
Abstract
Deletion or mutation of the gene encoding the deubiquitinating enzyme CYLD is a common genomic aberration in multiple myeloma (MM). However, the functional consequence of CYLD loss and the mechanism underlying its putative role as a tumor suppressor gene in the pathogenesis of MM has not been established. Here, we show that CYLD expression is highly variable in myeloma cell lines and primary MMs and that low CYLD expression is associated with disease progression from monoclonal gammopathy of undetermined significance to MM, and with poor overall and progression free-survival of MM patients. Functional assays revealed that CYLD represses MM cell proliferation and survival. Furthermore, CYLD acts as a negative regulator of NF-κB and Wnt/β-catenin signaling and loss of CYLD sensitizes MM cells to NF-κB-stimuli and Wnt ligands. Interestingly, in primary MMs, low CYLD expression strongly correlated with a proliferative and Wnt signaling-gene expression signature, but not with an NFκB target gene signature. Altogether, our findings identify CYLD as a negative regulator of NF-κB and Wnt/β-catenin signaling in MM and indicate that loss of CYLD enhances MM aggressiveness through Wnt pathway activation. Thus, targeting the Wnt pathway could be a promising therapeutic strategy in MM with loss of CYLD activity.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is a still incurable B-lineage malignancy characterized by uncontrolled expansion of malignant plasma cells in the bone marrow (BM). The transition of a normal plasma cell to a fully transformed, aggressive myeloma cell is a multistep process, which requires the acquisition of multiple structural and numerical genomic abnormalities. Most of this evolution takes place in the BM, indicating that the interaction with the BM microenvironment plays a critical role in the pathogenesis of MM.1, 2 The most common recurrent genomic aberrations in MM are found in both the pre-malignant monoclonal gammopathy of undetermined significance (MGUS) and MM, and involve translocation of the immunoglobulin heavy chain locus and one of the following partner genes: CCND1, MMSET, CCND3, c-MAF or MAFB. Most of the remaining cases of MM are associated with trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19 and 21.3, 4, 5, 6 Other recurrent genomic abnormalities in MM include activating RAS mutations, deletions of 6q, 13q, 8p, 16q or 17p, or changes involving MYC family genes, and are mostly associated with progression to advanced stage MM.3, 4, 5, 6 Deletion of all or part of chromosome 16q has been reported in ~15–20% of MM cases and is associated with an unfavorable prognosis.4, 6, 7 Gene expression profiling and gene mapping using single-nucleotide polymorphism arrays identified CYLD (16q12.1) as a potential determinant of poor prognosis in a subset of these cases.7
CYLD was originally identified as a tumor suppressor gene mutated in familial cylindromatosis (Brooke–Spiegler syndrome), an autosomal dominant disorder predisposing to benign tumors of skin appendages.8 Subsequent studies have linked loss of the tumor suppressor function of CYLD to the pathogenesis of several other tumors including melanoma, T-cell acute lymphoblastic leukemia, and colon and hepatocellular carcinoma.9, 10, 11 In MM, loss of CYLD, resulting from deletion (17% of primary MMs) or inactivating mutations (2.4% of primary MMs), is a common genomic aberration.4, 6, 7, 12, 13, 14 CYLD is a member of the USP family of deubiquitinating enzymes. A number of studies have shown that CYLD acts as a negative regulator of canonical, as well as non-canonical nuclear factor-ĸB (NF-ĸB) signaling by removing K63-linked polyubiquitin chains from TRAF2, TRAF6 and NEMO, which destabilizes these proteins.15 Other CYLD substrates important for NF-ĸB signaling include RIPK1, BCL3 and TAK1.16, 17, 18, 19 In addition, CYLD has been identified as a negative regulator of TGFβ-controlled Akt signaling,20 and as a target9 and negative regulator of Notch signaling.21 More recently, we reported that CYLD also acts as a negative regulator of proximal Wnt/β-catenin signaling by removing K63-linked polyubiquitin chains from the adapter protein Disheveled (Dvl). Accordingly, loss of CYLD was shown to cause hyperubiquitination of the DIX domain of Dvl, leading to enhanced Dvl polymerization and Wnt signaling.22 Importantly, human cylindroma skin tumors that arise from mutations in CYLD were indeed found to display hyperactive Wnt signaling, suggesting that the tumor growth instigated by loss of CYLD involves enhancement of Wnt responses.22
In the present study, we investigated the expression of CYLD in MM and explored its relation to disease progression and prognosis, as well as its functional impact on intracellular signaling and on MM growth and survival. Our results identify CYLD as a regulator of both NF-κB and Wnt/β-catenin signaling in MM and suggest that loss of CYLD enhances MM aggressiveness through aberrant Wnt pathway activation.
Results
HMCLs and primary MMs show highly variable levels of CYLD expression
Loss of the tumor suppressor function of CYLD in cancer can be caused by genetic inactivation of CYLD,8, 23, 24 epigenetic regulation by microRNAs or posttranslational regulation by phosphorylation and/or processing by proteases.9, 11, 24, 25, 26 To investigate CYLD expression and function in MM, we initially assessed CYLD messengerRNA (mRNA) and protein expression in a panel of human myeloma cell lines (HMCLs). As shown in Figure 1, HMCLs cell lines displayed a broad range of CYLD expression levels: whereas moderate to high levels of CYLD mRNA were found in ANBL-6, L363, RPMI8226, U266, XG-1 and XG-3, CYLD expression in the other cell lines was either low (Fravel, NCI-H929, LME-1, OPM-1 and UM-1) or undetectable (UM-3) (Figure 1a). CYLD protein levels largely, but incompletely, paralleled these mRNA expression data (Figure 1b): immunoblotting showed high expression levels in ANBL-6, L363, RPMI8226, U266 and XG-1, low levels in Fravel, NCI-H929, LME-1, OPM-1 and XG-3, and a complete absence of CYLD protein in UM-3. However, for XG-3, protein levels were relatively low compared with mRNA expression. Similar to HMCLs, primary MMs also displayed a broad range of CYLD mRNA and protein expression levels (Figures 1c and d). Notably, CYLD protein was not expressed in two of five samples tested (Figure 1d), but in one of these samples (pMM5) CYLD mRNA was readily detectable. Hence, CYLD expression in both HMCLs and primary MM cells is highly variable suggesting regulation at the transcriptional, as well as at the post-transcriptional level.

CYLD is variably expressed in MM cell lines and primary MM cells (a) qPCR analysis of CYLD mRNA expression in MM cell lines. TBP was used as an input control. (b) Western blot analysis of CYLD protein expression (upper panel). β-actin was used as a loading control. Quantification of CYLD band intensity normalized to β-actin (lower panel) (c) CYLD mRNA in a panel of five primary MM samples. TBP was used as an input control. (d) Western blot analyses of CYLD protein expression in five primary MM. β-actin was used as a loading control.
Loss of CYLD expression in MM is associated with disease progression and with a proliferation gene-expression signature
To obtain a more global view of CYLD mRNA expression in primary MMs and explore its relation to disease progression and MM molecular subgroup, we next analyzed a publically available microarray dataset containing 414 newly diagnosed MM patients27 for CYLD expression. Interestingly, this analysis revealed that CYLD expression was markedly reduced in a subset of patients with overt MM, compared with normal bone marrow plasma cells (BMPC) (Figure 2a). By contrast, in patients with MGUS and smoldering myeloma (sMM), CYLD expression levels were comparable to those in normal BMPCs (Figure 2a). These results indicate that disease progression from MGUS and sMM to full blown MM is accompanied by loss of CYLD expression in a subset of MM patients. Analysis of CYLD expression in distinct gene-expression profiling-defined MM molecular subgroups27 revealed that individual MM cases with reduced CYLD expression can be found in each subgroups. Importantly, however, CYLD expression in the proliferation (PR) subgroup, which is characterized by overexpression of numerous cell cycle and proliferation-related genes and associated with an adverse prognosis, was strongly decreased in comparison to all other molecular subgroups (P<0.001) (Figure 2b).

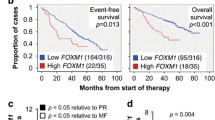
CYLD expression in MM in relation to disease stage, molecular subgroup classification, and prognosis. (a) CYLD mRNA expression analysis of a publically available micro-array dataset (TT2/3)27 containing healthy donors BMPCs (n=22), MGUS (n=44), sMM patients (n=12) and MM (n=414). MM vs BMPCs P<0.05, by Kruskal–Wallis test with Dunn correction for multiple comparisons. *P<0.05 (b) CYLD mRNA expression in distinct molecular MM subgroups.27 PR, proliferation; LB, Low-bone disease; HY, hyperdiploid; MS, MMSET; MF, MAFB. PR vs LB, HY, CD1, CD2, MS, P<0.001, PR vs MF P<0.05, by Kruskal-Wallis test with Dunn correction for multiple comparisons. (c) Low expression of CYLD is associated with poor prognosis of MM patients. The HOVON-65/GMMG-HD4 micro-array dataset28 containing newly diagnosed MM patients (n=327) was used to determine the prognostic value of CYLD expression (expression above or below the median). Kaplan–Meier curves are shown for overall survival (OS, left panel) and progression free survival (PFS, right panel). Low expression of CYLD is associated with inferior OS and PFS in MM patients, P<0.01 and P<0.05 by the log-rank test.
Next, we studied the relation between CYLD expression and disease outcome in a large group of newly diagnosed MM patients.28 For analysis, the cohort was divided into a CYLD high and a CYLD low group using median CYLD expression as cut-off. As shown in Figure 2c, there was a significant association between low expression of CYLD and inferior overall survival (OS, P<0.01) and progression-free survival (PFS, P<0.05), for example, the 5-year progression-free survival was 14% in the low vs 33% in the high CYLD expressing group. Similarly, analysis of a cohort of relapsed MM patients29 showed a significant association between low CYLD expression and inferior OS (P<0.05, Supplementary Figure 1, left panel) and a trend towards inferior PFS that did not reach significance (P=0.15, Supplementary Figure 1, right panel), possibly because of the almost invariable rapid disease progression of these patients.
Next, we performed multivariate Cox proportional hazard regression analysis to study the predictive value of low CYLD expression, independent of other prognostic factors. Interestingly, this analyses identified low CYLD expression as an independent prognostic factor for poor OS and PFS (both P<0.05) in newly diagnosed MM patients28 and OS (P<0.01) in relapsed MM patients29 (Supplementary Table 1). A similar trend was observed for PFS in relapsed MM patients,29 which did not reach statistical significance (P=0.065), as mentioned previously, likely because the almost universal rapid disease progression of these patients.
Taken altogether, these results show that loss of CYLD expression in primary MMs is associated with disease progression, with a proliferative MM phenotype, and with an unfavorable prognosis.
CYLD regulates growth and survival of MM cells
In MM, homozygous deletion or inactivating mutations of CYLD are among the most common genomic aberrations.7, 12, 13 The HMCL UM-3 models these patients, since array comparative genomic hybridization and karyotyping of this cell lines revealed loss of 16q accompanied by deletion of large part of the other CYLD allele (Figure 3a), explaining the complete absence of CYLD mRNA and protein expression (Figures 1a and b). To provide direct functional evidence for a tumor suppressor role of CYLD in MM, we explored the impact of re-introducing CYLD on the growth and survival of UM-3 cells (Figures 3b–d). On transduction of UM-3 cells with a bicistronic vector co-expressing CYLD and YFP, CYLD-transduced cells were outcompeted by CYLD negative cells, resulting in a progressive loss of YFP+ cells, whereas the percentage of cells transduced with an empty vector was stable in time (Figure 3c, left panel). This growth suppressive effect of CYLD was confirmed in experiments comparing growth of FACS-sorted UM-3 cells transduced with either a CYLD containing or a control vector (Figure 3c, right panel). Subsequent cell cycle analysis in UM-3 cells revealed that cells ectopically expressing CYLD incorporated approximately 50% less BrdU compared with control transduced cells, indicative of suppression of proliferation (Figure 3d). Moreover, in cells ectopically expressing CYLD there was an approximately two-fold increase in cells in sub-G1 phase of the cell cycle, indicative of increased cell death (Figure 3d). In addition to impairing cell growth of CYLD deficient UM-3 cells, ectopic expression of CYLD in LME-1 and XG-3 cells, which express CYLD at relative low levels (Figures 1a and b), also decreased growth (Supplementary Figure 2). Hence, CYLD acts as a tumor suppressor in MM by repressing proliferation and survival.

Reconstituted expression of CYLD in CYLD-deficient MM cells decreases growth and survival. (a) Karyotyping of UM-3 cells showing loss of 16q (left panel). Array comparative genomic hybridization (aCGH) analysis of UM-3 cells reveals homozygous deletion of the CYLD locus 16q21 (right panel), resulting in loss of CYLD mRNA and protein (Figure 1). (b) Western blot analysis of CYLD protein expression in UM-3 cells transduced with an empty or CYLD containing bicistronic vector. β-actin was used as a loading control. (c) Flow cytometry analysis of UM-3 cells transduced with an empty or CYLD containing bicistronic vector co-expressed with YFP. The percentage of YFP positive cells was followed in time and plotted as the percentage of YFP+ cells compared with t=0. Results are presented as mean±s.e.m. of three independent transductions. (left panel) Flow cytometry analysis of expansion of sorted UM-3 cells transduced with an empty or CYLD containing vector. The number of viable cells was determined at indicated time points and defined as being 7AAD negative. The number of living cells was normalized to t=0 and the mean±s.e.m. of three independent experiments performed in triplicate is shown. ***P <0.001, **P<0.01 using unpaired student’s t test (right panel). (d) Representative picture of flow cytometric analysis of BrdU incorporation of sorted CYLD or control vector transduced UM-3 cells (left panel) and quantification of the percentage of cells in the sub-G1 and S phase of the cell cycle (right panel). The mean±s.e.m. of three independent transductions is shown (lower panel). ***P<0.001, **P<0.01, *P<0.05 using unpaired student’s t test.
Loss of CYLD sensitizes MM cells to both NF-ĸB and Wnt ligands
CYLD is an established negative regulator of NF-κB signaling,15, 16, 17, 18, 19 and has been reported to negatively regulate Wnt/β-catenin signaling.22 Loss of CYLD expression might thus increase the sensitivity of MM cells to NF-ĸB and Wnt ligands expressed in the BM microenvironment. To explore this notion, we generated CYLD-deficient XG-1, XG-3 and LME-1 HMCLs using the CRISPR/CAS9 system and analyzed sensitivity to Wnt and NF-ĸB inducing ligands (Figure 4a, upper panel). Of note, XG-1, XG-3 and LME-1 do not carry any known genetic abnormalities in components of the NF-κB or Wnt/β-catenin pathway.30 Silencing of CYLD significantly increased nuclear localization of the p50 and p65/RelA NF-ĸB subunits, indicative of NF-ĸB signaling pathway activation. (Figure 4a, lower panel). Moreover, CYLD deficient cells displayed increased NF-ĸB activity in response to TNFα, which mimics paracrine signaling (Figure 4a, lower panel). Furthermore, silencing of CYLD significantly increased both basal and TNFα-induced NF-ĸB luciferase reporter activity (Figure 4b). Conversely, ectopic expression of CYLD in the CYLD-deficient HMCL UM-3 inhibited NF-ĸB signaling as evidenced by decreased basal and TNFα-induced levels of nuclear p65/RelA and a decrease in NF-ĸB reporter activity (Figures 4c and d). Hence, our data show that CYLD negatively regulates NF-ĸB signaling in MM and that loss of CYLD increases sensitivity to paracrine NF-ĸB ligands.

CYLD inhibits NF-κB signaling in malignant plasma cells. (a) Western blot analysis of CYLD expression after CRISPR/CAS9 mediated silencing of CYLD. β-actin was used as a loading control (upper panel). Western blot analysis of the subcellular localization of the p50 and p65/RELA NF-κB subunits after CYLD silencing and/or TNFα stimulation in the XG-1 and XG-3 HMCLs. Lamin A/C (nucleus) and β-tubulin (cytoplasm) were used as a loading and fractionation control (lower panel). (b) Analysis of NF-κB transcriptional activity in the XG-1 and XG-3 HMCLs after silencing of CYLD and/or stimulation with TNFα, using an NF-κB luciferase reporter. NF-κB luciferase activity was corrected for TK-Renilla luciferase activity (relative light units, RLU) and subsequently normalized to untreated conditions. The mean±s.e.m. of three independent experiments in triplo is shown. ***P<0.001 using one-way ANOVA with Bonferroni correction. (c) Westernblot analysis of CYLD expression and subcellular localization of p65/RelA in sorted CYLD or control vector transduced UM-3 cells after stimulation with TNFα. Lamin A/C (nucleus) and β-tubulin (cytoplasm) were used as a loading and fractionation control. (d) Analysis of NF-κB transcriptional activity in control or CYLD transduced UM-3 cells stimulated with TNFα, using a NF-κB luciferase reporter. NF-κB luciferase activity was corrected for TK-Renilla luciferase activity (relative light units, RLU) and subsequently normalized to untreated conditions. The mean±s.e.m. of three independent experiments in triplo is shown. *P<0.05 using one-way ANOVA with Bonferroni correction.
Interestingly, in addition to affecting NF-ĸB signaling, silencing of CYLD also profoundly affected Wnt/β-catenin signaling. CRISPR/CAS9-mediated silencing of CYLD in HMCLs resulted in increased stabilization and translocation of nuclear β-catenin, a hallmark of active Wnt/β-catenin signaling, in response to stimulation with recombinant Wnt3a (Figure 5a). This effect was strongly enhanced by simultaneous stimulation with R-spondin (Figure 5a), which binds LGRs on the MM plasma cell membrane leading to stabilization of Wnt receptors, resulting in greatly increased sensitivity to Wnt ligands (HvA and STP, manuscript in preparation).31, 32 In addition, CYLD-deficient HMCLs displayed increased TCF4/β-catenin-mediated luciferase reporter activity in response to stimulation with Wnt3a or Wnt3a and R-spondin, further supporting a role for CYLD in regulating Wnt/β-catenin signaling in MM (Figure 5b). Conversely, ectopic expression of CYLD in the CYLD deficient HMCL UM-3 resulted in suppression of Wnt/β-catenin signaling, evidenced by decreased levels of nuclear β-catenin (Figure 5c) and decreased Wnt reporter activity (Figure 5d).

CYLD represses Wnt signaling in malignant plasma cells. (a) Western blot analysis of the nuclear localization of β-catenin after CYLD silencing and/or stimulation with recombinant Wnt3a, Rspo2 or both in the XG-3 and LME-1 HMCLs. Lamin A/C (nucleus) and β-tubulin (cytoplasm) were used as a loading and fractionation control (lower panel). (b) Analysis of Wnt/β-catenin transcriptional activity in the XG-1 and LME-1 HMCLs after silencing of CYLD and/or stimulation with recombinant Wnt3a, Rspo2 or both, using the TopFlash luciferase reporter. TopFlash luciferase activity was corrected for TK-Renilla luciferase activity (relative light units, RLU) and subsequently normalized to untreated conditions. The mean±s.e.m. of three independent experiments in triplo is shown. ***P<0.001 using one-way ANOVA with Bonferroni correction. (c) Western blot analysis of CYLD expression and subcellular localization of β-catenin in sorted CYLD or control vector transduced UM-3 cells after stimulation with recombinant Wnt3a and Rspo2. Lamin A/C (nucleus) and β-tubulin (cytoplasm) were used as a loading and fractionation control. (d) Analysis of Wnt/β-catenin transcriptional activity in control or CYLD transduced UM-3 cells stimulated with recombinant Wnt3a and Rspo2, using the TopFlash luciferase reporter. TopFlash luciferase activity was corrected for TK-Renilla luciferase activity (relative light units, RLU) and subsequently normalized to untreated conditions. The mean±s.e.m. of three independent experiments in triplo is shown. **P<0.01 using one-way ANOVA with Bonferroni correction.
Taken altogether, these data demonstrate that loss of CYLD has profound effects on both NF-ĸB and Wnt/β-catenin signaling in MM cells and support the notion that CYLD loss during disease progression enhances sensitivity of MM cells to NF-ĸB stimuli and Wnt ligands emanating from the bone marrow microenvironment.
Loss of CYLD in primary MMs is associated with a Wnt gene-expression signature
The above findings suggest that loss of CYLD can unleash NF-ĸB and/or Wnt signaling which drives MM progression. To explore this notion, we analyzed the relation between CYLD mRNA expression and the strength of a Wnt and a NF-ĸB gene-expression signature in MM patients (Figure 6a).27 Interestingly, as shown in Figure 6a, CYLD expression showed a significant inverse correlation to a Wnt profile (left panel), whereas CYLD was positively correlated to NF-ĸB profile genes (right panel). Furthermore, analysis of the individual molecular MM subgroups revealed that the proliferation (PR) subgroup, which is characterized by low CYLD expression (Figure 2b), displayed a significantly stronger Wnt signature than the other molecular subgroups (Figure 6b, left panel), while the NF-ĸB signature in the PR subgroup was weak (Figure 6b, right panel). Thus, the PR group is characterized by active Wnt signaling, but shows reduced NF-ĸB signaling, suggesting that Wnt signaling might drive proliferation in these tumors. Consistent with this notion, we found a strong positive correlation (R=0.9, P<0.001) between the GEP-defined proliferation index27 and the Wnt expression signature (Figure 6c, left panel). By contrast, the proliferation index showed a weak but significant negative correlation to the NF-ĸB profile (Figure 6c, right panel). In support of these data, gene set enrichment analysis comparing CYLD low to CYLD high primary MM patients,28 revealed significant enrichment of genes involved in cell-cycle regulation and Wnt signaling in the CYLD low group (Supplementary Table 2). Noteworthy, there was a significant enrichment of other biological pathways and processes known to be regulated by CYLD, such as T-cell receptor signaling, B-cell receptor signaling, Cytoskeleton regulation and Colorectal Cancer, confirming specificity of our analysis (Supplementary Table 2). Altogether, these findings suggest a scenario in which loss of CYLD enhances autocrine and/or paracrine Wnt signaling, leading to enhanced proliferation and tumor progression.

Wnt- and NF-κB gene-expression profiles; correlation to CYLD expression, MM subgroup, and proliferation index. (a) Correlation between Wnt profile and CYLD expression in primary MMs;27 Spearman's rank correlation coefficient=−0.3, P<0.001 (left panel). Correlation between NF-κB profile and CYLD expression; Spearman's rank correlation coefficient=0.2, P<0.001 (right panel) (b) Wnt (left panel) and NF-κB (right panel) expression profile in different molecular MM subgroups. The Wnt expression profile was significantly higher in the proliferation subgroup (PR) in comparison to other subgroups, whereas the NF-κB expression profile was significantly lower in the PR subgroup in comparison to others subgroups ***P< 0.001, by Kruskal Walis test with Dunn correction for multiple comparisons. (c) Correlation between a Wnt expression profile and the proliferation index; Spearman's rank correlation coefficient=0.9, P<0.001 (left panel). Correlation between a NF-κB profile and the proliferation index; Spearman's rank correlation coefficient=−0.2, P<0.05 (right panel).
Discussion
CYLD was originally identified as a tumor suppressor gene mutated in familial cylindromatosis8 and has subsequently also been implicated in the pathogenesis of a number of other tumors.9, 10, 11 In MM, loss of CYLD by deletion or mutation is among the most common genomic aberrations, suggesting an important role for CYLD in the pathogenesis of MM.4, 5, 6, 7, 12, 13 In this study, we show that CYLD expression is highly variable in both HMCLs and primary MMs, which presumably involves regulation at both the transcriptional and the post-transcriptional levels (Figure 1). Furthermore, our analysis of CYLD expression in healthy donor plasma cells, MGUS, sMM and symptomatic myeloma patients revealed that CYLD expression is low in subset of MM patients, but not in MGUS or sMM patients, supporting a possible role of CYLD loss in disease progression. It should be noted, however, that the extent of CYLD loss in MM is most likely underestimated looking at mRNA levels, since small mutations disrupting CYLD function will be missed. Consistent with a role for CYLD in disease progression, CYLD levels were significantly reduced in the proliferation (PR) subgroup (Figure 2b), which is characterized by a high proliferation rate and an unfavorable prognosis.27 Furthermore, low CYLD expression was associated with inferior PFS and OS in primary MMs and was found to be an independent prognostic factor of clinical outcome. (Figure 2c, Supplementary Figure 1 and Supplementary Table 1). In support of these data, gene set enrichment analysis comparing CYLD low to CYLD high primary MM patients,28 revealed significant enrichment of genes involved in cell-cycle regulation (Supplementary Table 2). Conceivably, downregulation or complete loss of CYLD thus plays an instrumental role in aggressive disease behavior. In support of this notion, introduction of CYLD in UM-3 cells, which lack endogenous CYLD expression because a homozygous deletion of the CYLD gene (Figure 3a) and thereby models this recurrent genomic aberration in primary MMs,4, 6, 7, 12, 13, 14 resulted in growth inhibition (Figure 3c), increased cell death, and suppressed proliferation (Figure 3d). These findings provide proof of concept for a functional role of CYLD in the control of MM growth and survival. Further studies are needed to explore alternative causes of loss of CYLD activity in MM, other than by deletion/mutation. Potential mechanisms include transcriptional repression, for example, mediated by SNAIL, the Notch/Hes1 pathway, or by microRNAs,9, 11, 25 as well as phosphorylation of CYLD,26 which have all been implicated in loss of CYLD tumor suppressor function in cancer.
We observed that loss of CYLD has profound effects on both NF-ĸB and Wnt/β-catenin signaling in MM cells, sensitizing MM cells to NF-ĸB as well as Wnt ligands (Figures 4 and 5). Indeed, by removing K63-linked polyubiquitin chains from proteins involved in the NF-ĸB signaling cascade,15, 16, 17, 18, 19 CYLD acts as a negative regulator of NF-ĸB signaling in various biological processes including lymphoid development, inflammation, and cancer.25 Our current finding that CYLD regulates basal as well as autocrine- and/or paracrine-controlled NF-ĸB activity represents the first functional evidence that CYLD is also involved in the regulation of NF-ĸB signaling in MM (Figure 4). Interestingly, however, we observed that CYLD also functions as a potent negative regulator of Wnt signaling in MM cells, controlling sensitivity to Wnt ligands (Figure 5). This corroborates our previous study, showing that CYLD controls Wnt signaling through deubiquitination of Dvl, an adapter protein which transmits proximal Wnt signals and is dependent on K63-linked ubiquitination for its function.22 Our findings imply that down-regulation or loss of CYLD expression can potentially increase the sensitivity of MM cells to both NF-ĸB and Wnt ligands expressed in the BM microenvironment, thus unleashing NF-ĸB, as well as Wnt signaling. However, although we did find a significant negative correlation between CYLD expression and expression of a Wnt target genes in primary MM samples, we found a (weak) positive correlation to a NF-ĸB signature (Figure 6a). This suggests that loss of CYLD predominantly activates Wnt signaling in MM in vivo. Consistent with this scenario, the PR subgroup, which is characterized by low CYLD expression (Figure 2b), displayed a significantly stronger Wnt but weaker NF-ĸB-signature compared with all other molecular subgroups (Figure 6b). Moreover, gene set enrichment analysis identified significant enrichment of genes associated with Wnt signaling in MM patients with low CYLD expression compared with MM patients with high CYLD expression (Supplementary Table 2). A possible explanation for the positive correlation between CYLD expression and NF-ĸB activation could be that CYLD acts as a classical negative feedback target in MM as suggested by previous studies;33 however, we did not observe upregulation of CYLD after NF-ĸB (TNFα) activation in vitro (data not shown). Taken altogether, these findings support a scenario in which loss of CYLD enhances autocrine and/or paracrine Wnt signaling, thereby promoting proliferation and mediating tumor progression. Indeed, a very strong positive correlation (R=0.9, P<0.001) between the GEP-defined proliferation index and the Wnt expression signature (Figure 6c) suggests that Wnt signaling is instrumental for driving MM proliferation in vivo. Presumably, the Wnt/β-catenin-mediated expression of the cell cycle regulator AurKA/B plays an important role in this pathogenic effect.34
Previous studies from our own and other laboratories have established that aberrant activation of the canonical Wnt pathway is common in advanced MM and plays an important role in MM pathogenesis by promoting cell growth, survival, and drug resistance.27, 30, 35, 36, 37 Although activation of Wnt signaling in human cancer is typically caused by mutations in the adenomatous polyposis gene or in β-catenin (CTNNB1), resulting in ligand-independent Wnt pathway activation, these mutations are absent in MM. Instead, Wnt pathway activation in MM involves stimulation of an intact Wnt signaling pathway by autocrine and/or paracrine Wnt ligands, which are overexpressed by MM cells as well as by stromal cells in the MM microenvironment.30, 35, 36, 37, 38 Notably, we have recently established that the activity of these Wnt ligands is greatly enhanced by niche-derived R-spondins that bind to LGR4 on the MM cell surface, resulting in Wnt-receptor stabilization (HvA and STP, manuscript in preparation). In addition to CYLD loss, silencing of other negative regulators of Wnt signaling may also play a role in hyperactivating Wnt signaling and driving MM progression. Indeed, promoter methylation of Wnt antagonists, including WIF1, DKK3, SFRP1, 2, 4 and 5, has been reported in MM and was associated with disease progression.39, 40 Furthermore, DKK1, a major negative feed-back regulator of Wnt-signaling, is also a target of promoter methylation, especially in advanced stage MM.37
Our current finding that CYLD acts as a negative regulator of MM growth and Wnt/β-catenin signaling and that low CYLD expression in primary MMs is associated with a Wnt signaling and proliferative gene-expression signature, suggests that CYLD loss enhances MM aggressiveness through hyperactivation of the Wnt pathway rather than by affecting NF-ĸB signaling. Consequently, since Wnt signaling in MM is ligand-driven, targeting of the Wnt (co)receptors and/or use of Wnt secretion inhibitors is a potentially promising therapeutic strategy for MMs with loss of CYLD activity.
Materials and methods
Cell culture
HMCLs were cultured in IMDM (Invitrogen Life Technologies, Carlsbad, CA, USA) containing 10% FCS, 100 units/ml penicillin (Sigma Aldrich, St Louis, MO, USA), 100 μg/ml streptomycin (Sigma Aldrich), 20 μg/ml human apo-transferrin (Sigma Aldrich) and 50 μM β-mercaptoethanol (Sigma Aldrich). All cell lines were found to be mycoplasma negative by PCR. For LME-1, XG-1, XG-3 and ANBL-6 500 pg/mL IL-6 was added (Prospec, East Brunswick, NJ, USA). Recombinant proteins used were hTNFα (50 ng/mL, Prospec), hWnt3a (100 ng/mL, R&D systems, Minneapolis, MN, USA) and hR-spondin 2 (50 ng/mL R&D systems).
Transfection and transduction
sgCYLD (ATGGGAAGGACGATTCTGCC, chr16:50828286-50828305, hg19) was inserted in pLentiCrispr (addgene 52961) as previously described.41 Phoenix-GALV cells were transfected with LZRS-pBMN-IRES-YFP/LZRS-pBMN-CYLD-IRES-YFP using Genius DNA transfection reagent (Westburg, Leusden, The Netherlands). 2 μg/ml puromycin (Sigma Aldrich) was added to enrich virus producing cells. HMCLs were spinfected on retronectin (Takara Biomedicals, Tokio, Japan) and subsequently incubated for 16 h. Cells were analyzed on a FACSCanto II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and subsequently sorted.
Q-PCR
PCRs were conducted on a Lightcycler 480 (Roche). Primers used were: CYLD forward (5′– TGCAGGCTGTACGGATGGAACCT –3′); CYLD reverse (5′– TCCTGATGCAGCCTCCACCT –3′); TBP forward (5′– CCCATGACTCCCATGACC–3′); TBP reverse (5′– TTTACAACCAAGATTCACTGTGG–3′). Primers were manufactured by Biolegio (Nijmegen, The Netherlands)
Reporter assays
10.10e6 cells were electroporated with the TOPflash Wnt reporter or NF-ĸB reporter (10 μg) and pRL-TK-Renilla (2 μg). After 24 h of recovery, cells were stimulated overnight. Firefly luciferase and renilla luciferase activity were measured using the dual luciferase assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Renilla luciferase activity served as a control for transfection efficiency.
Immunoblotting and FACS analysis
Protein samples were separated by SDS-polyacrylamide gel electrophoresis and subsequently blotted. Nuclear/cytosolic fractions were prepared using the Nuclear/Cytosolic fractionation kit (Biovision, Milpitas, CA, USA). Antibodies used for immunoblotting were: mouse anti-β-tubulin (clone D66, Sigma Aldrich), mouse anti-β-actin (clone AC-15, Sigma Aldrich), mouse anti-β-catenin (clone 14, BD Biosciences, Franklin Lakes, NJ), polyclonal rabbit anti-Lamin A/C (Santa Cruz, Dallas, TX, USA), mouse anti-p65/RelA (clone C-20, Santa Cruz), polyclonal rabbit anti-p50/NFKB1 (Cell Signaling, Danvers, MA, USA) and mouse anti-CYLD (clone E-10, Santa Cruz). Quantification of westernblot gels was done using Image J software.
Growth assays and cell cycle analysis
FACS-sorted YFP-positive MM cells were plated at 15 000 cells/well in medium containing 5% FCS. Living cells were defined as 7AAD- in the lymphocytic gate. For cell cycle analysis, cells were incubated for 0.5–1 h with 20 μM BrdU (Sigma Aldrich) and sorted for YFP. Sorted cells were fixed in ice-cold 70% ethanol and incubated with 0.4 mg/mL pepsin/0.2 mM HCL for 30’ on RT and subsequently incubated in 2M HCL for 25’ at 37 °C. Thereafter, cells were washed with 0,05%Tween-20/0,5%BSA in PBS (PBS-TB) and with anti-BrdU FITC (clone B44, BD Biosciences). After washing, cells were stained with 0,1μM TO-PRO-3-Iodide (Invitrogen Life Technologies) in 0,02%NaN3/0,5%BSA/PBS containing 500 μg/mL RNAse-A (Bioke, Leiden, The Netherlands) for 15’ at 37 °C.
Patients and statistical analysis
Gene expression data publically available and deposited in the NIH Gene Expression Omnibus National Center for Biotechnology Information were analyzed. These concerned the U133 Plus 2.0 affymetrix oligonucleotide microarray data from 44 MGUS patients, 22 healthy donors and 559 newly diagnosed MM patients included in the total therapy 2/3 (TT2, TT3) trials,27 the gene expression and survival data of 327 MM patients included in randomized clinical trials, that is the Institutional Review Board-approved HOVON-65/GMMG-HD4 (ISRCTN64455289) trial for newly diagnosed patients with MM,28 and the gene expression and survival data of 264 relapsed MM patients enrolled in the APEX trial.29
The MM NF-κB profile was determined by Annuziata et al. (GSE8487).12 Genes were chosen if they correlated in expression across MM cell lines (r>0.5). NF-κB profile genes, using Affymetrix U133plus 2.0 data, relied on the following probe sets: 210538_s_at(BIRC3), 202644_s_at(TNFAIP3), 207535_s_at(NFKB2), 204116_at(IL2RG), 203927_at(NFKBIE), 205205_at(RELB), 201502_s_at(NFKBIA), 209619_at(CD74), 203471_s_at(PLEK), 210018_x_at(MALT1), 223709_s_at(WNT10A).
The MM Wnt profile was based on a study reported by Dutta-Simmons et al.34 Genes were chosen if they correlated in expression across the MM patients. Wnt profile genes, using Affymetrix U133plus 2.0 data, relied on the following probe sets: 207828_s_at(CENPF), 208079_s_at(AURKA), 225655_at(UHRF1), 218755_at(KIF20A), 223307_at(CDCA3), 202095_s_at(BIRC5), 218039_at(NUSAP1), 1555772_a_at(CDC25A), 203276_at(LMNB1), 210559_s_at(CDC2).
The probe set for CYLD expression was chosen based on the highest frequency of P calls (present calls) in MM patients data sets determined by Affymetrix GCOS-software.
The Kruskal–Wallis test with Dunn correction for multiple comparisons was used to compare CYLD expression in MM molecular subgroups and to analyze the difference in CYLD expression during disease progression. Cox proportional hazard regression was used to assess the influence of CYLD expression on survival outcome. Multivariate analysis was performed by backward-selection Cox regression analysis. Covariates differ per analyzed study, depending on information available. Survival curves were estimated by the product-limit methods of Kaplan and Meier and compared using the log-rank test using Prism 5.0 software. Spearman rank correlation coefficients were used to determine correlations between CYLD expression and NF-κB and Wnt profile and to analyze the correlation between Wnt and NF-κB profile and the proliferation index.27
Biological processes and pathways (KEGG) differentially represented between CYLD high and low MM patients were analyzed in the HOVON-65/GMMG-HD4 (GSE19784) data set. Differentially expressed genes between the 75 MM patients with the lowest CYLD expression and 75 patients with the highest CYLD expression were identified using an R/Bioconductor software package, limma. To determine the pathways/processes (KEGG) in which the differentially expressed genes are present, we used JEPETTO,42 a Cytoscape plugin for gene set enrichment.
Array comparative genomic hybridization and karyotyping
Array comparative genomic hybridization (aCGH) was done by means of the Agilent 180 K oligo-array platform (Amadid 023363) using Genomic Workbench software for data analysis (both Agilent Inc, Santa Clara, California, USA). The NCBI Build GRCh36.1/hg18 was used for locus identification. Chromosome analysis was carried out by QFQ-banding.
References
Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC . Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007; 7: 585–598.
Kuehl WM, Bergsagel PL . Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest 2012; 122: 3456–3463.
Kumar S, Fonseca R, Ketterling RP, Dispenzieri A, Lacy MQ, Gertz MA et al. Trisomies in multiple myeloma: impact on survival in patients with high-risk cytogenetics. Blood 2013; 119: 2100–2105.
Morgan GJ, Walker BA, Davies FE . The genetic architecture of multiple myeloma. Nat Rev Cancer 2012; 12: 335–348.
Lawasut P, Groen RW, Dhimolea E, Richardson PG, Anderson KC, Mitsiades CS . Decoding the pathophysiology and the genetics of multiple myeloma to identify new therapeutic targets. Semin Oncol 2013; 40: 537–548.
Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014; 25: 91–101.
Jenner MW, Leone PE, Walker BA, Ross FM, Johnson DC, Gonzalez D et al. Gene mapping and expression analysis of 16q loss of heterozygosity identifies WWOX and CYLD as being important in determining clinical outcome in multiple myeloma. Blood 2007; 110: 3291–3300.
Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 2000; 25: 160–165.
Espinosa L, Cathelin S, D'Altri T, Trimarchi T, Statnikov A, Guiu J et al. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell 2010; 18: 268–281.
Hellerbrand C, Bumes E, Bataille F, Dietmaier W, Massoumi R, Bosserhoff AK . Reduced expression of CYLD in human colon and hepatocellular carcinomas. Carcinogenesis 2007; 28: 21–27.
Massoumi R, Kuphal S, Hellerbrand C, Haas B, Wild P, Spruss T et al. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J Exp Med 2009; 206: 221–232.
Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007; 12: 115–130.
Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007; 12: 131–144.
Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol 2015; 33: 3911–3920.
Brummelkamp TR, Nijman SM, Dirac AM, Bernards R . Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003; 424: 797–801.
Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G . The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003; 424: 801–805.
Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fässler R . Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell 2006; 125: 665–677.
Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med 2007; 204: 1475–1485.
Wright A, Reiley WW, Chang M, Jin W, Lee AJ, Zhang M et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell 2007; 13: 705–716.
Lim JH, Jono H, Komatsu K, Woo CH, Lee J, Miyata M et al. CYLD negatively regulates transforming growth factor-β-signalling via deubiquitinating Akt. Nat Commun 2012; 10: 771.
Rajan N, Elliott RJ, Smith A, Sinclair N, Swift S, Lord CJ et al. The cylindromatosis gene product, CYLD, interacts with MIB2 to regulate notch signaling. Oncotarget 2014; 5: 12126–12140.
Tauriello DV, Haegebarth A, Kuper I, Edelmann MJ, Henraat M, Canninga-van Dijk MR et al. Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell 2010; 37: 607–619.
An CH, Kim SS, Kang MR, Kim YR, Kim HS, Yoo NJ et al. Frameshift mutations of ATBF1, WNT9A, CYLD and PARK2 in gastric and colorectal carcinomas with high microsatellite instability. Pathology 2010; 42: 583–585.
Massoumi R . CYLD: a deubiquitination enzyme with multiple roles in cancer. Future Oncol 2011; 7: 285–297.
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K . STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010; 39: 493–506.
Hutti JE, Shen RR, Abbott DW, Zhou AY, Sprott KM, Asara JM et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol Cell 2009; 34: 461–472.
Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S et al. The molecular classification of multiple myeloma. Blood 2006; 108: 2020–2028.
Sonneveld P, Schmidt-Wolf IG, van der Holt B, El Jarari L, Bertsch U, Salwender H et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/ GMMG-HD4 Trial. J Clin Oncol 2012; 30: 2946–2955.
Mulligan G, Mitsiades C, Bryant B, Zhan F, Chng WJ, Roels S et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2007; 109: 3177–3188.
Derksen PW, Tjin E, Meijer HP, Klok MD, MacGillavry HD, van Oers MH et al. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc Natl Acad Sci USA 2004; 101: 6122–6127.
de Lau W, Barker N, Low TY, Koo BK, Li VS et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011; 476: 293–297.
Carmon KS, Gong X, Lin Q, Thomas A, Liu Q . R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc Natl Acad Sci USA 2011; 108: 11452–11457.
Smith EM, Zhang L, Walker BA, Davenport EL, Aronson IL, Krige D et al. The combination of HDAC and aminopeptidase inhibitors is highly synergistic in myeloma and leads to disruption of the NFκB signalling pathway. Oncotarget 2015; 6: 17314–17327.
Dutta-Simmons J, Zhang Y, Gorgun G, Gatt M, Mani M, Hideshima T et al. Aurora kinase A is a target of Wnt/beta-catenin involved in multiple myeloma disease progression. Blood 2009; 114: 2699–2708.
Takada K, Zhu D, Bird GH, Sukhdeo K, Zhao JJ . Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci Transl Med 2012; 4: 148ra117.
Sukhdeo K, Mani M, Zhang Y, Dutta J, Yasui H, Rooney MD et al. Targeting the beta-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proc Natl Acad Sci USA 2007; 104: 7516–7521.
Kocemba KA, Groen RW, van Andel H, Kersten MJ, Mahtouk K, Spaargaren M et al. Transcriptional silencing of the Wnt-antagonist DKK1 by promoter methylation is associated with enhanced Wnt signaling in advanced multiple myeloma. PLOS One 2012; 7: e30359.
Van Den Berg DJ, Sharma AK, Bruno E, Hoffman R . Role of members of the Wnt gene family in human hematopoiesis. Blood 1998; 92: 3189–3202.
Chim CS, Pang R, Fung TK, Choi CL, Liang R . Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia 2007; 21: 2527–2536.
Jost E, Gezer D, Wilop S, Suzuki H, Herman JG, Osieka R, Galm O . Epigenetic dysregulation of secreted Frizzled-related proteins in multiple myeloma. Cancer Lett 2009; 281: 24–31.
Sanjana NE, Shalem O, Zhang F . Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014; 11: 783–784.
Winterhalter C, Widera P, Krasnogor N . JEPETTO: a Cytoscape plugin for gene set enrichment and topological analysis based on interaction networks. Bioinformatics 2014; 30: 1029–1030.
Acknowledgements
This study was supported by grants from the Dutch Cancer Society and EU-FP7 OVER-MYR.
Author contributions
HvA and KAK performed most in vitro experiments, analyzed the data, designed the figures and wrote the paper; AH-K provided technical assistance. CHM performed aCGH; AB, MvD and MP provided micro-array data and performed statistical analysis; PS supervised AB and MvD and reviewed the manuscript. MM provided CYLD reagents and expertize and reviewed the manuscript; MJK provided patient samples and reviewed the manuscript. MS and STP designed the research, supervised the study, analyzed the data. STP wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
About this article

{kind=link}
Cite this article
van Andel, H., Kocemba, K., de Haan-Kramer, A. et al. Loss of CYLD expression unleashes Wnt signaling in multiple myeloma and is associated with aggressive disease. Oncogene 36, 2105–2115 (2017). https://doi.org/10.1038/onc.2016.368
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.368
- Springer Nature Limited
This article is cited by
-
MALT1-dependent cleavage of CYLD promotes NF-κB signaling and growth of aggressive B-cell receptor-dependent lymphomas
Blood Cancer Journal (2023)
-
Down-regulation of cylindromatosis protein phosphorylation by BTK inhibitor promotes apoptosis of non-GCB-diffuse large B-cell lymphoma
Cancer Cell International (2021)
-
Deubiquitinases in hematological malignancies
Biomarker Research (2021)
-
MicroRNAs as the critical regulators of cisplatin resistance in gastric tumor cells
Genes and Environment (2021)
-
Reversal of CYLD phosphorylation as a novel therapeutic approach for adult T-cell leukemia/lymphoma (ATLL)
Cell Death & Disease (2020)