
Abstract
Caspase-2 has been embodied as an initiator or executioner protease in diverse apoptotic scenarios. However, accumulating evidence is challenging this view, pertaining to its true role. The enzyme’s catalytic activity is currently implicated in various functions required for correct cell proliferation, such as counteracting genomic instability, as well as suppressing tumorigenesis. Here, apart from summarizing the latest observations in caspase-2-related research, we make an attempt to reconcile these findings and discuss their implications for future directions.
Similar content being viewed by others

Introduction
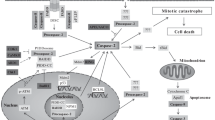
Genomic instability refers to the concept of karyotypic change over time and encompasses a range of processes including nucleotide instability, microsatellite instability and chromosome instability, all of which are characteristics of tumor genomes and indicators of cancer progression. Although the two former changes to the genome arise as a result of malfunctioning DNA repair mechanisms, chromosome instability is more complex and can occur because of defects in the regulation of virtually all parts of the chromosomal cycle.1 Unless the genome remains balanced or is corrected for by the cell through diverse checkpoint regulations and repair mechanisms, chromosome mis-segregation, including aneuploidy and polyploidy, may lead to genomic instabilities or, alternatively, render cells unviable through engagement of programmed cell death pathways.2, 3 Accordingly, caspase-2 has been implicated in the maintenance of genomic stability by being a member of the caspase enzyme family and, thereby, pivotal for the removal of abnormal cells by apoptosis. In experimental settings, silencing or inhibition of the enzyme is then followed by diminished appearances of endpoint apoptotic markers, such as the processing of effector caspases and cleavage of targeted substrates. It should be noted, however, that apoptotic outcomes in these situations can be compromised by the canonical caspase cascade.4 At a glance, the recent discovery of the involvement of caspase-2 in tumor suppression harmonizes with the concept of important functions of cell death regulators as gatekeepers for tumor cell development. Based on findings from the caspase-2 knockout mouse, however, the enzyme hardly qualifies as a key apoptotic enzyme and in other experimental settings in vitro only a few stress stimuli were reported to depend on caspase-2 for efficient cell elimination.4, 5 Moreover, recent results strongly suggest that caspase-2 emerges as a suppressor of tumor formation and progression solely following oncogenic pressure. When transformed with E1A and Ras oncogenes, caspase-2−/− mouse embryonic fibroblasts caused more aggressive tumors in nude mice compared with caspase-2+/+ mouse embryonic fibroblasts. Similarly, caspase-2-deficient Eμ-Myc transgenic mice displayed an accelerated tumorigenic onset,6, 7 as did MMTV/c-neu casp2−/− and Atm−/−casp2−/− mice.8, 9 Remarkably, this is not always the case, as loss of caspase-2 in a TH-MYCN neuroblastoma model proved the opposite.10 Common signatures in the described reports and others that are aiming to develop a more profound understanding of caspase-2 function during tumorigenesis emphasize cell proliferation, p53 regulation and aneuploidy tolerance (Figure 1). Briefly summarized, the current model for the enzyme is gradually shifting from an unmitigated apoptotic factor toward a regulatory involvement in genomic stability. The current review is an attempt to discuss these findings in relation to the role of caspase-2 in tumor suppression with the aim to position the enzyme function with respect to data that are becoming altogether more uniform.

Schematic presentation illustrating the involvement of caspase-2 in tumor suppression and aneuploidy. The PIDDosome-activated caspase-2 is important for prevention of aneuploidy but not tumorigenesis. Potential alternative mechanisms of caspase-2 regulation are described in the text.
Caspase-2 in genomic instability
It is stated that malignant transformations frequently result from deregulated genes associated with growth regulation, cell cycle progression and arrest as well as programmed cell death. Besides, although the distinct mechanism(s) remain elusive, several models are trying to describe the momentum of tumor onset and progression through genomic instability, which thereby becomes a dynamic process shaping the genomes of cancer cells in order to develop their survival advantage.1, 11, 12 An alternative view emphasizes genomic instability as pro-death, whereby surviving cells with tumorigenic capabilities are selected. Indeed, caspase-2 has been implicated in several mechanisms that are associated with tumor suppression and therefore we have previously discussed the enzyme in terms of being either versatile or a factor regulating an isolated process functionally connected to other cellular systems.13 Cells isolated from caspase-2-deficient mice were reported to multiply faster than their normal counterpart,6, 8, 14, 15 a trait that has been attributed to the failure of cells to enter a senescent state.16, 17 Dawar et al.18 observed that DNA damage and aneuploidy increase in bone marrow cells of caspase-2-deficient mice. In addition, the hematopoietic stem cell differentiation was impaired, and progenitor fractions were skewed toward myeloid progenitors. This was not the first case reported where blood cells were affected by the loss of caspase-2, as T cells from premalignant Atm−/−casp2−/− mice also displayed elevated levels of aneuploidy.14 Upon tumor formation, the dependency on caspase-2 becomes more pronounced, as was seen in casp2−/−/MMTV tumors, which displayed karyomegaly, abnormal mitoses and were often multinucleated.8 A tendency for genomic instabilities and allowance of aneuploidization is thus a prominent characteristic of primary cells lacking caspase-2, and especially during oncogenic pressure.9, 15, 16
The identification of the PIDDosome complex as a unique platform for caspase-2 activation was followed by a period of ambiguity as several experimental models failed to connect the finding to a distinct programmed cell death process.19, 20, 21, 22, 23 This uncertainty, however, began to dissipate following the establishment of Mdm2 as a specific caspase-2 substrate.24, 25 It was shown that PIDD-activated caspase-2 cleaves Mdm2, leading to the loss of the C-terminal RING domain responsible for p53 ubiquitination and, consequently, the reinforcement of tumor-suppressor stability. Recently, the model was further refined by data indicating that PIDDosome activation can occur following an increased number of mature centrosomes generated through aberrant cytokinesis,26 thus providing a detailed mechanism for observations associating loss of caspase-2 to aneuploidy. Several reports are in support of this view. For instance, Ho et al.27 concluded that caspase-2 is required for apoptosis induced by therapies targeting microtubule dynamics, such as vincristine and paclitaxel, in a PIDD-dependent manner. Further, in a systematic study using somatic mutation analysis of chromosomally unstable colorectal cancer genomes as a starting point, dysfunction of the transcriptional regulator B-cell CLL/lymphoma 9-like entailed ceased caspase-2 expression, p53 accumulation and aneuploidy.28 Interestingly, in both studies Bid processing and engagement of mitochondrial apoptosis was emphasized as central for the termination of mitotically stressed cells. Besides, there are mechanisms described that either are discrete or, alternatively, need further clarification in order to align to the proposed model. For instance, compared with controls, liver cells from caspase-2-deficient mice display karyomegaly (a trait often associated with aneuploidy), which is furthermore pronounced in response to paraquat treatment. This was accompanied by a reduced expression of genes important in combating oxidative stress, suggesting that caspase-2 may prevent aneuploidy through means other than tumor cell termination.29 In summary, although it is clear that loss of caspase-2 in both premalignant and tumorigenic settings can allow cells to complete the cell cycle despite being faulty, resulting in aneuploidy, the exact mechanisms may vary depending on the biological context.
Normally, following a prolonged mitotic arrest occurring in response to irreparable cell cycle errors, cells typically undergo one of two fates; either they die in mitosis via mitotic catastrophe, a mechanism with hallmarks of apoptosis,30, 31, 32 or they undergo ‘slippage’, whereby mitosis exit occurs without or by asymmetric cytokinesis and the G1 phase is entered, albeit in a polyploid or aneuploid state, respectively.33 It was recently demonstrated that aneuploidy, but not polyploidy, causes p53-dependent post-mitotic apoptosis in spindle assembly checkpoint (also referred to as the mitotic checkpoint) impaired cells.34 Notably, caspase-2 has been implicated in the model of mitotic catastrophe, which was dependent on mitochondrial release of pro-apoptotic factors.35, 36 In an alternate experimental model, however, polyploidization following treatment using the aureolic acid antibiotic mithramycin SK (plicamycin) appears to be coupled with a decrease in p53 and p21WAF1 and subsequent necrosis. Interestingly, p53−/− cells primarily died via caspase-2-mediated apoptosis,32 indicating that the ploidy status is what dictates how p53 will influence the function of caspase-2. If so, then it may partly explain why p53−/− cells died through apoptosis following mithramycin SK treatment. Lack of p53 would in this case be expected to have a negative impact on PIDD expression and in turn PIDDosome formation, which, according to Fava et al.26 may be pivotal for caspase-2-mediated cell cycle arrest. It should, however, be noted that mithramycin SK does not act on the level of centrosomes, but rather is targeting the Sp1 family of transcription factors.37, 38
Obviously, mitotic and post-mitotic cell death mechanisms need further clarification and, hence, more sophisticated nomenclature. Given that current data strongly imply caspase-2 as an endpoint regulator, which ultimately determines the fate of genetically unstable cells, does this mechanism correlate with the proposed tumor-suppressor function of the enzyme?
Caspase-2 in tumor suppression
The mutation(s) of critical genes can transform a progenitor cell. Further tumor evolution driven by additional rounds of genomic alterations may then result in heterogenic sub-populations of cancer cells with aggressive and therapeutically insensitive properties. Intriguingly, the tumor-suppressor activity and p53 linkage of caspase-2 does not seem to rely on PIDD, which, in contrast, upon loss associates with delayed disease onset in a tumor mouse driven by aberrant c-Myc expression.7 In addition, loss of neither the PIDDosome adaptor protein RAIDD nor the BH3-only protein Bid correlate with increased dissemination of tumor cells in animal models.7, 39 Thus, as the catalytic activity was found to be a necessity for caspase-2 tumor repressing functions,40 current data support a view of redundant activation mechanisms or, alternatively, separate, but simultaneous regulatory events may have to occur in order for caspase-2 to be activated in the process (Figure 1). Some reports have provided alternatives to PIDDosome-dependent mitotic enzyme regulation. For example, it was shown that the mitosis-promoting kinase, cdk1-cyclin B1, suppresses caspase-2 upstream of the mitochondria through phosphorylation of an evolutionarily conserved interdomain motif.41 It should also be noted that additional mechanisms for caspase-2 regulation has been described although not yet implicated in tumor suppression.42, 43, 44
As it is possible that the role the enzyme has in tumor formation/suppression varies between different biological situations, it may also be difficult to find one specific function of caspase-2 that would designate it as a 'true' tumor suppressor throughout all tumorigenic settings. Some mechanistic studies have demonstrated how caspase-2 displays its antitumorigenic potential in certain contexts. For instance, by cleaving RIP1, thereby abrogating transcription of the nuclear factor-κB (NF-κB) target gene survivin, HCT116 cells were pushed into apoptosis, consequently reducing the tumorigenicity.45 In a similar manner, Ren et al.40 reported that the catalytic sites Cys-320 and Ser-139 of caspase-2 are important for repressing NF-κB activity in SV40- and K-Ras transformed mouse embryonic fibroblasts, which further indicates the central role of this pathway for the proposed tumor-suppressor function of caspase-2. Another potential manner in which caspase-2 may act antitumorigenic is by modulating autophagy, a process considered to have dual functions as it can be either a way in which cells commit themselves to self-eating (and thus die), or a way of promoting their survival through degradation and recycling of cellular components. In a study by Tiwari et al.,46 it was concluded that caspase-2 acts as a negative regulator of the canonical autophagy pathway, as a loss of the protein increased levels of LC3-II, autophagosomes, autolysosomes and furthermore enhanced protein degradation. Similar findings were observed in rabies virus-infected cells, which induced incomplete autophagy by downregulating caspase-2.47 The mechanism by which caspase-2 inhibits autophagy could thus have an influence on tumor survival, as downregulation of autophagy could disable energy production that otherwise leads to tumor growth and therapeutic resistance.48
Recent research has proposed a possible way to effectively target and eliminate cancer cells, which have abolished tumor-suppressor genes in order to become immortalized.49 This novel approach is based on the fact that many cancer cells also co-delete genes adjacent to the tumor-suppressor genes, thus making them vulnerable to certain targeting treatments. For instance, cells that co-delete a gene encoding a metabolic protein will thereby become highly dependent on backup genes for other metabolic proteins. Were these proteins are to be specifically targeted, it is likely that the tumor cells would perish.
It was demonstrated by Dey et al.50 that pancreatic ductal adenocarcinoma cells, in which the copies of SMAD4 had been deleted, lost the genes encoding mitochondrial malic enzyme 2 (ME2) as well. This consequently led to the increased reliance on mitochondrial malic enzyme 3 (ME3), in order to convert malate into pyruvate. ME2 and ME3 are furthermore involved in the regeneration of reduced nicotinamide adenine dinucleotide phosphate, which is important for normal metabolic processes.
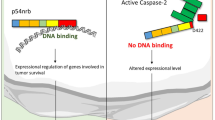
Interestingly, caspase-2 has previously been demonstrated to be regulated by metabolic processes, being able to cause apoptosis in Xenopus eggs during nutrient depletion.42 When glucose-6-phosphate (G6P) was added, simulating nutrient abundance, apoptosis was inhibited. Furthermore, it was suggested that the pentose-phosphate-pathway was seemingly the underlying process leading to abrogated cell death, through the production of nicotinamide adenine dinucleotide phosphate. When either intermediates of the pentose-phosphate-pathway, glucose-6-phosphate, malate or nicotinamide adenine dinucleotide phosphate were added to the egg extracts, caspase-2 processing, and subsequently apoptosis, were inhibited. This turned out to be caused by the inhibitory phosphorylation in the prodomain of caspase-2, mediated by CaMKII, although at the time the direct link between CaMKII and metabolic stimulation remained unknown (Figure 2). Later on, this was however solved, as it turned out that the addition of glucose-6-phosphate increased the levels of free coenzyme A in the cytosol. This coenzyme A would then bind to the calmodulin-binding domain of CaMKII, thus triggering the phosphorylation of caspase-2 at Ser135.51, 52 Could this potentially be a mechanism through which caspase-2 exerts its tumor-suppressing function, or, conversely, how cancer cells inhibit caspase-2?

Metabolic processes in oocytes negatively regulate caspase-2 while nutrient levels are adequate. Coenzyme A, a metabolic byproduct, directly binds calcium/calmodulin-dependent protein kinase II (CaMKII). In turn, calmodulin (CAM) binds CaMKII, leading to the activation of the complex, thereby resulting in the inhibitory phosphorylation of caspase-2 at Ser135. Hence, apoptosis is repressed.
Concluding remarks
The true function of caspase-2, as well as the context in which it becomes activated, has for a long time been an enigma. Although recognized as an initiator caspase in apoptosis, the view of this being the main function of the protease has been open for debate. Lately, an increasing number of studies point instead toward functions that may influence tumorigenesis when malfunctioning. These include executing apoptosis after mitotic failures, preventing the emergence of aneuploid cells, as well as counteracting oncogenic pressure (Figure 1). Furthermore, caspase-2 has been shown to be regulated by metabolic processes. Whether or not this may impact the overall survival of tumors is still unknown. Although the exact mechanisms of the protein are hard to define, as well as the machinery that causes the switch between various functions, it appears to be highly situation specific. Nevertheless, caspase-2 is strongly indicated to have a vital role in ensuring that only normal cells replicate. Future work has to be carried out in order to provide us with new and exciting knowledge about the still unknown function(s) of this enzyme.
References
Heng HH, Bremer SW, Stevens JB, Horne SD, Liu G, Abdallah BY et al. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev 2013; 32: 325–340.
Zhivotovsky B, Kroemer G . Apoptosis and genomic instability. Nat Rev Mol Cell Biol 2004; 5: 752–762.
Gordon DJ, Resio B, Pellman D . Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012; 13: 189–203.
Fava LL, Bock FJ, Geley S, Villunger A . Caspase-2 at a glance. J Cell Sci 2012; 125: 5911–5915.
Bouchier-Hayes L, Green DR . Caspase-2: the orphan caspase. Cell Death Differ 2012; 19: 51–57.
Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, Kumar S . A tumor suppressor function for caspase-2. Proc Natl Acad Sci USA 2009; 106: 5336–5341.
Manzl C, Peintner L, Krumschnabel G, Bock F, Labi V, Drach M et al. PIDDosome-independent tumor suppression by caspase-2. Cell Death Differ 2012; 19: 1722–1732.
Parsons MJ, McCormick L, Janke L, Howard A, Bouchier-Hayes L, Green DR . Genetic deletion of caspase-2 accelerates MMTV/c-neu-driven mammary carcinogenesis in mice. Cell Death Differ 2013; 20: 1174–1182.
Puccini J, Shalini S, Voss AK, Gatei M, Wilson CH, Hiwase DK et al. Loss of caspase-2 augments lymphomagenesis and enhances genomic instability in Atm-deficient mice. Proc Natl Acad Sci USA 2013; 110: 19920–19925.
Dorstyn L, Puccini J, Nikolic A, Shalini S, Wilson CH, Norris MD et al. An unexpected role for caspase-2 in neuroblastoma. Cell Death Dis 2014; 5: e1383.
Yao Y, Dai W . Genomic instability and cancer. J Carcinog Mutagen 2014; 5: 100016.
Lee JK, Choi YL, Kwon M, Park PJ . Mechanisms and consequences of cancer genome instability: lessons from genome sequencing studies. Annu Rev Pathol 2016; 11: 283–312.
Olsson M, Forsberg J, Zhivotovsky B . Caspase-2: the reinvented enzyme. Oncogene 2015; 34: 1877–1882.
Puccini J, Dorstyn L, Kumar S . Caspase-2 as a tumour suppressor. Cell Death Differ 2013; 20: 1133–1139.
Dawar S, Lim Y, Puccini J, White M, Thomas P, Bouchier-Hayes L et al. Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene 2017; 36: 2704–2714.
Dorstyn L, Puccini J, Wilson CH, Shalini S, Nicola M, Moore S et al. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ 2012; 19: 1288–1298.
Gitenay D, Lallet-Daher H, Bernard D . Caspase-2 regulates oncogene-induced senescence. Oncotarget 2014; 5: 5845–5847.
Dawar S, Shahrin NH, Sladojevic N, D'Andrea RJ, Dorstyn L, Hiwase DK et al. Impaired haematopoietic stem cell differentiation and enhanced skewing towards myeloid progenitors in aged caspase-2-deficient mice. Cell Death Dis 2016; 7: e2509.
Vakifahmetoglu H, Olsson M, Orrenius S, Zhivotovsky B . Functional connection between p53 and caspase-2 is essential for apoptosis induced by DNA damage. Oncogene 2006; 25: 5683–5692.
Tinel A, Tschopp J . The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004; 304: 843–846.
Manzl C, Krumschnabel G, Bock F, Sohm B, Labi V, Baumgartner F et al. Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol 2009; 185: 291–303.
Ribe EM, Jean YY, Goldstein RL, Manzl C, Stefanis L, Villunger A et al. Neuronal caspase 2 activity and function requires RAIDD, but not PIDD. Biochem J 2012; 444: 591–599.
Kim IR, Murakami K, Chen NJ, Saibil SD, Matysiak-Zablocki E, Elford AR et al. DNA damage- and stress-induced apoptosis occurs independently of PIDD. Apoptosis 2009; 14: 1039–1049.
Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ et al. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell 2011; 43: 57–71.
Terry MR, Arya R, Mukhopadhyay A, Berrett KC, Clair PM, Witt B et al. Caspase-2 impacts lung tumorigenesis and chemotherapy response in vivo. Cell Death Differ 2015; 22: 719–730.
Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 2017; 31: 34–45.
Ho LH, Read SH, Dorstyn L, Lambrusco L, Kumar S . Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene 2008; 27: 3393–3404.
Lopez-Garcia C, Sansregret L, Domingo E, McGranahan N, Hobor S, Birkbak NJ et al. BCL9L dysfunction impairs caspase-2 expression permitting aneuploidy tolerance in colorectal cancer. Cancer Cell 2017; 31: 79–93.
Shalini S, Puccini J, Wilson CH, Finnie J, Dorstyn L, Kumar S . Caspase-2 protects against oxidative stress in vivo. Oncogene 2015; 34: 4995–5002.
Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G . Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004; 23: 2825–2837.
Imreh G, Norberg HV, Imreh S, Zhivotovsky B . Chromosomal breaks during mitotic catastrophe trigger gammaH2AX-ATM-p53-mediated apoptosis. J Cell Sci 2011; 124: 2951–2963.
Bataller M, Mendez C, Salas JA, Portugal J . Mithramycin SK modulates polyploidy and cell death in colon carcinoma cells. Mol Cancer Ther 2008; 7: 2988–2997.
Ohashi A . Different cell fates after mitotic slippage: from aneuploidy to polyploidy. Mol Cell Oncol 2015; 3: e1088503.
Ohashi A, Ohori M, Iwai K, Nakayama Y, Nambu T, Morishita D et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat Commun 2015; 6: 7668.
Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004; 23: 4362–4370.
Vitale I, GM, Castedo M, Kroemer G . Caspase 2 in mitotic catastrophe: the terminator of aneuploid and tetraploid cells. Mol Cell Oncol 2017 (in press).
Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM . Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J Clin Invest 1991; 88: 1613–1621.
Albertini V, Jain A, Vignati S, Napoli S, Rinaldi A, Kwee I et al. Novel GC-rich DNA-binding compound produced by a genetically engineered mutant of the mithramycin producer Streptomyces argillaceus exhibits improved transcriptional repressor activity: implications for cancer therapy. Nucleic Acids Res 2006; 34: 1721–1734.
Peintner L, Dorstyn L, Kumar S, Aneichyk T, Villunger A, Manzl C . The tumor-modulatory effects of caspase-2 and Pidd1 do not require the scaffold protein Raidd. Cell Death Differ 2015; 22: 1803–1811.
Ren K, Lu J, Porollo A, Du C . Tumor-suppressing function of caspase-2 requires catalytic site Cys-320 and site Ser-139 in mice. J Biol Chem 2012; 287: 14792–14802.
Andersen JL, Johnson CE, Freel CD, Parrish AB, Day JL, Buchakjian MR et al. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J 2009; 28: 3216–3227.
Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC et al. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell 2005; 123: 89–103.
Lavrik IN, Golks A, Baumann S, Krammer PH . Caspase-2 is activated at the CD95 death-inducing signaling complex in the course of CD95-induced apoptosis. Blood 2006; 108: 559–565.
Mendelsohn AR, Hamer JD, Wang ZB, Brent R . Cyclin D3 activates caspase 2, connecting cell proliferation with cell death. Proc Natl Acad Sci USA 2002; 99: 6871–6876.
Guha M, Xia F, Raskett CM, Altieri DC . Caspase 2-mediated tumor suppression involves survivin gene silencing. Oncogene 2010; 29: 1280–1292.
Tiwari M, Sharma LK, Vanegas D, Callaway DA, Bai Y, Lechleiter JD et al. A nonapoptotic role for CASP2/caspase 2: modulation of autophagy. Autophagy 2014; 10: 1054–1070.
Liu J, Wang H, Gu J, Deng T, Yuan Z, Hu B et al. BECN1-dependent CASP2 incomplete autophagy induction by binding to rabies virus phosphoprotein. Autophagy 2017; 13: 739–753.
Yang ZJ, Chee CE, Huang S, Sinicrope FA . The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10: 1533–1541.
Biffi G, Tuveson DA . Cancer: double trouble for tumours. Nature 2017; 542: 34–35.
Dey P, Baddour J, Muller F, Wu CC, Wang H, Liao WT et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017; 542: 119–123.
McCoy F, Darbandi R, Lee HC, Bharatham K, Moldoveanu T, Grace CR et al. Metabolic activation of CaMKII by coenzyme A. Mol Cell 2013; 52: 325–339.
McCoy F, Darbandi R, Chen SI, Eckard L, Dodd K, Jones K et al. Metabolic regulation of CaMKII protein and caspases in Xenopus laevis egg extracts. J Biol Chem 2013; 288: 8838–8848.
Acknowledgements
The work in the authors’ laboratories is supported by grants from the Swedish and Stockholm Cancer Societies, the Swedish Childhood Cancer Foundation, the Swedish Research Council. BZ was supported by the Russian Science Foundation (14-25-00056-II) and the Russian President Fund (grant number NSH-7082.2016).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Forsberg, J., Zhivotovsky, B. & Olsson, M. Caspase-2: an orphan enzyme out of the shadows. Oncogene 36, 5441–5444 (2017). https://doi.org/10.1038/onc.2017.169
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2017.169
- Springer Nature Limited
This article is cited by
-
Exploring caspase functions in mouse models
Apoptosis (2024)
-
Effects of intravitreal injection of siRNA against caspase-2 on retinal and optic nerve degeneration in air blast induced ocular trauma
Scientific Reports (2021)
-
Uncovering the PIDDosome and caspase-2 as regulators of organogenesis and cellular differentiation
Cell Death & Differentiation (2020)
-
Characterization of the responses of the caspase 2, 3, 6 and 8 genes to immune challenges and extracellular ATP stimulation in the Japanese flounder (Paralichthys olivaceus)
BMC Veterinary Research (2019)
-
A caspase-2-RFXANK interaction and its implication for MHC class II expression
Cell Death & Disease (2018)