyclobutane pyrimidine dimers, which might lead to mutations in the epidermal cells, leading to the cancer cell progression.7 Damage of cells at the molecular stage and DNA level instigates transcription factor pathways, which in turn regulate a number of UV response gene expressions involved in the course of cell proliferation, cell differentiation and cell survival, and thus have a major role in tumor progression.8 DNA damage response is a network of signaling pathways that ensures genomic integrity and stability by recognizing and repairing the DNA damage.9 It is well established that stalled forks at replication and factors that induce bulky adducts, such as UV-B radiation, activate ataxia–telangiectasia and Rad3-related protein (ATR).10 Repair of DNA damage induced by UV is of high importance to UV-induced skin cancer. Nucleotide excision repair (NER) mechanism either repairs the damage completely before replication or synthesis of DNA may occur after replication using repair-specific DNA polymerase, which is error free in human cells to avoid the impending mutation at sites damaged by UV. NER is an exceedingly conserved strategy to repair bulk DNA damages, such as cyclobutane pyrimidine dimers induced by cosmological UV-B radiation, and are repaired efficiently without mistake, but flawed repair of these lesions may result in mutations of oncogenes.3
Accumulating data have indicated that epidermal growth factor receptor (EGFR) has a significant role in signal transduction induced by UV rays.11 Reactive oxygen species generated by UV mediates EGFR activation, which in turn inhibits tyrosine phosphatase activities.12 Hence, in epidermal keratinocytes, the phosphorylation of multiple tyrosine residues of EGFR increases. EGFR activation results from autophosphorylation of intracellular C-terminal domain at specific tyrosine residues, which act as docking sites for various signals, which might in turn activate different signal transduction cascades resulting eventually in cellular responses.11, 13 Thus, reactive oxygen species function upstream of EGFR. Tumorigenesis was suppressed on pharmacological inhibition of EGFR activation by UV in a genetically initiated skin tumorigenesis mouse model.14 P21-activated kinase-1 (Pak1) is a serine/threonine signaling kinase and a well-known regulator of cytoskeletal remodeling that has a vital role in cell survival functions.15, 16 An alteration in Pak1 expression has been renowned in various types of cancers.17 Small Rho GTPases Cdc42 and Rac, protein kinase A, phosphoinositide-dependent kinase-1, EGFR and AKT are a wide range of upstream signaling molecules that regulate Pak1 activity via phosphorylation or protein–protein interaction.18 In addition, Pak1 dysregulation has been shown to be associated with DNA damage.19
Despite all this information, the molecular events that contribute to UV-induced skin carcinogenesis are feebly understood mainly owing to the lack of mechanistic insights. It is also unclear how the normal keratinocytes are transformed into malignant ones and what progressive change occurs in the biology of skin cells, which remains the focus of this study. In the current study, we looked at the Pak1 expression in various premalignant lesions of NMSC and observed that increased expression of phospho-Pak1 and Pak1 is related to histological evidence of chronic sun damage. We also elucidate the molecular mechanism of Pak1 activation by UV-B in keratinocyte cell lines and animal models.
Results
Pak1 expression is deregulated in human premalignant sun-induced skin lesions and UV-B- exposed mouse skin tissues
To study the role of Pak1 in the initiation and progression of chronic sun-induced skin lesions to more invasive forms such as NMSC, phospho-Pak1 and Pak1 expression was evaluated in formalin-fixed, paraffin-embedded human sunlight-induced skin lesions (n=16, details about specific samples used are given in Supplementary Table 2) by immunohistochemistry (IHC). Results from our studies showed that phospho-Pak1 expression was higher in sunlight-induced skin lesions as compared with Pak1. The mean Q-score of phospho-Pak1 in skin lesion samples studied was 8.37, as compared with 5.18 for Pak1, probably indicating that Pak1 expression and its activation could serve as a marker for sunlight-induced lesions. The mean and median along with their respective s.d. for the Q-scores are shown in Figure 1a.

IHC analysis of Pak1 expression in sun-induced skin lesions and UV-B-exposed mouse skin tissues. (a) Representative images of phospho-Pak1 and Pak1 IHC in human sun-induced lesions. (b) Representative images of phospho-Pak1 and Pak1 IHC in mouse UV-B-exposed and -unexposed skin tissue. The mean and median along with their respective s.d. for the Q-scores is shown in the table.
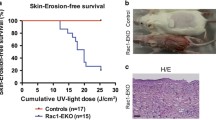
To support our clinical findings of upregulated and activated Pak1 in human skin samples, we performed IHC studies on skin samples collected from controlled mouse experiments exposed to 25 weeks of UV-B. IHC data show that Pak1 levels are not only high but also activated as documented by strong positivity for phospho-Pak1 in UV-B-exposed skin samples compared with the age-matched controls. The mean Q-score of phospho-Pak1 and Pak1 in UV-B exposed skin samples studied was 12.0 and 1.5, respectively, as compared with 1.5 and 0.25, respectively, for the unexposed skin samples (Figure 1b). This Q-score value was found to be statistically significant, probably indicating that Pak1 expression and its activation could serve as a marker for UV-B-induced lesions.
Pak1 is activated in response to UV-B radiation in in vitro and in vivo models
UV-B radiation was reported to induce keratinocyte proliferation and survival.20 As the role of Pak1 in normal cell survival and proliferation is well defined and based on our above clinical data, we hypothesized that Pak1 might be activated in response to UV-B. UV-B radiation significantly induced Pak1 activity in normal cell line human epidermal keratinocyte adult, normal human fibroblasts AG1522, human keratinocyte cell lines A431 and HaCaT and in murine epidermal cell line JB6 in a dose- and time-dependent manner (Figures 2a–d, upper panels). Further, we determined the Pak1 activation status in a physiological in vivo whole-animal setting by UV-B radiation, and results showed that Pak1 activity was significantly increased in mouse skin tissue (Figure 2e, upper panel). As Pak1 kinase activity depends on its activation by phosphorylation,15 we also observed a significant increase in Pak1 phosphorylation at serine 144 (Figures 2a–e, lower panels) and threonine 212 site by UV-B radiation (Supplementary Figure S1).

(a–d) Pak1 is activated by UV-B: HEKa, AG1522, HaCaT, A431 and JB6 cells were irradiated with a stipulated dose of UV-B. After incubation for 30 min, Pak1 kinase activity was checked using PAKtide (Tricine gel) and MBP (myelin basic protein) as substrates. Phospho-PAKtide and phospho-MBP bands were analyzed with autoradiogram images; Ponceau or Memcode image shows equal amount of substrates used for the kinase assay, and subsequent immune blotting was carried out with anti-Pak1 antibody (upper panel) and was detected using alkaline phosphatase (AP) or horseradish peroxidase (HRP) substrate. Western blots of equal amounts of total protein from control (Con) and UV-B-irradiated cells (lower panel) were carried out using phospho-Pak1 (phosphorylation at serine 144) and Pak1 antibody. (e) Pak1 activity in skin tissue lysates of Swiss Albino mice on whole-body UV-B irradiation (400 mj/cm2 and 200 mj/cm2). HEKa, human epidermal keratinocyte adult.
UV-B alters the localization of Pak1
Pak1 is found both in the cytoplasmic and nuclear compartments, and earlier studies21 have demonstrated that activated Pak1 translocates into the nuclear compartment. This incited us to investigate the effect of Pak1 phosphorylation and its activation on its subcellular localization in control and UV-B-irradiated A431 and HaCaT cells by confocal microscopy. Significant increase in the levels of nuclear phospho-Pak1 was observed upon UV-B stimulation, an indication of activated Pak1 being translocated to the nucleus from the cytoplasm (Figures 3a and b). In support of this notion, cytoplasmic and nuclear proteins extracted from UV-B-stimulated A431 and HaCaT cells also showed similar results (Figures 3c and d). Taken together, these results revealed that UV-B stimulation activates Pak1, and activated Pak1 in turn translocates from the cytoplasmic to the nuclear compartment.

Pak1 translocates to the nucleus upon UV-B irradiation in keratinocytes. (aand b) Confocal microscopy images of A431 and HaCaT cells either untreated (Cntrl) or treated (UV-B) with 200 and 40 mj/cm2dose of UV-B, and harvested at 30 min for immunofluorescence. Nuclear staining (blue) by 4',6-diamidino-2-phenylindole (DAPI). (c and d) A431 and HaCaT cells were untreated or treated with 200 and 40 mj/cm2 dose of UV-B, respectively, harvested at 30 min, and lysates from cytoplasmic and nuclear extracts were analyzed by western blotting with Pak1-specific antibody. Poly (ADP-ribose) polymerase (PARP), paxillin or tubulin were used as markers for nuclear and cytoplasm lysates, respectively.
ATR, a novel transcriptional target of Pak1
ATR has been shown to be specifically associated and activated with DNA damage induced by UV-B.10 Nuclear Pak1 was shown to modulate the expression of various genes—that is, nuclear factor of activated T-cell 1, tissue factor, phosphofructokinase-muscle isoform tissue factor pathway inhibitor 1 and fibronectin—in various cellular frameworks.22, 23, 24 These two observations combined with the fact that Pak1 is activated immediately after UV-B radiation and translocated to the nuclear compartment raised the possibility that Pak1 might regulate ATR. To explore whether ATR is the downstream molecular target that is regulated by Pak1, we made stable Pak1 overexpression clones and stable clones of lentiviral Pak1 short hairpin RNA (shRNA) in both HaCaT and A431 cell lines (Figures 4a–d, upper panels) and looked for ATR levels in both the systems. Results showed that there is a significant change in the protein and mRNA levels of ATR upon modulating Pak1 levels (Figures 4a–d, lower panels). Further, we checked for ATR levels in Pak1-knockout clones of HaCaT cell line and observed that there is a convincing decrease in mRNA and protein levels of ATR (Supplementary Figure S2A).

ATR, a novel molecular target of Pak1. (aandb) Quantitative PCR (qPCR) and western blot analysis of ATR in stable Pak1 overexpression clones in HaCaT and A431. Pak1 expression (upper panel) and upregulation of ATR (lower panel). (c and d) qPCR and western blot analysis of ATR in stable Pak1 shRNA knockdown clones in HaCaT and A431. Pak1 expression (upper panel) and downregulation of ATR (lower panel).
Consistent with this, we found that Wt-Pak1 augmented the 2.1- kb ATR promoter activity (−2032 to +121) (Figure 5a, upper and lower panels). Further, to check whether kinase activity of Pak1 is essential for ATR promoter regulation, we used (constitutively active) Pak1-T423E construct and performed ATR promoter luciferase assay. Results showed a significant increase in ATR promoter activity with the expression of constitutively active Pak1-T423E (Figure 5a, upper and lower panels). This was further confirmed with the use of Pak1 inhibitors—Frax597 and IPA3—which resulted in radical decrease of ATR levels with both the inhibitors (Supplementary Figures S2B and C). Further, to check the role of nuclear Pak1 in ATR promoter activation, we used a mutant construct of Pak1 NLS22 and observed that this Pak1 NLS mutant was not capable of increasing the activity of ATR promoter (Figure 5b, upper and lower panels). These results clearly indicate that Pak1 kinase activity and its localization into the nucleus is required for the ATR promoter activation.

Pak1 modulates ATR transcription. (a) A431 and HEK-293 cells were co-transfected transiently with 0.5 μg of ATR FL promoter with or without wild-type Pak1 (Wt) or active Pak1 (T423E). Western blot showing endogenous ATR expression on transfection of wild-type Pak1 (Wt) or active Pak1 (T423E) transiently. (b) A431 and HEK-293 cells were transiently co-transfected with 0.5 μg of ATR FL promoter with or without Pak1 (Wt) or Pak1 that lacks signal for nuclear localization (Pak1 NLS). (c) A431 cells were transiently transfected with 0.5 μg of ATR FL promoter and irradiated with UV-B 200 mj/cm2 after downregulation of Pak1 with Pak1 siRNA. (d) An illustrative representation of ATR luciferase reporter plasmids with insert 2 -kb DNA and truncated fragments of ATR promoter. Arrow indicates the transcription start site. The number of bases upstream and downstream of the transcription start site (upper) are indicated by numbers, luciferase activity of ATR promoter FL, Del1, Del2 and Del3 in A431 and HEK-293 cell lines and identification of core promoter region essential for ATR regulation by Pak1 (lower).
Further, we checked the effect of UV-B on ATR promoter activity and observed that there is an increase in the promoter activity with UV-B. In addition to this, we also looked at the role of Pak1 in UV-B-induced ATR promoter activity and observed no significant increase in the promoter activity of ATR upon silencing Pak1 (Figure 5c). Next, to gain deeper insights into the modulation of ATR by Pak1, we made different deletion constructs of ATR to identify the core promoter region of ATR that is regulated by Pak1. Results showed that the core sequence of the promoter was localized in the region from −531 to +121, and the same could be sufficient for ATR gene activation by Pak1, as the enhanced promoter activity by Pak1 was found to be similar in all deletion constructs (Figure 5d, upper and lower panels). Further, we performed chromatin immunoprecipitation-based promoter walk with four different primer sets that cover the 2.1- kb region and examined the conscription of Pak1 onto the 2.1- kb ATR promoter region. Our results showed that Pak1 was recruited to the 652- bp ATR core promoter at a region (F4) between −531 and +121 (Figure 6a), and the recruitment was enhanced upon UV-B treatment as analyzed by quantitative PCR.

Pak1 modulates ATR via C-Fos. (a) Schematic representation of the 2 -kb ATR promoter, showing analyzed regions. The recruitment of Pak1 to ATR promoter in A431 cells was shown by chromatin immunoprecipitation (ChIP) analysis. Ctrl and UV represent untreated and UV-B (200 mj/cm2) treated, respectively. (b) HEK-293 cells were co-transfected with 0.5 μg of 2- kb pGL4 ATR Del3 luciferase reporter and Pak1 or C-Fos or Pak1 and C-Fos. (c) ChIP analysis showing that the recruitment of Pak1 to human ATR promoter is mediated by C-Fos in A431 cells. ChIP was carried out with an anti-Pak1 antibody after downregulation of C-Fos with C-Fos siRNA followed by PCR amplification using the specific primers for F4. Western blot showing the siRNA-mediated downregulation of c-Fos in A431 cells. (d) Schematic representation of the 0.5- kb ATR core promoter, showing the C-Fos-binding regions deleted (Del1, Del2 and Del1 and 2). A431 and HEK-293 cells were co-transfected with 0.5 μg of pGL4 ATR Del3 luciferase reporter or pGL4 ATR Del3 C-Fos binding regions deleted (Del1, Del2 and Del1 and 2) luciferase reporter or pGL4 basic vector plasmid with or without Pak1. (e) EMSA analysis of c-Fos binding to human ATR promoter using biotin-labeled probe that mimics c-Fos-binding sites using A431 cell nuclear lysate.
Pak1 regulates ATR via the C-Fos pathway
Our observations that ATR promoter activity is modulated by Pak1 encouraged us to study the possible regulatory mechanism of ATR transcription by Pak1. Pak1, which lacks the transcriptional activity on its own, was shown to modulate gene expression via several transcription factors.25 To identify the transcription factors that mediate the induction of ATR by Pak1, we next analyzed the sequence of ATR core promoter using various transcription factor prediction tools. We observed that C-Fos is one such potential transcriptional factor that binds to the ATR promoter. It was reported that C-Fos is induced by UV-B radiation.26 As UV-B role in induction of C-Fos is already established and the fact that Pak1 is activated by UV-B, we assumed that C-Fos could be an appropriate candidate to study Pak1-mediated ATR regulation. On the basis of this, we rationaled that Pak1 might be regulating ATR via C-Fos.
To examine the possibility of involvement of C-Fos in ATR regulation by Pak1, we initially checked for the ATR core promoter activity by transfecting C-Fos and Pak1. As expected, Pak1 alone induced the ATR promoter activity, whereas a synergistic increase in the activity of ATR promoter was observed upon transfection with both C-Fos and Pak1 (Figure 6b). Further, to establish the C-Fos importance in upregulation of ATR by Pak1, C-Fos expression was silenced using siRNA and the recruitment of Pak1 onto the ATR core promoter was examined. Significant decrease in the Pak1 recruitment to the ATR promoter was observed upon C-Fos silencing (Figure 6c). Further, to narrow down the C-Fos-binding region on the ATR promoter in the transcriptional regulation of ATR by Pak1, we scanned the 652- bp ATR core promoter using the prediction tool ConSite and identified two potential C-Fos-binding sites : TGACTCAC27 at positions −376 to −368 and −314 to −306 in the core promoter region of ATR. Next, to investigate the functional C-Fos-binding region on the ATR promoter, deletion constructs were made by deleting the two binding sites alone or in combination and promoter luciferase activity was assessed. Results showed that deletion of binding sites 1 and 2 individually (C-Fos del1 and C-Fos del2) did not show any effect on ATR promoter activity by Pak1, whereas the promoter construct in which both the C-Fos-binding sites were mutually deleted (C-Fos del3) showed no enhanced promoter activity with Pak1, thereby indicating that both C-Fos-binding sites are important for ATR activation by Pak1 (Figure 6d). Further, the recruitment of C-Fos to the ATR promoter was studied by performing electrophoretic mobility shift assay (EMSA) using the oligonucleotides containing the C-Fos consensus sequence for both the predicted sites, and binding of the C-Fos was analyzed. The formation of DNA–protein complexes and the observed complex was significantly enhanced with UV-B treatment, as observed in results (Figure 6e).
Pak1 confers efficient survival and DNA damage response to keratinocytes in response to UV-B
It is already established that Pak1 promotes cell survival functions.15 Above results clearly showed that Pak1 is rapidly activated following UV-B exposure and the fact that UV-B radiation induces keratinocyte proliferation and survival20 encouraged us to examine the role of Pak1 in cell proliferation on UV-B irradiation. For this, Pak1-modulated stable clones of A431 were exposed to UV-B and cell growth rate was determined. Results showed that UV-B irradiation caused a decrease in cell viability compared with control cells, which were recovered partially on overexpression of Pak1, whereas UV-B irradiation of Pak1-downregulated knockdown (KD) clones resulted in reduced proliferation compared with nontarget shRNA-expressing NT clones (Figure 7a, upper and lower panels). Further, to check the long-term effects of Pak1 after UV-B irradiation, we performed clonogenic cell survival assay in Pak1-modulated clones either with or without UV-B and counted the number of colonies after 20 days. The results showed that Pak1-overexpressing clones formed larger number of colonies as compared with vector control clones upon UV-B irradiation, whereas Pak1-KD clones showed reduced ability to form colonies upon UV-B irradiation (Figure 7b, upper and lower panels; Supplementary Figures S3 and S4). These results are in corroboration with the previous findings that cell survival in short-term culture was reduced after UV irradiation.28 However, Pak1 regulation modulates the cell survival functions of both the clones and accredit the significance of exogenous Pak1 in conferring the long-term protective function against UV-B-irradiation-induced cytotoxic effect.

Pak1 protects keratinocytes from apoptosis and UV-B-induced DNA damage. (a) Cell proliferation assay upon UV-B treatment. (b) Clonogenic assay in which colony formation efficiency was calculated on UV-B treatment. (c) TUNEL assay graph represents % of TUNEL-positive cells and (d) Comet assay graph represents % of tail moment. Top panel represents stable Pak1 overexpression clones and bottom panel represents Pak1 shRNA knockdown clones of A431. (e) Western blot and Annexin V assay of HaCaT Pak1-knockout clones.
On the basis of the above results, we next focused on the role of Pak1 in UV-induced apoptosis by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay. Pak1 overexpression clones showed a decrease in the percentage of TUNEL-positive cells, whereas an increase in the percentage of TUNEL-positive cells was observed upon UV-B irradiation in Pak1-knockdown clones (Figure 7c, upper and lower panels). This was further supported by the DNA damage alkaline comet assay results, which showed an increase in the tail moment in Pak1-knockdown clones upon exposure to UV-B, whereas Pak1-overexpressing clones were more resistant to the effect of UV-B (Figure 7d, upper and lower panels; Supplementary Figures S5 and S6). Further, we analyzed the functional role of Pak1 upon UV-B using HaCaT Pak1-knockdown clones (Supplementary Figures S7). To strengthen the fact that Pak1 confers protection on UV-B-induced DNA damage, we developed Pak1-knockout cells (Pak1-null cells) in HaCaT background and studied the response of cells on UV-B-induced damage. Pak1-knockout cells showed higher Annexin-V-positive cells compared with the control cells (Figure 7e, upper and lower panels). Taken together, these data suggest that Pak1 confers an efficient protective role in DNA damage response induced by UV-B.
Discussion
The aim of this study was to define the role of Pak1 in UV-B-induced premalignant skin lesions. Strong clinical and experimental evidence supports UV-B-induced premalignant skin lesions as precursors of SCC of the skin. To our knowledge, this is the first study to evaluate the functional and clinical significance of a signaling molecule—Pak1—in sun-induced skin lesions and UV-B-exposed mouse models. At present, p53 mutations and sunburn cells (apoptotic keratinocytes) are well-known markers of DNA lesions induced by UV-B and act as precursors for malignant transformation of epidermal keratinocytes.29 With this study, we propose that increased Pak1 expression and its activation in sun-induced skin lesions could serve as an early warning sign of progression toward NMSC, if unattended.
Several upstream regulators of Pak1 are known to be involved in UV-B signaling. Recently, Pak1 was shown to be a therapeutic target in BRAF wild-type melanomas.30 However, the direct role of Pak1 and its contribution to the signaling and etiology of NMSC has not yet been studied. A number of genes such as ectodysplasin A2 receptor, β2 pleckstrin homology-like domain, family A member 3 (PHLDA3), G-to-S phase expressed protein 1, DNA damage-inducible transcript 4, adrenergic receptor and (sestrin) p53-induced protein were reported to mediate p53-dependent DNA damage signaling31 and are known to be regulated by Pak1 in response to ionizing radiation, providing evidence that Pak1 signaling is also involved in DNA damage response. In addition, Pak1 per se has been shown to be involved in DNA repair pathways for ionizing radiation-induced DNA damage via MORC2.32 In this study, we provide clear molecular evidence that Pak1 participates in UV-B-induced DNA damage response via ATR. It was previously shown that UV damage increases the transcriptional activity of ATR.33 Interestingly, the ATR promoter region that they used for luciferase reporter assay to show ATR induction by UV-B coincided with the region we identified, that is, Del3 ATR promoter construct (−531 to +121), where Pak1 is recruited and is the core promoter region observed in our results. There was a significant increase in the recruitment of Pak1 to the ATR promoter upon UV-B radiation, indicating the pivotal role of Pak1 in regulating the transcriptional activity of ATR by UV-B. Thus, increased ATR will participate in pathways mediating the DNA damage response and thereby protect cells from UV-induced DNA damage. This also indirectly signifies the inherent protective function of Pak1 in response to the DNA-damaging agent UV-B.
The role of C-Fos in the cellular defense against the genotoxic effect of UV radiation was well established.34 As Pak1 cannot bind to DNA on its own, it may use the transcriptional factor C-Fos as a mediator to induce the transcriptional activity of ATR in response to UV-B. Functional studies with Pak1-modulating clones revealed that the stimulus leading to keratinocytes' sensitivity to UV-B irradiation is circumvented on Pak1 overexpression, providing evidence that Pak1 protects the cells against UV-B-induced apoptosis and DNA damage. Previously, Pak1 was shown to protect the cells from apoptosis by phosphorylating the death agonist bad.35 It is imperative to note that the very low transfection efficiency in HaCaT cells36 pressed us to perform further functional and mechanistic studies in human keratinocyte A431 cells and that these cells have greater tolerance to UV-B-induced damage and other biological assays such as cell viability as compared with HaCaT cells.37 In a way this means that the Pak1-mediated functional mechanism of UV-B regulation might hold true in vivo even in the case of HaCaT Cells, but at a relatively lower UV-B dose. We maintained the defined minimal erythemal dose reported in the literature for most of the cellular and functional assays, although the UV-B dosage varied between different cell lines depending on their tolerance.38
In our study, we observed an increase in Pak1 phosphorylation and activation as early as 15 to 30 min after UV-B irradiation, indicating that Pak1 activation might be an early and indirect membranous event. Previous studies using EGFR inhibitors revealed that Pak1 activation is mediated via the EGFR pathway, which is an upstream activator of Pak1.39 However, our results (data not shown) showed that the Pak1 kinase activity induced by UV-B was not completely diminished by blocking the EGFR pathway, indicating that there could be other possible mechanisms of Pak1 activation by UV-B.
Results from our clinical samples showed higher Pak1 expression in the dermal and epidermal layers in sun-induced skin lesions in contrast to normal skin where Pak1 expression was observed only in basal layers of the epidermis. This corroborates with the previously reported studies that in NMSC Pak1 is high in both the dermal and epidermal layers, whereas in the normal skin it is present only in the epidermal layer.40 This probably indicates that Pak1 expression and localization could be an indicator of progression marker from normal skin to sun-induced skin lesions to NMSC.
Cells have evolved complex defense system to counteract the harmful effects of UV-B irradiation. The DNA repair and recovery from lower levels of UV-B damage is the first line of defense against UV-B by all the eukaryotes.41 They respond to DNA damage by increased activation or expression of many early responsive genes, such as p53, c-fos, NF-kB and c-jun. Interestingly, some of these genes, such as NF-kB, are also activated by signals that arise outside the nucleus, indicating a degree of overlap between nuclear and non-nuclear signaling events. At the plasma membrane, UV-irradiated cells activate Fas or TNF receptors, which recruit and activate upstream caspase leading to apoptotic death.42 In the cytoplasm, various mitogen-activated protein kinase family members are activated, which participate in either prosurvival (for example, ERK) or proapoptotic (for example, JNK and p38-mitogen-activated protein kinase) functions.43 Finally, reactive oxygen species generated in the cytoplasm can induce different proapoptotic or antiapoptotic events, by mediating its effect through oxidative DNA damage, activation of kinase signaling cascade or activation of different genes.
Despite having increasing literature evidence that UV-B targets both cell membrane and nuclear components, it is unclear how these relate to each other and whether they are mutually exclusive or work in a synchronized manner. It is also reported that there exists a signal transfer process from sunlight photoproducts inside the nucleus to the cytoplasm.44 In addition, the concept of signaling loop also explains the activation of cytoplasmic and membrane kinases in response to DNA damage caused by radiation.45 Thus, it is the net sum of all of these events that determine the eventual fate of UV-irradiated cell. In conclusion, our study demonstrates that Pak1 is activated in response to UV-B and this activated Pak1 in turn activates the transcription of DNA repair kinase ATR, which eventually participates in DNA damage response and protects the cells. Despite all these protective mechanisms deliberated by Pak1 via ATR, if the damaged DNA is not properly repaired persisting DNA damage can result in mutations in oncogenes and tumor suppressor genes, leading to permanent alterations in signaling pathways that control cell survival, proliferation and differentiation. All this depends on the dose and the number of cells that are targeted for the subsequent UV-B exposure and that these cellular level events may significantly affect the time needed to accumulate alterations in genes needed for skin cancer to arise, ultimately resulting in cancer.
Materials and methods
Cell culture
The immortalized epidermal human keratinocyte (HaCaT) and human epidermoid carcinoma A431 cell lines were purchased from NCCS (Pune, India). The mouse epidermal cell line JB6 was maintained in minimum essential medium. Human epidermal keratinocyte adult was purchased from Invitrogen (Waltham, MA, USA). Human skin fibroblast AG1522 cells were obtained from Dr Sonia M de Toledo (The State University of New Jersey, New Brunswick, NJ, USA). Mouse epidermal cell line JB6 cells were obtained from Dr Rana P Singh (JNU, Munirka, New Delhi).
UV irradiation of cells
For UV radiation, cells were given prewarmed phosphate-buffered saline wash and then UV-B irradiated. UV gene linker is used as a source of UV irradiation (UV-B) that is equipped with an energy output control (UVP, model CL-1000).
Animal studies
Animal studies were conducted as per the standard procedures and principles approved by Animal ethics committee of IIT Madras.
Western blot
Cell extracts were then resolved by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, proteins were transferred to nitrocellulose membranes, which is blocked in 5% skim milk for 1 h, and specific antibodies were used for probing.
Pak1 in vitro kinase assay
Pak1 in vitro kinase assay was performed by immunoprecipitating the endogenous Pak1 from a 100 -μg aliquot of cell lysates from UV-B-irradiated keratinocytes. PAKtide/MBP was used as a substrate.
Stable Pak1 overexpression and Pal1-knockdown clones
Pak1 gene cloning and stable overexpression
Retroviral-mediated stable clones overexpressing Pak1 were developed according to the prior protocols (Garry P Nolan, Stanford University, Stanford, CA, USA). The cells were selected for stable integration using puromycin. All the primer sequences used in this study are enlisted in Supplementary Table 1.
Pak1-knockdown clones
Pak1-knockdown clones were generated by transducing HaCaT and A431 cell lines with lentiviral particles containing plasmid constructs coding for an shRNA sequence against p21-activated kinase.
Pak1-knockout clones
Pak1-knockout clones were generated by transducing HaCaT cell line with Alfa Pak1 CRISPR/Cas9 KO Plasmid (h), sc-400857 with FugeneHD as per the manufacturer’s protocol (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
TUNEL assay
Clones were cultured on 60- mm cell culture plates at a seeding density of 1 × 106 cells per plate. After 24 h of serum starvation, the cells were UV-B irradiated. After the recovery time of 30 min, trypsinized cells were used for the TUNEL assay, that is, performed with the APO-BRDU Kit (BD Pharmingen, San Deigo, CA, USA) as per the instructions provided.
ATR promoter cloning and luciferase activity
Human ATR promoter (FL) (from −2032 to +120) was cloned from human total genomic DNA by PCR amplification with a set of primers followed by insertion into Kpn1 and XhoI sites of pGL4 basic vector. The 5′ truncations of 2.2- kb FL fragments (Del) were generated by PCR. The c-Fos-binding site deletion constructs (c-Fos Del) were made using NEB Q5 polymerase, with specific primers.
Electrophoretic mobility shift assay
UV-B-treated and UV-B-untreated A431 nuclear lysates were extracted using the Nonidet P-40 lysis method. The Light Shift Chemiluminescent EMSA Kit (Pierce Biotechnology, Rockford, IL, USA) was used to perform the EMSA assay.
Immunohistochemistry
For IHC, sun-induced skin lesions (Supplementary Table 2) were procured from the Pathology department of Sri Ramachandra University after obtaining ethical clearance. Progesterone receptor/estrogen receptor-positive breast cancer tissue samples were used as a positive control and secondary antibody-only control was used as a negative control. UV-B-exposed and UV-B-unexposed skin samples from hairless SKH mouse model were procured from Dr KathleenTober (The Ohio State University, Columbus, OH, USA). IHC was carried out using mouse anti-phospho-Pak1 antibody, P3237, for human sections and rabbit anti-phospho-Pak1 antibody, SAB4300127, for mouse sections; rabbit anti-Pak1 antibody was also used.
Statistics
Experiments were carried out in triplicate. Data are presented in means plus or minus the standard errors of the mean. The differences between UV-B-irradiated and control groups were analyzed using the Graph Prism Program. T-test was used to calculate the P-value of two groups compared. Values of P⩽0.05 were considered to be statistically significant.
References
Panda S . Nonmelanoma skin cancer in India: current scenario. Indian J Dermatol 2010; 55: 373–378.
Corona R . Epidemiology of nonmelanoma skin cancer. Ann Ist Super Sanita 1996; 32: 37–42.
Ichihashi M, Ueda M, Budiyanto A, Bito T, Oka M, Fukunaga M et al. UV-induced skin damage. Toxicology 2003; 189: 21–39.
Marks VJ . Actinic keratosis. A premalignant skin lesion. Otolaryngol Clin North Am 1993; 26: 23–35.
Bhat Mr, Dandakeri S, Gatti R, Hulmani M, Kambil S . Disseminated discoid lupus erythematosus leading to squamous cell carcinoma. Indian J Dermatol 2012; 57: 158–161.
Criscione VD, Weinstock MA, Naylor MF, Luque C, Eide MJ, Bingham SF et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer 2009; 115: 2523–2530.
Anna B, Blazej Z, Jacqueline G, Andrew CJ, Jeffrey R, Andrzej S . Mechanism of UV-related carcinogenesis and its contribution to nevi/melanoma. Expert Rev Dermatol 2007; 2: 451–469.
Cooper SJ, Bowden GT . Ultraviolet B regulation of transcription factor families: roles of nuclear factor-kappa B (NF-kappaB) and activator protein-1 (AP-1) in UVB-induced skin carcinogenesis. Curr Cancer Drug Targets 2007; 7: 325–334.
Ghosal G, Chen J . DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res 2013; 2: 107–129.
Ward IM, Minn K, Chen J . UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem 2004; 279: 9677–9680.
Xu Y, Voorhees JJ, Fisher GJ . Epidermal growth factor receptor is a critical mediator of ultraviolet B irradiation-induced signal transduction in immortalized human keratinocyte HaCaT cells. Am J Pathol 2006; 169: 823–830.
Xu Y, Shao Y, Voorhees JJ, Fisher GJ . Oxidative inhibition of receptor-type protein-tyrosine phosphatase by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes. J Biol Chem 2006; 281: 27389–27397.
Sato K . Cellular functions regulated by phosphorylation of EGFR on Tyr845. Int J Mol Sci 2013; 14: 10761–10790.
El-Abaseri TB, Hansen LA . EGFR activation and ultraviolet light-induced skin carcinogenesis. J Biomed Biotechnol 2007; 2007: 97939.
Bokoch GM . Biology of the p21-activated kinases. Annu Rev Biochem 2003; 72: 743–781.
Radu M, Semenova G, Kosoff R, Chernoff J . PAK signaling during the development and progression of cancer. Nat Rev Cancer 2014; 14: 13–25.
Kumar R, Gururaj AE, Barnes CJ . P21-activated kinases in cancer. Nat Rev Cancer 2006; 6: 459–471.
Tse EYT, Ching YP . The role of p21-activated kinases in hepatocellular carcinoma metastasis. J Mol Signal 2014; 9: 7.
Gan J, Zhang Y, Ke X, Tan C, Ren H, Dong H et al. Dysregulation of PAK1 is associated with DNA damage and is of prognostic importance in primary esophageal small cell carcinoma. Int J Mol Sci 2015; 16: 12035–12050.
Carr TD, DiGiovanni J, Lynch CJ, Shantz LM . Inhibition of mTOR suppresses UVB-induced keratinocyte proliferation and survival. Cancer Prev Res (Phila) 2012; 5: 1394–1404.
Holm C, Rayala S, Jirström K, Stål O, Kumar R, Landberg G . Association between Pak1 expression and subcellular localization and tamoxifen resistance in breast cancer patients. J Natl Cancer Inst 2006; 98: 671–680.
Singh RR, Song C, Yang Z, Kumar R . Nuclear localization and chromatin targets of p21-activated kinase 1. J Biol Chem 2005; 280: 18130–18137.
Sánchez-Solana B, Motwani M, Li D-Q, Eswaran J, Kumar R . P21-activated kinase-1 signaling regulates transcription of tissue factor and tissue factor pathway inhibitor. J Biol Chem 2012; 287: 39291–39302.
Jagadeeshan S, Krishnamoorthy YR, Singhal M, Subramanian A, Mavuluri J, Lakshmi A et al. Transcriptional regulation of fibronectin by p21-activated kinase-1 modulates pancreatic tumorigenesis. Oncogene 2015; 34: 455–464.
Frost JA, Swantek JL, Stippec S, Yin MJ, Gaynor R, Cobb MH . Stimulation of NFkappa B activity by multiple signaling pathways requires PAK1. J Biol Chem 2000; 275: 19693–19699.
Schreiber M, Baumann B, Cotten M, Angel P, Wagner EF . Fos is an essential component of the mammalian UV response. EMBO J 1995; 14: 5338–5349.
Rauscher FJ, Sambucetti LC, Curran T, Distel RJ, Spiegelman BM . Common DNA binding site for Fos protein complexes and transcription factor AP-1. Cell 1988; 52: 471–480.
Tai M-H, Weng C-H, Mon D-P, Hu C-Y, Wu M-H . Ultraviolet C irradiation induces different expression of cyclooxygenase 2 in NIH 3T3 cells and A431 cells: the roles of COX-2 are different in various cell lines. Int J Mol Sci 2012; 13: 4351–4366.
Doré JF, Pedeux R, Boniol M, Chignol MC, Autier P . Intermediate-effect biomarkers in prevention of skin cancer. IARC Sci Publ 2001; 154: 81–91.
Ong CC, Jubb AM, Jakubiak D, Zhou W, Rudolph J, Haverty PM et al. P21-activated kinase 1 (PAK1) as a therapeutic target in BRAF wild-type melanoma. J Natl Cancer Inst 2013; 105: 606–607.
Motwani M, Li D-Q, Horvath A, Kumar R . Identification of novel gene targets and functions of p21-activated kinase 1 during DNA damage by gene expression profiling. PLoS One 2013; 8: e66585.
Li D-Q, Nair SS, Ohshiro K, Kumar A, Nair VS, Pakala SB et al. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Rep 2012; 2: 1657–1669.
Zheng L, Liao X-H, Wang N, Zhou H, Ma W-J, Zhang T-C Construction and functional analysis of luciferase reporter plasmids containing ATM and ATR gene promoters. In: Zhang T-C, Nakajima M (eds). Advances in Applied Biotechnology. Springer: Berlin, Heidelberg, Germany, 2015, pp 627–634.
Haas S, Kaina B . c-Fos is involved in the cellular defence against the genotoxic effect of UV radiation. Carcinogenesis 1995; 16: 985–991.
Schürmann A, Mooney AF, Sanders LC, Sells MA, Wang HG, Reed JC et al. P21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol Cell Biol 2000; 20: 453–461.
Yong W, Peng D, Wang L, Dong Z, He B . Screening of HaCaT clones for CCL20 gene knockout and preliminary exploration of gene-targeting vector transfection approaches in this cell line. Med Sci Monit Basic Res 2015; 21: 21–28.
Zhou MJ, Zheng L, Guo L, Liu WL, Lv C, Jiang LH et al. Differential responses to UVB irradiation in human keratinocytes and epidermoid carcinoma cells. Biomed Environ Sci 2012; 25: 583–589.
Gill P, Kalia S . Assessment of the feasibility of using sunlight exposure to obtain the recommended level of vitamin D in Canada. CMAJ Open 2015; 3: E258–E263.
Yang Z, Bagheri-Yarmand R, Wang RA, Adam L, Papadimitrakopoulou VV, Clayman GL et al. The epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 (Iressa) suppresses c-Src and Pak1 pathways and invasiveness of human cancer cells. Clin Cancer Res 2004; 10: 658–667.
Chow HY, Jubb AM, Koch JN, Jaffer ZM, Stepanova D, Campbell DA et al. P21-activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res 2012; 72: 5966–5975.
Auclair Y, Rouget R, Drobetsky EA . ATR kinase as master regulator of nucleotide excision repair during S phase of the cell cycle. Cell Cycle 2009; 8: 1865–1871.
Sage E, Drouin R, Rouabhia M (eds). Nuclear and non-nuclear signals leading to UV-induced apoptosis. In: From DNA Photolesions to Mutations, Skin Cancer and Cell Death. Royal Society of Chemistry: Cambridge, MA, USA, 2005, pp 247–267.
Sage E, Drouin R, Rouabhia M (eds). Opposing roles of UV-induced apoptosis in early skin cancer. In: From DNA Photolesions to Mutations, Skin Cancer and Cell Death. Royal Society of Chemistry: Cambridge, MA, USA, 2005, pp 269–280.
Bender K, Göttlicher M, Whiteside S, Rahmsdorf HJ, Herrlich P . Sequential DNA damage-independent and -dependent activation of NF-kappaB by UV. EMBO J 1998; 17: 5170–5181.
Brzóska K, Szumiel I . Signalling loops and linear pathways: NF-kappaB activation in response to genotoxic stress. Mutagenesis 2009; 24: 1–8.
Acknowledgements
We thank Dr Barathidasan, Veterinary Pathologist from Mahatma Gandhi Medical College and Research Institute (MGMCRI), Pondicherry for scoring the mice slides. We also thank Rahul and Venu for help with IHC and animal studies. We thank DRDO-LSRB, Government of India, for the financial support to SKR (Grant No: DLS/81/48222/XXIII/LSRB/2009), and NCI R01CA133629 (to TMO) and Indian Institute of Technology Madras (IITM) for all other facilities.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
About this article

Cite this article
Beesetti, S., Mavuluri, J., Surabhi, R. et al. Transcriptional regulation of ataxia–telangiectasia and Rad3-related protein by activated p21-activated kinase-1 protects keratinocytes in UV-B-induced premalignant skin lesions. Oncogene 36, 6154–6163 (2017). https://doi.org/10.1038/onc.2017.218
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2017.218
- Springer Nature Limited