
Abstract
Loss of HOXA5 expression occurs frequently in breast cancer and correlates with higher pathological grade and poorer disease outcome. However, how HOX proteins drive differentiation in mammalian cells is poorly understood. In this paper, we investigated cellular and molecular consequences of loss of HOXA5 in breast cancer, and the role played by retinoic acid in HOXA5 function. Analysis of global gene expression data from HOXA5-depleted MCF10A breast epithelial cells, followed by validation, pointed to a role for HOXA5 in maintaining several molecular traits typical of the epithelial lineage such as cell–cell adhesion, tight junctions and markers of differentiation. Depleting HOXA5 in immortalized MCF10A or transformed MCF10A-Kras cells reduced their CD24+/CD44lo population, enhanced self-renewal capacity and reduced expression of E-cadherin (CDH1) and CD24. In the case of MCF10A-Kras, HOXA5 loss increased branching and protrusive morphology in Matrigel, all features suggestive of epithelial to basal transition. Further, orthotopically implanted xenografts of MCF10A-Kras-scr grew as well-differentiated pseudo-luminal carcinomas, while MCF10A-Kras-shHOXA5 cells formed aggressive, poorly differentiated carcinomas. Conversely, ectopic expression of HOXA5 in aggressive SUM149 or SUM159 breast cancer cells reversed the cellular and molecular alterations observed in the HOXA5-depleted cells. Retinoic acid is a known upstream regulator of HOXA5 expression. HOXA5 depletion in MCF10A cells engineered to express doxycycline-induced shHOXA5 slowed transition of cells from a less differentiated CD24−/CD44+ to the more differentiated CD24+/CD44+ state. This transition was promoted by retinal treatment, which upregulated endogenous HOXA5 expression and caused re-expression of occludin and claudin-7 (CLDN7). Expression of CDH1 and CD24 was transcriptionally upregulated by direct binding of HOXA5 to their promoter sequences as demonstrated by luciferase and ChIP analyses. Thus, loss of HOXA5 in mammary cells leads to loss of epithelial traits, an increase in stemness and cell plasticity, and the acquisition of more aggressive phenotypes.
Similar content being viewed by others

Introduction
Members of both the HOX and retinoic acid receptor (RAR) families are known to be associated with cellular differentiation.1, 2, 3, 4 HOX proteins are transcription factors that orchestrate body segmentation during embryogenesis.5, 6 Perturbed HOX gene expression has been implicated in multiple cancer types.7 Further, continued expression of several HOX members into adulthood suggests a necessity for their biological functions in maintaining tissue homeostasis and differentiation.8, 9 Our laboratory has reported the tumor suppressive function of HOXA5 in promoting apoptosis in breast cancer cells in a p53-dependent and p53-independent manner.10, 11 Retinoids have a strong influence on mammary gland differentiation.12 Retinoids bind to RARs and transcriptionally activate the HOXA5 promoter.13, 14 Therefore, we sought to understand the function of HOXA5 alone and together with retinoids in mammary epithelial cell differentiation, and further define the molecular basis of its tumor suppressive properties during breast cancer progression.
Research in the past 20 years has identified multiple markers common to both normal and breast cancer stem/progenitor cells. Among them, the two most common are the cell surface markers, CD24 and CD44.15 Interestingly, the progenitor cell profile of CD24lo/CD44hi is often displayed by mesenchymal cells16, 17, 18 that also express low levels of the adhesion molecule, CDH117, 19 and exhibit a cytokeratin profile resembling that of basal epithelium.20 This is in contrast to differentiated epithelial cells, which express high levels of CD24 and low CD44.21 CD24 is a glycosyl-phosphatidyl-inositol-linked cell surface molecule that has been implicated in lineage development of hematopoietic cells.22 Although CD24 is a marker of the more differentiated epithelial cell population,23, 24 whether it has biological functions in regulating cell differentiation and specification is unknown. Also, little is known about its transcriptional regulation.25, 26
Retinoic acid is produced in the stem cell niche and is implicated in stem cell differentiation and induction of lineage progression in hematopoietic cells in vitro.12, 27 However, the precise pathways utilized by the RAR acting through HOXA5 to mediate cell differentiation are not yet clear.
In this paper, we report that ectopic expression of HOXA5 in mammary epithelial cells promotes transitioning of their progenitor population to a more differentiated state. Loss of HOXA5 in tumor cells is accompanied by loss of CDH1 and CD24. We demonstrate that loss of CDH1 and CD24 promotes the process of dedifferentiation and cellular transformation mediated by loss of HOXA5. We conclude that one mechanism by which HOXA5 acts as a tumor suppressor is through its direct action on cell adhesion and lineage determination factors in progenitor cells.
Results
Depletion of HOXA5 results in perturbation of epithelial characteristics
Previous studies, including ours, have provided evidence for loss of HOXA5 in primary breast cancers and its possible function as a tumor suppressor gene in breast cancer cells.10, 11, 13, 28, 29 To gain further insight into pathways through which HOXA5 functions as a tumor suppressor gene, microarray analysis of gene expression was performed with MCF10A immortalized breast epithelial cells and with two sublines stably depleted of HOXA5, MCF10A-sh528 and MCF10A-sh529 (Figures 1a and b). Pairwise differential expression analysis performed using Limma across biological replicates identified 43 genes, which were differentially regulated with a false discovery rate of <0.01 (Figure 1c). To validate a subset of highly differentially expressed genes associated with epithelial integrity, we performed quantitative RT–PCR of MCF10A-sh528 and MCF10A-sh529, and the same cell lines reconstituted with HOXA5 (Figure 1d) for E-cadherin (CDH1), a cell adhesion protein; CD24, a differentiation marker; ALDH1A3, a retinal biosynthesis enzyme; claudin-1 (CLDN1), a tight junction protein; DSG3, a desmosomal protein; and ITGA10, an integrin, along with relevant vector-transfected cells (Figure 1e). Forced expression of HOXA5 in HOXA5-depleted cell cultures led to a substantial recovery of mRNA expression of each of the validated genes (Figure 1e), suggesting first, that loss of gene expression was a direct consequence of the loss of HOXA5 and second, that HOXA5 expression correlates with the expression of genes that are necessary for the maintenance of epithelial cell identity. In line with this reasoning, the more elongated and disorganized morphology of both HOXA5-depleted MCF10A-sh528 and -sh529 cell lines (Supplementary Figure 1A), and a significantly increased capacity to invade through Matrigel (Supplementary Figure 1B), supported compromised epithelial cell identity. We also used transformed MCF10A-Kras cells since they expressed HOXA5 and reliably formed tumors in immunodeficient mice. Depletion of HOXA5 in MCF10A-Kras increased its invasiveness (Supplementary Figure 1C). Conversely, ectopic expression of HOXA5 in SUM149, LM2 (a lung metastatic derivative of MDA-MB-231) and SUM159 resulted in strikingly impeded invasiveness (Supplementary Figures 1D–F). In line with these traits, MCF10A-sh528, MCF10A-sh529 and MCF10A-Kras-sh528 cells showed a higher efficiency in wound healing/migration capability compared to their -scr cell controls (Supplementary Figures 1G and H). The converse was evident in SUM149-HOXA5, LM2-HOXA5 and SUM159-HOXA5 cells (Supplementary Figures 1I–K). We ascertained that altered proliferation (MTT and BrdU uptake analysis; Supplementary Figures 1L–P) of the cell lines, with the exception of 231-LM2, did not dictate the changes observed in invasion and motility upon HOXA5 depletion or overexpression.

Depleting HOXA5 perturbs epithelial characteristics. (a) Quantitative RT–PCR analysis of HOXA5 mRNA expression level in MCF10A-scr and HOXA5-depleted MCF10A-sh528 and MCF10A-sh529 cells. (b) Western blot analysis of HOXA5 protein level in MCF10A-scr and HOXA5-depleted sh528 and sh529 cells. β-Actin serves as the loading control. (c) Hierarchical clustering heatmap showing genes differentially expressed between MCF10A-scr cells (n=2, biological duplicates) and HOXA5-depleted MCF10A-sh528 and MCF10A-sh529 cells (n=2 each, biological duplicates) that have an false discovery rate<0.01. (d) Western blot analysis of HOXA5 protein level in MCF10A-scr and HOXA5-depleted sh528 and sh529 cells, transfected with a HOXA5-expressing plasmid or vector. (e) Quantitative RT–PCR validation of a selection of five underexpressed (CDH1, CD24, ALDH1A3, CLDN1 and DSG3) and one overexpressed gene (ITGA10) identified in HOXA5-depleted MCF10A cells by the array. Columns 1–3 show RT–qPCR validation in MCF10A-scr, -sh528 and -sh529 cells. Columns 4–7 show expression in the same cells transfected with HOXA5 plasmid or vector (restoration of HOXA5 expression; n=4, two biological duplicates). *P<0.05, **P<0.01 and ***P<0.001.
Depletion of HOXA5 alters cell–cell adhesion
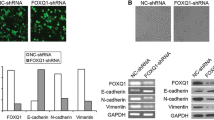
Gene set enrichment analysis (GSEA)30 of the microarray data revealed that MCF10A-scr cells were enriched for expression of genes involved in cell–cell adhesion and tight junctions (Figure 2a). Analyzing molecular changes associated with epithelial integrity by RT–PCR, we observed that the loss of expression of CDH1 was accompanied, in HOXA5-depleted cells, by an elevation of N-cadherin (CDH2; Figure 2b) and a decrease in expression of P-cadherin (CDH3; Figure 2b), which is known to disrupt the normal suppressor function of CDH1 by decreasing the interaction between CDH1 and intracellular catenins.31 Upon HOXA5 depletion, integral components of tight junctions in epithelial cells such as occludin and CLDN7 were also downregulated (Figure 2b). Two other regulators of epithelial integrity acting in opposing ways were also investigated. The transcription factor FOXA1, shown to potently neutralize several epithelial-mesenchymal transition (EMT)-related, CDH1-repressed mechanisms32 was underexpressed, while FOXQ1 (Figure 2b), which represses CDH1 by binding to the E-box in its promoter region33, 34 was overexpressed. HOXA5-depleted MCF10A-sh528 and MCF10A-sh529 cells expressed significantly lower protein levels of CDH1, occludin, CLDN7 and CLDN1 than controls (Figure 2c). The loss of CDH1 and CLDN1 expression could be restored by re-expression of HOXA5 in the HOXA5-depleted cells (Figure 2d), suggesting that HOXA5 may play a key role in regulating the expression of both genes. Immunofluorescence analysis provided additional evidence for membrane loss of CDH1 and occludin, concordant with the disorganized growth pattern and elongated morphology of the HOXA5-depleted MCF10A cells (Figure 2e; Supplementary Figure 2).

Loss of HOXA5 disrupts cell–cell adhesion molecules that govern epithelial integrity. (a) Gene set enrichment analysis of MCF10A-scr and HOXA5-depleted cells indicates that cell–cell junction genes are enriched for expression in MCF10A-scr cells. (b) RT–PCR analysis shows reduced expression of EMT markers (CDH2 and CDH3), tight junction molecules (occludin and CLDN7) and increased expression of luminal fate marker, FOXA1 and reduced expression of FOXQ1 in MCF10A-scr, HOXA5-depleted MCF10A-sh528 and MCF10A-sh529 cells. (c) Western blot analysis of HOXA5 and HOXA5-regulated genes CDH1, occludin, CLDN7 and CLDN1 in MCF10A-scr, -sh528 and -sh529 cells. β-Actin serves as the loading control. (d) Western blot analysis shows decrease in CDH1 and CLDN1 protein upon HOXA5 depletion in MCF10A cells; and reversal by ectopic expression of HOXA5 in the knockdown clones. (e) Representative immunofluorescence images of CDH1 (green), occludin (yellow), actin stress fiber, phalloidin (red) and nuclear marker, DAPI (blue) with expanded views (× 2) of CDH1 and occludin in MCF10A-scr cells to clearly visualize localization.
HOXA5 expression inhibits outgrowth of organoids in three-dimensional (3D) cultures
Transformed breast epithelial cells form protrusive and microinvasive structures in 3D cultures, a phenotype that is strongly associated with epithelial–mesenchymal transitional state of the cells.34, 35 231-LM2 breast cancer cells expressed low or undetectable levels of CD24 and CLDN1; expression of both molecules was restored when HOXA5 was ectopically expressed (Figure 3a). Since the proliferation of 231-LM2 cells was significantly reduced upon HOXA5 expression (Supplementary Figure 1O), we determined whether this occurs due to apoptosis by flow cytometry analysis using annexin V and 7-aminoactinomycin (7-AAD). Compared to 231-LM2-vec cells, 231-LM2-HOXA5 cells exhibited an increased proportion of early (Q4, 7.3% versus 10.6%) and late (Q2, 4.5% versus 9%) apoptotic cells (Figure 3b). In Matrigel, 231-LM2-vec cells formed multiple branching and stellate structures unlike the rounded morphology of 231-LM2-HOXA5 organoids (Figure 3c, quantified in 3d), further corroborated by phalloidin staining of the organoids (Figure 3e). The organoids were positive for cleaved caspase-3, showed increased apoptotic blebs, and were characterized by de-nucleated cells and GFP+ cytoplasm (Figure 3f). The smaller number of 231-LM2-HOXA5 spheroids observed (Figure 3d) could be the result of apoptosis. On the other hand, depleting HOXA5 in MCF10A-Kras cells led to a significant increase in branching structures (Figure 3g, quantified in 3h), characterized by protruding outgrowths into the gel (Figure 3i) and the loss of CDH1 expression (Figure 3j). Collectively, the results led us to conclude that ectopic expression of HOXA5 impedes protrusive and microinvasive outgrowth of breast cancer cells in 3D cultures, with concomitant upregulation of CDH1 and promotion of focal apoptosis.

Loss of HOXA5 promotes formation of protrusive and disorganized organoids in 3D Matrigel culture. (a) Western blot analysis shows increased expression of CD24 and CLDN1 protein in 231-LM2-HOXA5 cells compared to 231-LM2-vec controls. (b) Flow cytometry analysis of annexin V (x axis) against 7-AAD (y axis) shows a higher apoptotic cell population in 231-LM2-HOXA5 cells compared to 231-LM2-vector. Quantification is shown in the bar graphs. Representative (c) phase-contrast images show growth in Matrigel (12 days) of 231-LM2-vec and 231-LM2-HOXA5 organoids and (d) quantitative analysis of spheroids or branched structures formed (n=4). Confocal images of the same cells stained with (e) phalloidin (red), (f) cleaved caspase-3 (red), GFP (green) and nuclear stain DAPI (blue). Representative (g) phase-contrast images of MCF10A-Kras-scr or HOXA5-depleted MCF10A-Kras-sh528 organoids grown in Matrigel:Collagen I matrix (14 days) with (h) quantitative analysis of spheroids or branched structures formed (n=4) and confocal images of the same cells stained with (i) phalloidin (red), (j) CDH1 (green) and DAPI (blue). **P<0.01 and ***P<0.001.
HOXA5 loss leads to an increase in stemness of epithelial cells
Human mammary epithelial cells often acquire more stem-like characteristics as they undergo EMT.16, 17 Our observations raised the possibility that similar changes in differentiation status may be brought about by depletion of HOXA5 in MCF10A cells. Flow cytometry analysis showed that, compared to MCF10A-scr cells, the CD24+/CD44+ population decreased in MCF10A-sh528 and in MCF10A-sh-529 cells (Figure 4a), accompanied by an increase in the number of mammospheres formed (Supplementary Figure 3A). These results suggested that loss of HOXA5 skewed the MCF10A cell population to a more progenitor-like state. Similar results were obtained in the MCF10A-Kras cells (Figure 4b; Supplementary Figure 3B). Conversely, SUM149-HOXA5 cells showed an increase in the CD24+/CD44+ population (Figure 4c) and a decrease in the number of mammospheres formed (Supplementary Figure 3C). Similarly, SUM159-HOXA5 cells showed an increase in the CD24+/CD44+ population (Supplementary Figure 3D).

HOXA5 reduces self-renewal capacity of cancer cells and regulates expression of CDH1 and CD24 in breast cancer cell lines. Flow cytometry analysis of CD24 (x axis) against CD44 (y axis) shows: (a) a reduction of CD24+/CD44+ population from 65.8% in MCF10A-scr to 20.2% in MCF10A-sh428 and 42.9% in MCF10A-sh529; (b) a reduction of CD24+/CD44lo population from 22.7% in MCF10-Kras-scr to 3% in MCF10A-K-ras-sh528 cells; and (c) an increase in CD24+/CD44+ population from 47.9% in SUM149-vector to 75.1% in SUM149-HOXA5 cells. Quantification is shown in the bar graphs. Formation and serial passage of mammospheres in: (d) MCF10A-Kras-scr and MCF10A-Kras-sh528 cells and (e) SUM-149-vector and SUM149-HOXA5 cells; cells were passaged every 2 weeks. Quantitative analysis of mammospheres formed in each passage is shown in the bar graphs. HOXA5 and CDH1 protein expression in: (f) MCF10A-Kras-scr and HOXA5-depleted MCF10A-Kras-sh528; and (g) SUM149-vec and HOXA5 overexpressing SUM149-HOXA5 cells. β-Actin serves as the loading control. *P<0.05, **P<0.01 and ***P<0.001.
To further investigate whether expression of CD24 determines the cell’s ability to form mammospheres, we sorted MCF10A-Kras and SUM149 cells using the CD24 and CD44 markers. Sorted CD24−/CD44hi MCF10A-Kras cells showed higher mammosphere-forming efficiency than CD24+/CD44lo cells (Supplementary Figure 3E), a result that was replicated in SUM149 cells (Supplementary Figure 3F). These findings suggest that expression of CD24 is one determinant of the intrinsic capability of breast cancer cells to form mammospheres.
Serial passaging of the mammospheres36 was performed to assess self-renewal capacity of the MCF10A-Kras-scr and -sh528 cells. Depleting HOXA5 in MCF10A-Kras cells markedly increased the efficiency of mammosphere formation over two passages (Figure 4d). In contrast, SUM149-HOXA5 cells formed a small number of mammospheres in the first passage that could not be propagated any further (Figure 4e). Supporting these observations, HOXA5 depletion in MCF10A-KRas-sh528 cells led to loss of CD24 and CDH1 mRNA expression, and CDH1 protein was undetectable. (Supplementary Figure 3G; Figure 4f). Conversely, compared to the SUM149-vec control cells, CDH1 protein levels increased by two-fold in SUM149-HOXA5 cells (Figure 4g). Collectively, these findings suggest that HOXA5 is functionally capable of altering properties of the stem/progenitor population to promote a more differentiated state in breast cancer cells. One pathway to differentiation by HOXA5, the data suggest, occurs partially through upregulation of CD24.
HOXA5 delays tumor initiation and controls tumor differentiation
Next, we performed orthotopic transplantation of SUM149 tumor cells in the mammary fat pad (mfp) in limiting dilution to evaluate their stemness in vivo. The schema for injecting serially diluted tumor cells into mfp of immunodeficient mice is shown in Figure 5a. The frequency of palpable tumors was assessed from the end of week 3. Compared to SUM149-vec xenografts, SUM149-HOXA5 cells exhibited a 17-fold reduction in the frequency of stem cells (Figure 5b), and a reduced rate of tumor growth (Figure 5c); the tumors were also histologically less aggressive (Supplementary Figure 4A). Conversely, depletion of HOXA5 in MCF10A-Kras cells increased the frequency of stem cells by 10-fold (Figure 5d). MCF10A-Kras-sh528 xenografts appeared significantly earlier and grew more rapidly than vector-control cells (Figure 5e). Histologically, MCF10A-Kras-scr tumors were well differentiated and were characterized by the formation of pseudo-ductal structures with stronger counterstaining with eosin (Figure 5f, left panels). MCF10A-Kras-sh528 tumors, on the other hand, failed to form duct-like structures and the tumor cells exhibited dedifferentiated morphology (Figure 5f, right panels). Immunohistochemical characterization of the xenografts revealed positive staining for (1) HOXA5 in nuclei, (2) CDH1 at adhesion junctions and (3) cytoplasmic CK18 in cells surrounding the pseudo-glandular structures in MCF10A-Kras xenografts, while the expression of these markers was lost in MCF10A-Kras-sh528 tumors (Figure 5g). Thus, loss of HOXA5 resulted in the formation of more aggressive and dedifferentiated tumors in vivo.

Overexpression of HOXA5 impedes tumor initiation, and loss of HOXA5 perturbs tumor differentiation. (a) Figure of sites of orthotopic injection of serially diluted vector-control or HOXA5 overexpressing or depleted tumor cells into the fourth and fifth mammary fat pads (mfp) of NOG or NOD/SCID mice. (b) Table shows tumor incidence 3 weeks after mfp injection of SUM149-vector or SUM149-HOXA5 cells (n=7 mice for each dilution). (c) Growth of SUM149-vector versus SUM149-HOXA5 xenografts (2.5 × 104 cells; n=7 mice). (d) Table shows tumor incidence 5 weeks after mfp injection of MCF10A-Kras-scr or MCF10A-Kras-sh528 cells (n=5 mice for each dilution). (e) Growth of MCF10A-Kras-scr versus MCF10A-Kras-sh528 xenografts over 7 weeks following injection of 2.5 × 104 cells (n=9 mice). (f) Representative images of H&E-stained sections of MCF10A-Kras-scr and MCF10A-Kras-sh528 tumors. (g) Representative images of immunohistochemistry (IHC) for HOXA5, CDH1 and CD18 in formalin-fixed paraffin-embedded (FFPE) sections of MCF10A-Kras-scr and MCF10A-Kras-sh528 tumors. ***P<0.001.
The ability of the tumor cells to grow as xenografts lies primarily in the stem/progenitor population. In fact, in MCF10A-Kras, the stem cell frequency in sorted CD24−/CD44hi was six-fold higher than in the CD24+/CD44lo cells (Supplementary Figure 4B). Thus, our in vivo data also strongly support the notion that HOXA5 promotes tumor cell differentiation.
These findings led us to examine the correlation between HOXA5 and clinical factors in human breast cancer. Meta-analysis of publicly available NKI and Desmedt data sets revealed an inverse correlation between HOXA5 expression and grade of breast cancer in both the data sets (Supplementary Figures 4C and D). In the same data sets, low HOXA5 expression also correlated with worse relapse-free survival outcome (Supplementary Figure 4E).
Differentiation of the CD24− population by retinal treatment is partially mediated by HOXA5
Our findings clearly highlight HOXA5′s function in maintaining the epithelial state, but a possible dynamic role for HOXA5 during cell fate transition is yet to be addressed. Pathway analysis of the differentially expressed genes in HOXA5-depleted MCF10A cells revealed changes in genes involved in retinoic acid biosynthesis, such as ALDH1A3 and DHRS3 (Figures 1b and d). Retinoic acid signaling and HOX genes have been implicated in tissue differentiation and development.37, 38 RARs are direct transcriptional regulators of HOXA5.13, 14 We developed an inducible HOXA5-knockdown model in MCF10A cells that would allow us to study the dynamics of this transition. Doxycycline-induced treatment in MCF10A-tet-sh528 resulted in a sequential loss of expression of CD24 and CDH1 mRNA (Supplementary Figures 5A–C), and HOXA5 and CDH1 proteins (Figure 6a), and loss of the CD24+ population over time (Figure 6b). The CD24−neg population in MCF10A-tet-sh528 cells was sorted by flow cytometry and the cells were treated with retinal (ATAL) to induce differentiation, with or without the addition of doxycycline (schema in Figure 6c). In the sorted CD24− population from MCF10A-tet-sh528 cells, treatment with retinal (1μm) for 7 days resulted in a striking enrichment of CD24+/CD44+-differentiated cells (Figure 6d). Repression of HOXA5 with doxycycline resulted in partial abrogation of the retinal-mediated increase in the CD24+/CD44+ population (Figure 6d). Expression of CD24, CDH1, occludin and CLDN7, but not ALDH1A3 was induced by retinal, and these genes were repressed when HOXA5 was simultaneously silenced in MCF10A-tet-sh528 cells (Figure 6e). Western blot analysis of CDH1, ZO-1, occludin, CLDN7, CK18 and the cell cycle arrest protein, p21, showed elevated expression upon retinal treatment, which was reversed by depleting HOXA5 (Figure 6f). Further substantiation was sought through additional assays of stem cell differentiation in MCF10A-tet-sh528 cells. Retinal treatment inhibited mammosphere formation while concurrent knockdown of HOXA5 in the presence of retinal significantly restored the ability of the cells to form mammospheres (Supplementary Figure 4D). Taken together, these results indicate that retinal-induced differentiation in MCF10A cells is, in part, driven by HOXA5.

Retinal-induced cell differentiation in MCF10A is compromised when HOXA5 is repressed. (a) Western blot analysis of HOXA5 and CDH1 expression in MCF10A-tet-sh528 cells with dox-induced depletion of HOXA5 over 96 h. β-Actin serves as the loading control. (b) Flow cytometry analysis of CD24 expression in MCF10A-tet-HOXA5 with doxycycline-induced depletion of HOXA5 shows decrease in CD24+ population (7-AAD negative) from 47 to 24% over 96 h. (c) Flow-sorted CD24− population was treated with DMSO, 1 μm retinal (ATAL) or 1 μm ATAL+100 nm doxycycline (DOX) and cultured for 1 week. (d) Flow cytometry analysis of CD24 (x axis) and CD44 (y axis). Quantitative analysis is shown in the bar graph. (e) Quantitative RT–PCR analysis of HOXA5, CD24, CDH1, occludin, CLDN7 and ALDH1A3 in MCF10A-tet-sh528 cells treated with DMSO, 1 μm retinal (ATAL) or 1 μm ATAL+100 nm doxycycline (DOX) and cultured for 1 week. (f) Western blot analysis of HOXA5, CDH1, p21, ZO-1, occludin, CLDN7 and CK18 in cells treated with DMSO, ATAL and ATAL+ doxycycline as in (d). β-Actin serves as the loading control. *P<0.05, **P<0.01 and ***P<0.001.
HOXA5 is a direct transcriptional regulator of CDH1 and CD24
How does HOXA5 regulate the multiple traits typical of luminal epithelial cells in the mammary gland? We addressed this question for the CDH1 and CD24 genes. We first performed promoter-linked luciferase assays. Transient expression of wild-type (WT)-HOXA5 with the luciferase constructs in 293T cells resulted in a significant induction of luciferase activity driven by the promoters of both CDH1 (Figure 7a) and CD24 (Figure 7b). Next, we performed chromatin immunoprecipitation (ChIP) assays on MCF10A-sh528 cells overexpressing myc-tagged HOXA5 to determine the occupancy of HOXA5 at putative HOX-binding sites located in each of the gene promoters. In both cases, we observed statistically significant enrichment of HOXA5 at their respective binding regions in chromatin compared to vector controls, confirming HOXA5 occupancy (Figures 7c and d). In contrast, no significant ChIP enrichment of HOXA5 was detected at regions ~2000 bp upstream or downstream of the promoters of each gene (Figures 7c and d). In sum, these results suggested that HOXA5 is a direct transcriptional regulator of two luminal-specific genes, CDH1 and CD24.

HOXA5 transcriptionally regulates CDH1 and CD24 through promoter occupancy. Promoter-linked luciferase activity was analyzed in 293 T cells 24 h after co-transfection of: (a) a luciferase construct containing the promoter sequence (−1000 bp) of CDH1 or vector control or (b) a luciferase construct containing the CD24 promoter (−1100 bp to +150 bp) and vector control, each with wild-type HOXA5 or homeodomain-truncated (ΔHD) HOXA5 and β-galactosidase plasmid to control for transfection efficiency. HOXA5 upregulated luciferase activity of both promoters; activity was significantly attenuated when truncated HOXA5 lacking the HD was used. (c) ChIP analysis of the CDH1 promoter was performed in vector or myc-tagged-HOXA5-expressing MCF10A-sh528 cells using α-myc antibody and isotype IgG control. qPCR shows enrichment of occupancy of HOXA5 at the −1003 to −881 bp region of CDH1, a region containing a putative HOX-binding TAAT sequence, upstream of its transcription start site, but no HOXA5 occupancy at distant (1500–2000 bp) upstream (−2426 to −2421 bp) and downstream (+1333 to +1443 bp) sequences. No enrichment was observed in the IgG ChIP. (d) ChIP analysis of CD24 promoter was performed in vector or myc-tagged-HOXA5-expressing MCF10A-sh528 cells using α-myc antibody and isotype IgG control. Bar graph shows enrichment of occupancy of HOXA5 at the −1128 to −1016 bp region of CD24, containing a putative HOX-binding TAAT sequence upstream of CD24 transcription start site, but not at the upstream (−3470 to −3300 bp) and downstream (+785 to +926 bp) regions. No enrichment was observed in the IgG ChIP. (e) ChIP analysis of CD24 promoter in DMSO, ATAL or ATAL+DOX-treated MCF10A cells using α-HOXA5 antibody or isotype IgG control. Bar graph shows enrichment of occupancy of HOXA5 at the −1128 to −1016 bp region of CD24 promoter, but not when HOXA5 was depleted. There was no enrichment at the upstream (−3470 to −3300 bp) and downstream (+785 to +926 bp) regions. ***P<0.001.
Next, we investigated whether endogenous HOXA5 induced by retinal treatment in MCF10A cells could bind to the CD24 promoter. ChIP analysis showed that HOXA5, induced by retinal, bound to the promoter region of CD24 in MCF10A-tet-shHOXA5 cells. Doxycycline-mediated HOXA5 depletion significantly reduced its occupancy at the same region (Figure 7e). This enrichment, however, was not detected at the distant regions upstream or downstream from the putative HOXA5-binding region. These findings provide evidence to support the transcriptional role of retinal-induced HOXA5 in regulating CD24 expression.
Discussion
Our investigation has revealed a role for HOXA5 in regulating cell differentiation, cell specification and tumor initiation in breast cancer, and has provided mechanistic insights into how HOXA5 maintains homeostasis in mammary epithelial cells. CDH1 and CD24 were identified as key molecules directly transcriptionally upregulated by HOXA5. We present evidence that HOXA5 regulates important epithelial traits in mammary cells. HOXA5 loss in tumor cells leads to reduced CDH1 expression and subsequent development of compromised epithelial integrity and acquisition of properties of invasion and migration. In turn, reduced CD24 expression leads to enrichment of CD24−/CD44+ population causing an expansion of breast cancer stem cells and rapid tumor initiation (Supplementary Figure 6).
Although HOXA5 orthologs have been implicated in cellular differentiation, most of the evidence was gathered from non-mammalian models.28 Many of the HOXA5 null mice died within a week of birth due to respiratory tract defects, and surviving female mice exhibited abnormal mammary development but failed to develop mammary tumors.39 Technical limitations have hindered the exploration of the mechanism of regulation of mammary cell differentiation by HOXA5. Our studies were facilitated by generating MCF10A mammary epithelial cells with Tet-inducible shHOXA5. Using these cells and other breast cancer models, our data are supportive of a role for HOXA5 in specifying cell fate brought about by upregulating critical cell surface proteins that maintain the structural integrity of epithelial cells. Expression of CDH1, occludin and claudins decreased when cells were stably depleted of HOXA5. Occludin, CLDN1 and CLDN7 are important components of tight junctions, a barrier expressed primarily in differentiating ductal cells. However, their expression is consistently lost in many breast carcinomas and breast cancer cell lines.40, 41, 42, 43, 44 Hence, these findings provide evidence for one of the many complex functions of HOXA5: that of regulating structural integrity of epithelial cells and thereby, likely, cellular patterning and development.
We show that HOXA5 regulates CD24 expression in MCF10A cells. On the basis of the loss of CD24, a cell marker of differentiation, we predicted that HOXA5-depleted MCF10A cells will convert to a more primitive and basal phenotype. Using a combination of models for depletion of HOXA5 in MCF10A-Kras, and overexpression in SUM149 breast cancer cell lines, we have shown that HOXA5 reduces the cancer stem/progenitor population. Observations of a HOXA5-mediated increase in the more differentiated CD24+/CD44+ population, reduction of self-renewal capacity, accompanied by elevation of CDH1 expression provided strong evidence that HOXA5 impedes stemness of breast cancer cells. Our results support the theory that breast cancer cells might acquire an enhanced ability to form tumors when they revert to a more undifferentiated state.45 In fact, tumor-initiating cells were previously generated through dedifferentiation of epithelial cells in a murine intestinal model.46 Our results support several tenets of this reversion theory: expression of HOXA5 markedly reduced the number of tumor-initiating cells in limiting dilution assays performed in immunodeficient mice. Also, depletion of HOXA5 in MCF10A-Kras cell line resulted in the formation of histologically dedifferentiated xenografts in mice. This observation is in agreement with the correlation seen in human breast cancer between low HOXA5 expression on the one hand and high pathological grades and poor survival outcome on the other. These observations strongly support a role for HOXA5 in maintaining homeostasis and suppressing tumor initiation in breast cancer.
Retinoid signaling has been shown to regulate human hematopoietic stem cells in culture,12, 27 while its inhibition delays the differentiation of human hematopoietic stem cells and prolongs their capacity for cell renewal. Tumor sphere formation by breast cancer cells was suppressed by retinoic acid, which behaved as a regulator of stem cell differentiation.47 Retinoic acid when used in combination with cytotoxic agents reduced the frequency of breast cancer stem cells in culture.48, 49 Further, the RARα agonist AM580 (a benzoic derivative of retinoic acid) has been reported to promote differentiation of mammary tumors arising in MMTV-neu and MMTV-wnt1 transgenic mice.50 Nevertheless, what drives the transition of cells from a primitive to the differentiated axis remains poorly understood. The inducible HOXA5-knockdown model system in MCF10A cells allowed us to demonstrate the role of HOXA5 in retinal-mediated transition from the CD24-neg to CD24-pos axis, which was characterized by elevated expression of epithelial-specific junctional proteins. Retinal treatment significantly increased the CD24+/CD44+ population through upregulation of HOXA5 in MCF10A, consequently decreasing their mammosphere-initiating capability. Our findings are consistent with the known role of the retinoic acid pathway in cellular differentiation,12, 27, 37 and an essential function for HOXA5 in this process.
Although cell surface molecules are useful for delineating specific cell lineages, there are not many studies focusing on the transcriptional cues for cell state transition. We have shown that HOXA5 transcriptionally regulates CDH1 and CD24 through promoter binding. We have thus identified novel molecular pathways that are functioning during cell fate transition.
In summary, we have shown that loss of HOXA5 in non-transformed breast epithelial cells results in reduced CDH1 and CD24 expression and dedifferentiation, but does not drive tumorigenesis. We have also provided evidence to support the notion that loss of HOXA5 and ensuing loss of epithelial phenotype in premalignant breast epithelial cells augments tumor-initiating potential and thereby promotes cancer progression. Finally, restoration of HOXA5 in malignant cells is capable of driving epithelial differentiation and thereby can impede aggressive phenotypes and stemness in breast tumor cells.
Materials and methods
Detailed materials and methods for each assay are provided in Supplementary Information online.
Cell lines and reagents
293 T and MCF10A obtained from the American Type Culture Collection (Manassas, VA, USA) were used in their early passages. MCF10A-ras was generated by stably expressing mutant KrasG12V in MCF10A cells. SUM149 and SUM159 cells were obtained from Dr S Ethier (Wayne State University, Detroit, MI, USA), and the lung metastatic subline of MDA-MB-231 cells (LM2) was obtained from Dr Joan Massagué (Memorial Sloan Kettering, New York, NY, USA). All trans-retinal (#R2500) was from Sigma-Aldrich (St Louis, MO, USA). All cell lines were tested for mycoplasma every 6 months.
Plasmid constructs
HOXA5 shRNAs (TRCN0000017528 and TRCN0000017529) were purchased from Open Biosystems (GE Dharmacon, Lafayette, CO, USA). Tet-inducible-shHOXA5 was generated by cloning sh528 into pLKO-Tet-On plasmid. HOXA5 cDNA with a myc-tag was cloned into pLHCX plasmid. CDH1 and CD24 gene promoters were cloned into pGL2 plasmid.
Mammosphere formation assay
Tumor sphere assays were performed as previously described.36 Quantification of the mammospheres was performed with ImageJ software (NIH, Bethesda, MD, USA).
3D growth assay
3D culture of the breast cancer cell lines was performed as previously described.35 For MCF10A-Kras, 2 × 103 cells were seeded into 100 μl of 50% Matrigel (#354234, Corning, New York, NY, USA) and 50% Collagen I (1 mg/ml #354236, Corning) mixture, plated in eight-well chamber slides and maintained in 400 μl of MCF10A complete media. For 231-LM2, 2.5 × 103 were seeded into 100 μl Matrigel plated in eight-well chamber slides.
Cell proliferation assay
Cell proliferation assay was performed as previously described.51
Wound healing assay
Cells were grown to confluence, a wound was created with a p200 tip and allowed to heal for 20–48 h. The area of migration was quantified from images by ImageJ software (NIH).
BrdU incorporation assay
BrdU incorporation assay was performed using cell proliferation ELISA kit (#ab126556, Abcam, Cambridge, UK) according to manufacturer’s instructions.
Cell invasion assay
Approximately, 1.5–5 × 104 cells were seeded in 500 μl basal media in the Boyden chamber (#354480, BD, Franklin Lakes, NJ, USA). After 16–24 h, the membrane was fixed with 10% formalin for 30 min and the cells were stained with 0.05% crystal violet. Cell numbers were determined from four independent fields in each chamber.
Quantitative RT–PCR
Quantitative SYBR Green RT–PCR (iQ SYBR Green Supermix, BIO-RAD, Hercules, CA, USA) was performed on total RNA as previously described.51 Gel-based RT–PCR products were resolved in a 2% ethidium bromide agarose gel.
Western blot analysis
Western blotting was performed using antibodies to: HOXA5 (#sc-365784, Santa Cruz, Santa Cruz, CA, USA); CDH1 (#13-1700, Invitrogen, Carlsbad, CA, USA); claudin-1 (#71-7800), claudin-7 (#34-9100), occludin (#71-1500), ZO-1 (#33-9100) and CK18 (#180158Z) from Zymed (Thermo Fisher Scientific, South San Francisco, CA, USA); p21 (Santa Cruz); β-actin (Sigma-Aldrich A2228).
Immunohistochemistry
Immunohistochemistry was performed as previously described.51 The sections were incubated overnight with primary antibodies: HOXA5 antibody (1:50), CDH1 antibody (1:500) and CK18 (1:500).
Immunofluorescent staining and confocal microscopy
For 2D images, cells were fixed and stained with corresponding antibodies using the manufacturer’s protocol. Confocal immunofluorescence staining of 3D organoids was performed as described.52 Images were obtained with a Zeiss LSM 780 microscope (Carl Zeiss, Oberkochen, Germany) using a × 40 c-Apo water objective.
Fluorescence-activated cell sorting and quantification
One million cells were stained with 5 μl of CD24-FITC (BD Pharmigen, San Jose, CA, USA; clone ML5), 5 μl CD44-PE (BD Pharmigen; clone 515) and 5 μl of 7-AAD (BD Pharmingen, San Jose, CA, USA) to exclude dead cells. Samples were run on the BD FACSCalibur system (Becton Dickinson, Franklin Lakes, NJ, USA) and data were analyzed using FlowJo software (Ashland, OR, USA). Cells were sorted on a BD FACSAria SORP and analyzed on a BDLSRII with BD FACSDiva Software (BD Biosciences, San Jose, CA, USA).
Xenograft and limiting dilution assay
The indicated number of cells were mixed in Matrigel and injected into mfp of NOG mice together with a mixture of irradiated and non-irradiated (1:1) immortalized human fibroblasts (25 000 cells per 100 μl Matrigel per fat pad).53, 54
Promoter luciferase assay
Luciferase activity (#E1501, Promega, Madison, WI, USA) was measured 24 h after transfection of cells and normalized to β-galactosidase activity (#117758241001, Roche, Basel, Switzerland).55
ChIP assay
ChIP assay was performed as previously described.56 Sonicated chromatin was enriched with either 1 μg of α-myc or 3 μg of α-HOXA5 antibody or the same amount of IgG isotype control, column-purified and analyzed.
Gene expression array and analysis
mRNA from MCF10A cells and MCF10A-shHOXA5 clones was hybridized onto Illumina’s HumanHT-12v4 BeadChip (Illumina, San Diego, CA, USA). After pre-processing using Illumina GenomeStudio software (Illumina), quantile normalization was applied to log-transformed intensities for analyses.
Statistical analysis
Quantitative data were expressed as mean±s.d. of biological triplicates. Statistical tests were analyzed by two-tailed Student’s t-test or one-way analysis of variance with Bonferroni correction, and considered statistically significant with a P-value of *P<0.05, **P<0.01 and ***P<0.001; GraphPad Prism (v5.0, GraphPad Software, San Diego, CA, USA).
References
Pratt MA, Langston AW, Gudas LJ, McBurney MW . Retinoic acid fails to induce expression of Hox genes in differentiation-defective murine embryonal carcinoma cells carrying a mutant gene for alpha retinoic acid receptor. Differentiation 1993; 53: 105–113.
Kondo T, Takahashi N, Muramatsu M . The regulation of the murine Hox-2.5 gene expression during cell differentiation. Nucleic Acids Res 1992; 20: 5729–5735.
Zwartkruis F, Kruyt F, van der Saag PT, Meijlink F . Induction of HOX-2 genes in P19 embryocarcinoma cells is dependent on retinoic acid receptor alpha. Exp Cell Res 1993; 205: 422–425.
Marshall H, Morrison A, Studer M, Popperl H, Krumlauf R . Retinoids and Hox genes. FASEB J 1996; 10: 969–978.
Dubrulle J, McGrew MJ, Pourquie O . FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell 2001; 106: 219–232.
Alexander T, Nolte C, Krumlauf R . Hox genes and segmentation of the hindbrain and axial skeleton. Annu Rev Cell Dev Biol 2009; 25: 431–456.
Shah N, Sukumar S . The Hox genes and their roles in oncogenesis. Nat Rev Cancer 2010; 10: 361–371.
Yan W, Chen Y, Yao Y, Zhang H, Wang T . Increased invasion and tumorigenicity capacity of CD44+/CD24- breast cancer MCF7 cells in vitro and in nude mice. Cancer Cell Int 2013; 13: 13–62.
Taylor HS, Vanden Heuvel GB, Igarashi P . A conserved Hox axis in the mouse and human female reproductive system: late establishment and persistent adult expression of the Hoxa cluster genes. Biol Reprod 1997; 57: 1338–1345.
Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E et al. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 2000; 405: 974–978.
Chen H, Chung S, Sukumar S . HOXA5-induced apoptosis in breast cancer cells is mediated by caspases 2 and 8. Mol Cell Biol 2004; 24: 924–935.
Chute JP, Muramoto GG, Whitesides J, Colvin M, Safi R, Chao NJ et al. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci USA 2006; 103: 11707–11712.
Chen H, Zhang H, Lee J, Liang X, Wu X, Zhu T et al. HOXA5 acts directly downstream of retinoic acid receptor beta and contributes to retinoic acid-induced apoptosis and growth inhibition. Cancer Res 2007; 67: 8007–8013.
Laursen KB, Mongan NP, Zhuang Y, Ng MM, Benoit YD, Gudas LJ . Polycomb recruitment attenuates retinoic acid-induced transcription of the bivalent NR2F1 gene. Nucleic Acids Res 2013; 41: 6430–6443.
Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS . Stem cells in normal breast development and breast cancer. Cell Prolif 2003; 36: 59–72.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–715.
Radisky DC, LaBarge MA . Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell 2008; 2: 511–512.
May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA . Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 2011; 13: 202.
Klopp AH, Lacerda L, Gupta A, Debeb BG, Solley T, Li L et al. Mesenchymal stem cells promote mammosphere formation and decrease E-cadherin in normal and malignant breast cells. PLoS One 2010; 5: e12180.
Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, Lovgren K et al. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res 2008; 10: R53.
Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH et al. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res 2006; 8: R59.
Kay R, Rosten PM, Humphries RK . CD24, a signal transducer modulating B cell activation responses, is a very short peptide with a glycosyl phosphatidylinositol membrane anchor. J Immunol 1991; 147: 1412–1416.
Petkova N, Hennenlotter J, Sobiesiak M, Todenhofer T, Scharpf M, Stenzl A et al. Surface CD24 distinguishes between low differentiated and transit-amplifying cells in the basal layer of human prostate. Prostate 2013; 73: 1576–1590.
Sleeman KE, Kendrick H, Ashworth A, Isacke CM, Smalley MJ . CD24 staining of mouse mammary gland cells defines luminal epithelial, myoepithelial/basal and non-epithelial cells. Breast Cancer Res 2006; 8: R7.
Thomas S, Harding MA, Smith SC, Overdevest JB, Nitz MD, Frierson HF et al. CD24 is an effector of HIF-1-driven primary tumor growth and metastasis. Cancer Res 2012; 72: 5600–5612.
Vesuna F, Lisok A, Kimble B, Raman V . Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 2009; 11: 1318–1328.
Ghiaur G, Yegnasubramanian S, Perkins B, Gucwa JL, Gerber JM, Jones RJ . Regulation of human hematopoietic stem cell self-renewal by the microenvironment's control of retinoic acid signaling. Proc Natl Acad Sci USA 2013; 110: 16121–16126.
Henderson KD, Andrew DJ . Regulation and function of Scr, exd, and hth in the Drosophila salivary gland. Dev Biol 2000; 217: 362–374.
Raman V, Tamori A, Vali M, Zeller K, Korz D, Sukumar S . HOXA5 regulates expression of the progesterone receptor. J Biol Chem 2000; 275: 26551–26555.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550.
Ribeiro AS, Paredes J . P-cadherin linking breast cancer stem cells and invasion: a promising marker to identify an "intermediate/metastable" EMT state. Front Oncol 2015; 4: 371.
Wang H, Meyer CA, Fei T, Wang G, Zhang F, Liu XS . A systematic approach identifies FOXA1 as a key factor in the loss of epithelial traits during the epithelial-to-mesenchymal transition in lung cancer. BMC Genomics 2013; 14: 680.
Zhang H, Meng F, Liu G, Zhang B, Zhu J, Wu F et al. Forkhead transcription factor foxq1 promotes epithelial-mesenchymal transition and breast cancer metastasis. Cancer Res 2011; 71: 1292–1301.
Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, Yu Q . FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res 2011; 71: 3076–3086.
Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol 2007; 1: 84–96.
Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 2003; 17: 1253–1270.
Strickland S, Mahdavi V . The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell 1978; 15: 393–403.
Wang YA, Shen K, Wang Y, Brooks SC . Retinoic acid signaling is required for proper morphogenesis of mammary gland. Dev Dyn 2005; 234: 892–899.
Aubin J, Lemieux M, Tremblay M, Berard J, Jeannotte L . Early postnatal lethality in Hoxa-5 mutant mice is attributable to respiratory tract defects. Dev Biol 1997; 192: 432–445.
Tokes AM, Kulka J, Paku S, Szik A, Paska C, Novak PK et al. Claudin-1, -3 and -4 proteins and mRNA expression in benign and malignant breast lesions: a research study. Breast Cancer Res 2005; 7: R296–R305.
Kominsky SL, Argani P, Korz D, Evron E, Raman V, Garrett E et al. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 2003; 22: 2021–2033.
Swisshelm K, Machl A, Planitzer S, Robertson R, Kubbies M, Hosier S . SEMP1, a senescence-associated cDNA isolated from human mammary epithelial cells, is a member of an epithelial membrane protein superfamily. Gene 1999; 226: 285–295.
Kramer F, White K, Kubbies M, Swisshelm K, Weber BH . Genomic organization of claudin-1 and its assessment in hereditary and sporadic breast cancer. Hum Genet 2000; 107: 249–256.
Martin TA, Mansel RE, Jiang WG . Loss of occludin leads to the progression of human breast cancer. Int J Mol Med 2010; 26: 723–734.
Keller PJ, Lin AF, Arendt LM, Klebba I, Jones AD, Rudnick JA et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res 2010; 12: R87.
Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013; 152: 25–38.
Ginestier C, Wicinski J, Cervera N, Monville F, Finetti P, Bertucci F et al. Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle 2009; 8: 3297–3302.
Ansari KI, Hussain I, Kasiri S, Mandal SS . HOXC10 is overexpressed in breast cancer and transcriptionally regulated by estrogen via involvement of histone methylases MLL3 and MLL4. J Mol Endocrinol 2012; 48: 61–75.
Sun R, Liu Y, Li SY, Shen S, Du XJ, Xu CF et al. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials 2015; 37: 405–414.
Arab HH, Wani G, Ray A, Shah ZI, Zhu Q, Wani AA . Dissociation of CAK from core TFIIH reveals a functional link between XP-G/CS and the TFIIH disassembly state. PLoS One 2010; 5: e11007.
Jin K, Park S, Teo WW, Korangath P, Cho SS, Yoshida T et al. HOXB7 is an ERalpha cofactor in the activation of HER2 and multiple ER target genes leading to endocrine resistance. Cancer Discov 2015; 5: 944–959.
Debnath J, Muthuswamy SK, Brugge JS . Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30: 256–268.
Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci USA 2004; 101: 4966–4971.
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007; 1: 555–567.
Jin K, Kong X, Shah T, Penet MF, Wildes F, Sgroi DC et al. The HOXB7 protein renders breast cancer cells resistant to tamoxifen through activation of the EGFR pathway. Proc Natl Acad Sci USA 2012; 109: 2736–2741.
Thiaville MM, Stoeck A, Chen L, Wu RC, Magnani L, Oidtman J et al. Identification of PBX1 target genes in cancer cells by global mapping of PBX1 binding sites. PLoS One 2012; 7: e36054.
Acknowledgements
We thank Akhilesh Pandey and Edward Gabrielson for guidance; Alan Rein for critically reviewing the manuscript; and help from members of the Sukumar lab. This work was supported by the Susan G. Komen Foundation Leadership Grant # SAC110050, the Department of Defense Center of Excellence-W81XWH-04-1-0595 and the SKCCC Core Grant P30 CA006973 to SS and The AVON Research Foundation Center of Excellence Grant to SS/WWT. AJE is supported by awards from the American Cancer Society (RSG-12-141-01-CSM) and from the Breast Cancer Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Teo, W., Merino, V., Cho, S. et al. HOXA5 determines cell fate transition and impedes tumor initiation and progression in breast cancer through regulation of E-cadherin and CD24. Oncogene 35, 5539–5551 (2016). https://doi.org/10.1038/onc.2016.95
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.95
- Springer Nature Limited
This article is cited by
-
TRAF7-targeted HOXA5 acts as a tumor suppressor in prostate cancer progression and stemness via transcriptionally activating SPRY2 and regulating MEK/ERK signaling
Cell Death Discovery (2023)
-
Deregulation of miR-375 Inhibits HOXA5 and Promotes Migration, Invasion, and Cell Proliferation in Breast Cancer
Applied Biochemistry and Biotechnology (2023)
-
HOXA5 inhibits the proliferation of extrahepatic cholangiocarcinoma cells by enhancing MXD1 expression and activating the p53 pathway
Cell Death & Disease (2022)
-
HOXA5 counteracts the function of pathological scar-derived fibroblasts by partially activating p53 signaling
Cell Death & Disease (2021)
-
HOXA5 inhibits the proliferation and neoplasia of cervical cancer cells via downregulating the activity of the Wnt/β-catenin pathway and transactivating TP53
Cell Death & Disease (2020)