
Abstract
Long-term survival remains low for most patients with glioblastoma (GBM), which reveals the need for markers of disease outcome and novel therapeutic targets. We describe that ODZ1 (also known as TENM1), a type II transmembrane protein involved in fetal brain development, plays a crucial role in the invasion of GBM cells. Differentiation of glioblastoma stem-like cells drives the nuclear translocation of an intracellular fragment of ODZ1 through proteolytic cleavage by signal peptide peptidase-like 2a. The intracellular fragment of ODZ1 promotes cytoskeletal remodelling of GBM cells and invasion of the surrounding environment both in vitro and in vivo. Absence of ODZ1 by gene deletion or downregulation of ODZ1 by small interfering RNAs drastically reduces the invasive capacity of GBM cells. This activity is mediated by an ODZ1-triggered transcriptional pathway, through the E-box binding Myc protein, that promotes the expression and activation of Ras homolog family member A (RhoA) and subsequent activation of Rho-associated, coiled-coil containing protein kinase (ROCK). Overexpression of ODZ1 in GBM cells reduced survival of xenografted mice. Consistently, analysis of 122 GBM tumour samples revealed that the number of ODZ1-positive cells inversely correlated with overall and progression-free survival. Our findings establish a novel marker of invading GBM cells and consequently a potential marker of disease progression and a therapeutic target in GBM.
Similar content being viewed by others


Introduction
Glioblastoma (GBM) is the most common brain tumour in adults and is associated with reduced life expectancy, ranging between 12 and 15 months.1 This aggressiveness is mostly due to the rapid growth and the invasive capacity of tumour cells. Even after complete resection of the tumour, local invasiveness eventually leads to regrowth of a recurrent tumour.2 Current therapeutic regimens do not adequately address the disseminated disease burden and rapid growth associated with infiltrative GBMs. Thus, there is an urgent need to develop novel treatments to specifically target the invasive and proliferative capacities of this tumour. Gene expression profiling of GBMs identified molecular subclasses with prognostic value that were designated proneural, proliferative and mesenchymal.3 Kaplan–Meier plots showed that median survival of the proneural subclass was markedly longer than any of the other two subtypes. It has been demonstrated that GBM contains hierarchies with highly tumorigenic cells that display stem cell features.4 These GBM stem-like cells (GSCs) are governed by molecular mechanisms active in brain development, including Notch, Wnt, BMP, TGFβ and receptor tyrosine kinase pathways.5 Moreover, there are many examples of genes that play essential roles in embryonic development and are also involved in promoting or facilitating cancer in adult tissues.6, 7, 8 As a representative example, the Hedgehog family of proteins plays an instructional role during the development of many metazoans and is implicated in stem cell maintenance and tissue repair, but also confers growth promoting and survival capabilities to cancer cells.9 Teneurins are philogenetically conserved type II transmembrane proteins.10 ODZ1 (Teneurin-1, TNM1), which is located on the X chromosome, is mainly expressed in the brain during the embryonic development.11 This transmembrane protein has an intracellular region with two nuclear localization signals, which may exert transcriptional regulation functions.12, 13 So far, expression analyses by using a network of differentially expressed genes or oligo-based DNA arrays, have shown increased ODZ1 levels in prolactin pituitary tumour metastasis14 and papillary thyroid carcinoma.15 However, no direct evidence exist that ODZ1 is involved in any process aimed at inducing tumorogenesis or facilitating tumour progression. The Drosophila ortholog of ODZ1 interacts with α-spectrin, a cytoskeleton protein that binds to filamentous F-actin, suggesting a mechanism whereby ODZ1 organize the cytoskeleton.16 Recently, we showed that activation of Rho GTPases Rac and RhoA contributed to the invasive capacity of a subpopulation of GSCs isolated from the peritumoral tissue.17 Rho GTPases are key regulators of cytoskeleton dynamics and cell polarity, cell cycle progression, cell migration and metastasis,18 which indicates their potential use as therapeutic targets in cancer. Although there is little understanding on the activity of RhoA in primary GSCs, reduced activation of this GTPase correlates with decreased invasive capacity of GSCs.19, 20 Targeting regulators of cytoskeleton dynamics and invasion might provide effective therapeutic opportunities in GBM.
In the current study, we found that ODZ1 is needed for GBM cells to migrate and invade the surrounding environment. Xenograft animal models and tumour specimens from GBM patients indicate that the presence of ODZ1 increases the spreading and growth of the tumour and reduces survival. We also showed that ODZ1 exerts these tumour-facilitating activities by inducing the expression of RhoA and activation of downstream ROCK kinases.
Results
Identification of ODZ1-deficient GSCs
We found that two GSC samples, G104 and G59, did not show the typical morphological changes when they were induced to differentiate (adhesion to the substrate, cytoplasmic projections) but remained forming neurospheres (Figure 1a). However, the expression pattern of differentiation was indistinguishable from other GSC cultures, as determined by analysing the levels of both GSC and lineage markers (Supplementary Figures S1 and S2). GSCs and GBM tissue from the G104 sample carried a 6 Mb deletion of the long arm of chromosome X that included four genes, SH2D1A, ODZ1, SMARCA1 and BCORL (Figures 1b–d). Among those, ODZ1 appeared to be the most interesting gene because it has been associated with cytoskeleton organization and cell shape. Data from the cBioPortal for cancer genomics21, 22 revealed a frequency of ODZ1 gene deletion lower than 1 % in GBM samples. Protein expression analyses confirmed the lack of ODZ1 in G104 but also showed very low levels of ODZ1 in G59 cells (Figure 1e). Since ODZ1 gene is not deleted in G59 GSCs (Figure 1d), we studied a potential repression by promoter methylation. Treatment of G59 with the demethylating agent 5-aza-2'-deoxycytidine increased more than threefold the expression of ODZ1 (Figure 1f). Although we did not find a canonical CpG island in the ODZ1 promoter, there were three CpG dinucleotides within a fragment of 60 nucleotides located 344 bp upstream of the transcription start site. Analysis of the methylation state at each CpG site in G59 and G63 cells (see Figure 1e for comparing levels of ODZ1) revealed an inverse correlation between the level of methylation and the expression of ODZ1 (Supplementary Figure S3). Thus, it is likely that methylation contributes to downregulation of ODZ1 in these cells. Unless otherwise indicated, G104 and G59 cells, named here as ODZ1-deficient GSCs, were used as cell models. Results shown correspond to G104 and were all confirmed in G59 cells.

Transfection of ODZ1-deficient GSCs with ODZ1 restores the morphology of differentiating cells. (a) Neurospheres were disgregated and cultured in the presence of FBS. Morphological changes of differentiating cells were visualized after 4 days of culture. Scale bar: 30 μm. (b) Representation of the X chromosome showing the size of the deleted genomic fragment and the deleted genes in G104 GSCs. (c) Amplification of genes included in the chromosomal deletion found in G104 cells. Actin is included as a positive amplification control. (d) Amplification of different exons of the ODZ1 gene from GSC-derived genomic DNA. Stag2 was used as a positive control of amplification. (e) ODZ1 protein expression in undifferentiated GSC samples. α-Tubulin was used to assure equal loading. (f) G59 GSCs were treated with the DNA demethylating agent 5-aza-2'-deoxycytidine (3 μM 5-Aza) for 3 days and the ODZ1 expression was determined by quantitative RT-PCR. Histograms show the mean±s.d. *P<0.01, Student’s t-test. Data representative of three separate experiments. (g) ODZ1 protein levels in ODZ1-transfected GSCs. The levels of α-tubulin were analysed to assure equal loading. Transfected cells were cultured in differentiation medium and 4 days later their morphology (h) and the distribution of F-actin (i) were assessed by using phase contrast or fluorescence microscopy, respectively. (h, Scale bar: 50 μm; i, Scale bar: 5 μm). (j) GSC transfectants cultured in differentiation medium were counted as a function of their morphology. NP, no protrusions; SP, small protrusions; LP, long protrusions. Histograms show the mean±s.d. *P<0.01, **P<0.05, Student’s t-test. Data representative of three separate experiments counting at least 100 cells per condition each time.
ODZ1 expression promoted spreading of GSCs in vitro and in vivo
Comparison between ODZ1-deficient cells before and after transfection with ODZ1 (Figure 1g) by gene expression microarrays revealed that cell morphology and cellular assembly and organization are among the top network functions associated with the expression of ODZ1 (Supplementary Table S1). Interestingly, ODZ1-transfected cells recovered the phenotypic features of differentiated cells, showing adhesion to the substrate and protrusion formation with F-actin location at the edges of projections (Figures 1h–j). Similar pattern was observed in cells cultured on a laminin-coated surface (Supplementary Figure S4a). We also observed that cells were able to migrate out of neurosphere when they expressed ODZ1 (Supplementary Figure S4b). Then, we transfected ODZ1-expressing GSCs with shRNAs targeting this gene. One of them, shRNA-2, significantly downregulated the endogenous levels of ODZ1 protein (Figures 2a and b) and promoted a sixfold increase in the number of non-attached cells that formed neurospheres (Figures 2c and d). Similar effects were observed with the less efficient shRNA-3 (data not shown). Moreover, GSCs with downregulated levels of ODZ1 tended to be less dispersed and more aggregated when injected into chicken embryos (Figure 2e). Conversely, ODZ1-deficient cells transfected with ODZ1 acquired the ability to propagate through the surrounding tissue in the embryo (Figure 2f). Consistent with this model, analysis of ODZ1 protein expression in tumour samples from GBM patients revealed the increase of ODZ1 levels in the invading area (Figures 2g and h). When sections of paraffin-embeded neurospheres were immunostained with anti-ODZ1, the staining was mainly localized in cells at the periphery of neurospheres (Figure 2i) likely revealing those cells with higher migratory capacity. Overall, these data strongly suggest that ODZ1 expression enables GBM cells to invade the surrounding environment.

ODZ1 expression is sufficient to induce cell spreading in vitro and in vivo. (a,b) Western blot and quantification of ODZ1 after transfection of G63 GSCs with specific shRNAs. The levels of α-tubulin were analysed to assure equal loading. *P<0.001, Student’s t-test. Data representative of three separate experiments. (c) GSCs were transfected with ODZ1-specific shRNA (sh-2) and the number of non-attached living cells were quantitated. sh-C, irrelevant shRNA. Histograms show the mean±s.d. *P<0.001, Student’s t-test. Data representative of three independent experiments. (d) GSCs transfected with ODZ1-specific shRNA-2 and EGFP formed neurospheres under differentiation conditions. (e) CFDA SE-labeled GSCs with downregulated levels of ODZ1 were injected into the chicken embryo limb and 24 h later cell spreading was determined by confocal microscopy. (f) ODZ1-deficient GSCs were transfected with ODZ1 or empty vector and xenografted in chicken embryo as above. Dotted lines delimitate the area of tumour cell spreading. Paraffin sections from ODZ1-deficient (g) and ODZ1-expressing (h) GBM tissue specimens were immunostained with anti-ODZ1 antibodies. The following histological areas were identified: the tumour core, with many ODZ1(-) cells; the tumour border, with a similar cellular density but a higher proportion of ODZ1(+) cells and the infiltrated parenchyma, with low cellularity but most cells showing a faint staining for ODZ1. (i) Sections of paraffin-embeded neurospheres were immunostained with anti-ODZ1. Note that immunostaining is mainly localized at the periphery of neurospheres.
ODZ1 expression in GBM tumours reduced survival in patients and xenografted mice
RG1 cell line is highly efficient in promoting rapid development of GBM tumours in xenografted mice,23 and we detected low expression levels of ODZ1 (Figure 3a). ODZ1-transfected RG1 cells promoted larger tumours than their control counterparts (Figure 3b), and the survival of these animals was significantly reduced (Figure 3c). In a second model, ODZ1-deficient GSCs were transfected with either the entire ODZ1 (about 300 kDa) or its cytoplasmic fragment (Figures 3d and e), the 45 kDa N-terminal region of the protein (icODZ1). We showed that icODZ1 was sufficient to recover the morphology of differentiated GSCs (Figure 3f). These transfectants were xenografted into the brain of immunodeficient mice. Immunohistochemical analysis of brain slices confirmed larger tumours in mice grafted with ODZ1- and icODZ1-containing cells as determined by using different markers including vimentin that specifically stained human tissue, human glial fibrillary acidic protein (GFAP) that is expressed in tumour cells, and Ki67 as a proliferation marker (Figure 3g). Then, we analysed the expression of ODZ1 in a tissue microarray that included 122 tumour samples from GBM patients (Supplementary Table S2). The anti-ODZ1 antibody that we generated clearly distinguished between tissues that express or do not express ODZ1 (Figures 3h and i). Interestingly, using 35 % as a cutoff point, we found that the number of ODZ1-positive cells was inversely correlated with overall survival and disease-free (or progression-free) survival, defined as the time from resection to the first radiological recurrence (Figures 3j and k). The increase in estimated median disease-free survival for patients with lower proportion of ODZ1-positive cells (<35 %) was about 45 % compared with patients having higher levels of positive tumour cells (7 vs 3.9 months). The increase in median overall survival was 24 % (12.5 vs 9.5 months). These results were consistent with those determined by analysing data of the Repository for Molecular Brain Neoplasia Database (Rembrandt) (Supplementary Figure S5a). Analysis of a public dataset3 revealed that grade IV gliomas (GBMs) have significantly higher ODZ1 levels than grade III gliomas (Supplementary Figure S5b). Overall, animal models and patient studies demonstrate that ODZ1 expression in GBM tumours clearly correlates with poorer survival.

ODZ1-expressing tumours are more aggressive and reduce survival. (a) ODZ1 mRNA levels in different GBM cell lines by RT-PCR. β-Actin expression was used for signal normalization. (b) Representative T2-weighted nuclear magnetic resonance images showing tumours in the brain of mice xenografted with ODZ1-transfected RG1 GBM cells. (c) Kaplan–Meier survival curves of mice harboring RG1-derived tumours with high or low levels of ODZ1. (d) Expression of ODZ1 and icODZ1 in transfected GSCs. The levels of α-tubulin were analysed to assure equal loading. (e) Schematic representation of the ODZ1 protein showing the intracellular (IC) transmembrane (TM) and extracellular (EC) parts of the protein. NLS, nuclear localization signal. (f) ODZ1-deficient cells were transfected with icODZ1 and cultured under differentiation conditions. Cell morphology was assessed after 4 days of culture. Scale bar: 50 μm. (g) ODZ1-deficient GSCs transfected with ODZ1 or icODZ1 were xenografted in mice and brain sections were obtained for immunohistochemical analysis. Note the intracranial tumour staining (brown) by using markers that reveal the presence of tumour cells. HE: hematoxylin-eosin staining. (h) Anti-ODZ1 antibodies identify negative or positive staining in GBM specimens. (i) Representative examples showing different levels of ODZ1 staining on a tissue microarray with specimens from 122 patients with GBM. Kaplan–Meier curves comparing overall survival (j) and disease-free survival (k) between two groups of patients with low and high proportion of ODZ1-positive GBM cells.
The intracellular fragment of ODZ1 is released through proteolytic cleavage following differentiation of GSCs
The signals for cleavage and the proteases that release icODZ1 still remain to be identified. As shown in Figure 4a the expression of ODZ1 mRNA increased 3 to 4-fold after differentiation, and most of the produced protein was cleaved to generate icODZ1 (Figure 4b). The increased levels of ODZ1 following differentiation was confirmed by immunofluorescence, which also revealed the nuclear translocation of icODZ1 (Figure 4c). ODZ1-deficient cells were transfected with the full-length protein and most of the ODZ1 staining was localized within the nucleus in differentiated cells (Figure 4d). Then, we treated cells with (Z-LL)2 Ketone, an inhibitor of SPP and the homologous SPP-like (SPPL) proteases, that selectively cleave type II integral membrane proteins. (Z-LL)2 Ketone blocked the nuclear localization of icODZ1, giving an ER/Golgi and cell surface staining, similar to the reported distribution of full-length ODZ1 overexpressed in HT1080 cells.12 However, L685,458, an inhibitor of γsecretase, which cleaves type I transmembrane proteins, even at a high concentration (10 μM), had no effect on ODZ1 nuclear localization (Figures 4d and e). Moreover, the SPP/SPPL inhibitor blocked ODZ1-mediated migration of GSCs in a Boyden chamber-based assay (Figure 4f). A search of our gene expression database of GSCs before and after differentiation24 revealed that SPPL2a showed the highest levels among members of this protease family and its expression was even increased in differentiated cells. Based on these data, we knockdown SPPL2a by siRNA in GSCs (Figure 4g). As shown in Figures 4h and i the nuclear staining of icODZ1 observed in control cells or after knocking down another protease such as S2P, a zinc metalloprotease, was drastically reduced in the absence of SPPL2a.

icODZ1 is translocated to the nucleus following differentiation of GSCs. (a) RNA was obtained from two GSC cultures before and after differentiation, and the expression of ODZ1 was determined by qRT-PCR. Values show the mean±s.d. *P<0.01, Student’s t-test. (b) ODZ1 protein expression in GSCs before and after differentiation. Note the increase in the levels of the intracellular fragment. α-Tubulin was analysed to assure equal loading. (c) Immunofluorescence of GSCs and differentiated GSCs with anti-ODZ1 antibodies. Scale bar, 5 μm. (d) Immunofluorescence of differentiated ODZ1-deficient cells overexpressing full-length ODZ1 in the absence or in the presence of protease inhibitors by using anti-ODZ1. Scale bar, 10 μm. NOTE: morphological changes following treatment with the inhibitor were observed in some GSC models but not in others, whereas reduction of nuclear levels of ODZ1 was always detected. (e) Quantification of the fluorescence intensities of immunofluorescence images exemplified in (d) by using ImageJ software. RFI, relative fluorescence intensity. (f) The same cells as in (d) were analysed for their capacity to migrate with or without the protease inhibitor (ZLL)2-Ketone by using a modified Boyden chamber. (g) mRNA expression levels of SPPL2a and S2P proteases following transfection with specific siRNAs. C, irrelevant siRNA. Values show the mean±s.d. *P<0.01, Student’s t–test. (h) Subcellular localization of ODZ1 in differentiated GSCs transfected with the indicated siRNAs. Scale bar, 10 μm. (i) Quantification of the fluorescence intensities of immunofluorescence images exemplified in (h).
The intracellular fragment of ODZ1 is a key mediator in promoting changes in cell shape and invasion capacity of GSCs
ODZ1-deficient GSCs were transfected with ODZ1, icODZ1 or the extracellular plus the transmembrane regions (ecODZ1) (Figure 5a). As shown in Figure 5b, both ODZ1 and icODZ1 promoted long actin-filled projections following differentiation, but cells transfected with ecODZ1 behaved as the control cells and showed little changes in morphology. icODZ1 also promotes localization of CAP/Ponsin to focal adhesions (Figure 5c), a protein that has been shown to interact with the intracellular region of ODZ112 and to reorganize F-actin at cell-extracellular matrix contacts.25 An epithelial-to-mesenchymal-transition-like programme highlighted by a T-cadherin to N-cadherin switch has been described in GBM cells associated with invasion and a worse prognosis.26 Interestingly, N-cadherin along with other mesenchymal markers such as Vimentin and Snail were upregulated whereas T-cadherin was downregulated in icODZ1-transfected GSCs (Figures 5d and e). Moreover, the acquisition of a mesenchymal gene expression pattern associated with a concomitant downregulation of genes that define the proneural GBM subtype3 DLL3, OLIG2, ASCL1 and NCAM1 (Figure 5f). Consistent with the chemoresistance associated with the mesenchymal phenotype, cells expressing icODZ1 were more resistant to temozolomide, a nitrosourea commonly used in GBM patients (Figure 5g). Wound healing assays showed that cells expressing ODZ1 or icODZ1 but not ecODZ1 efficiently repopulated the scratch area (Figures 6a and b). Moreover, invasion assays in 3D collagen matrices demonstrated that icODZ1 efficiently promoted migration of GSCs (more than sixfold increase relative to control) (Figures 6c and d). Thus, these results suggest that icODZ1 provides a migration advantage for GSCs to invade the surrounding environment.

ODZ1 promotes actin cytoskeleton remodeling and induces a chemotherapy resistant mesenchymal-like phenotype. (a) RT-PCR showing the expression of ODZ1 in ODZ1-deficient GSCs transfected the intracellular fragment (icODZ1), the extracellular plus transmembrane fragment (ecODZ1) or the entire ODZ1 with oligonucleotides specific for the ic or ec regions of the cDNA. β-Actin expression was used for signal normalization. (b) Representative confocal images of the different GSC transfectants showing F-actin staining. Scale bar, 10 μm. (c) ODZ1-deficient GSCs expressing icODZ1 accumulates the focal adhesion protein ponsin at projection tips as determined by immunofluroescence. Scale bar: 10 μm. (d) icODZ1-dependent mRNA expression levels of the epithelial-like marker T-Cadherin (T-Cad) and the mesenchymal specific markers N-Cadherin (N-Cad), Vimentin (Vim) and Snail as determined by qRT-PCR. Histograms show the mean±s.d. *P<0.01, **P<0.001, Student’s t-test. Data representative of three separate experiments. (e) N-Cadherin protein expression in transfectant GSCs. The levels of α-tubulin were analysed to assure equal loading. (f) Fold-change (FC) in the expression of genes associated with the proneural GBM subtype in icODZ1-transfected cells relative to control transfected cells. Data from the expression microarray. logFC indicates the log2 of FC. (g) icODZ1-transfected GSCs were treated with 100μM Temozolomide (TMZ) and 48 h later cell viability was determined by using Alamar Blue bioassay. Values show the mean±s.d. *P<0.001, Student’s t-test. Data representative of three independent experiments.

ODZ1 promotes cell migration and invasion. (a) Micrographs of a wound-healing assay showing the migration of different transfectant GSCs towards the scratched area. (b) Quantification of migration as percentage of wound closure (marked by the dashed lines). Histograms show the mean±s.d. *P=0.01 compared with pCMV6 control, Student’s t-test. Data representative of three separate experiments. (c) Representative images of a cell invasion assay in a 3D collagen matrix. Nuclei were stained with DAPI. (d) Quantification of the invasion assay as percentage of cells detected at 30 μm from the bottom. Histograms show the mean±s.d. *P<0.05, **P<0.001 compared with pCMV6 control, Student’s t-test. Data representative of three separate experiments.
The intracellular fragment of ODZ1 induces the transactivation of RhoA through E-box binding proteins
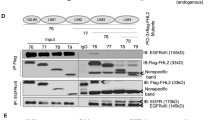
Rho GTPases, mainly Rac1, RhoA and Cdc42, regulate migration and invasion by controlling cytoskeletal dynamics.27 RhoA expression is upregulated at the mRNA and protein levels in a number of human malignancies28 and it has been associated with tumour progression.29 We showed that induced expression of icODZ1 was followed by a specific increase in the mRNA levels of RhoA but not Rac1 or Cdc42 (Figure 7a). ODZ1-deficient cells transfected with icODZ1 translocates this protein fragment to the nucleus (Figure 7b), where it may regulate transcription.12, 13 In a RhoA promoter-luciferase reporter assay, icODZ1 induced a fivefold increase in luciferase activity (Figure 7c). Total RhoA protein expression and RhoA GTPase activity were also upregulated by icODZ1 (Figures 7d and e). Activation of RhoA was further determined by analysing phosphorylation of MLC2, a downstream mediator of RhoA-ROCK signalling. As shown in Figure 7f, icODZ1 and to a lesser extent ODZ1 but not ecODZ1 increased the level of pMLC2. It has been shown that Myc induces RhoA transcription by recruiting a transcription complex to the non-canonical E-boxes E5 and E6 in the RhoA promoter.30 Chromatin immunoprecipitation (ChIP) assays showed significant increases in the binding of Myc to RhoA promoter in the presence of ODZ1 (more than twofold) and icODZ1 (fourfold) (Figure 7g). Moreover, as shown in Figure 7h, anti-Myc antibodies co-immunoprecipitated Myc and icODZ1 in cells transfected with entire ODZ1 or icODZ1, but no co-immunoprecipitation was obtained from cells transfected with ecODZ1. Luciferase reporter assays showed that overexpression of Myc increased the RhoA promoter activity (Figure 7j), confirming previous data. Transfection of ODZ1-containing cells with a Myc-specific shRNA that efficiently downregulated Myc mRNA (Figure 7i) reduced the RhoA promoter activity induced by icODZ1 (Figure 7j). In order to eliminate the risk of knocking down genes that are not intended for suppression, we have also used a Myc inhibitory mutant (dominant negative) carrying a deletion in the transactivation domain.31 Consistently, this blockade strategy resulted in reduction of RhoA promoter activity triggered by icODZ1 (Figure 7k). In support of the role of E-boxes as key promoter elements for ODZ1-induced expression of RhoA, mutation of the E6 binding site abolished activation of the RhoA promoter by icODZ1 (Figure 7l). In line with this, mobility shift assays confirmed that E6 efficiently bound E-box binding proteins and this protein-DNA complex was increased in the presence of icODZ1 (Figure 7m). These data show that icODZ1 induces the expression of RhoA through recruitment of Myc to the E6 box within the RhoA promoter.

ODZ1 triggers the transcriptional activation of RhoA. (a) The mRNA levels of Rho GTPases were analysed by qRT-PCR in GSCs transfected with an icODZ1-containing doxycycline-inducible construct. *P<0.01. (b) Nuclear localization of the intracellular fragment of ODZ1 protein after transfection, as determined by immunofluorescence. Scale bar: 5 μm. (c) ODZ1-deficient GSCs were cotransfected with a RhoA promoter-luciferase reporter construct and icODZ1 and luciferase activity was determined 48 h later. *P<0.01. (d,e) GTP-bound RhoA and total RhoA protein expression were determined in ODZ1-deficient cells transfected with icODZ1. C, empty vector; IC, icODZ1. *P<0.01, **P<0.001. (f) ODZ1 transfectants were analysed for the expression of phosphorylated MLC2 by immunofluorescence. Scale bar: 10 μm. Cell fluorescence intensities (mean±s.d.) were determined by analysing 12 cells for each of three independent experiments. (g). A ChIP assay was performed to study the ODZ1-mediated binding of Myc to the promoter of RhoA. Immunoprecipitates were analysed by qPCR using primers specific to the target site or an irrelevant region. *P<0.01. (h). ODZ1-Myc binding complexes were assessed in ODZ1-transfectants by immunoprecipitation with anti-Myc antibodies and subsequent blotting with anti-ODZ1 antibodies. The input represents the starting material. (i) Downregulation of Myc in cells transfected with shRNAs as determined by qRT-PCR. *P<0.01. (j) RhoA promoter-luciferase activity in the presence of Myc-specific shRNAs. *P<0.001. (k) RhoA promoter-luciferase activity in the presence of a dominant negative form of Myc. *P<0.001. (l) Activity of the RhoA promoter carrying mutated E5 or E6 binding sites. *P<0.02. (m) Mobility shift assay using E-boxes, E5 and E6 from the RhoA promoter, as DNA probes. All histograms show the mean±s.d., and statistical significance is determined by using the Student’s t-test. All data are representative of three separate experiments.
RhoA-ROCK pathway mediates the invasive and proliferative activities of ODZ1
The presence of ROCK inhibitor H1152 gave rise to a 4-fold decrease in the invasion capacity of icODZ1-expressing cells (Figures 8a and b). Moreover, we also analysed the localization of Ponsin at focal adhesions, as a marker of cell-matrix contacts, in the presence of H1152, a RhoA inhibitor or by using siRNAs against ROCK1 and ROCK2 (Supplementary Figure S6a-d). In all cases, Ponsin accumulation to projection tips was efficiently blocked by the inhibition strategies.

ODZ1 promotes invasion and proliferation through RhoA-ROCK axis. (a) Invasion assay of GSC transfectants in a 3D collagen matrix with or without ROCK inhibitor H1152. DAPI-stained cells were visualized by confocal microscopy at different focal planes. (b) Quantification of the invasion assay as percentage of cells detected at 20 μm from the bottom. Histograms show the mean±s.d. *P=0.0005, Student’s t-test. Data representative of three separate experiments. (c) GSC transfectants were cultured with or without H1152 and proliferation was assessed by Alamar Blue assay. Histograms show the mean±s.d. *P=0.001 compared with untreated control, Student’s t-test. Data representative of three independent experiments. (d) GSC transfectants were cultured with or without Rho inhibitor I and proliferation was assessed as above. Histograms show the mean values±s.d. *P=0.005 compared with untreated control, Student’s t-test. Data representative of three separate experiments. (e) Gene expression microarray data showing the fold change of CDKN/Cip family of CDK inhibitors in ODZ1-transfected cells compared with control cells. Dashed line marks the fold change cut-off (log2FC>0.5) used in the analysis.
We also described that the proliferation rate of icODZ1-expressing GSCs was about twice higher than control or ecODZ1-transfected cells. ODZ1 also promoted a clear increase in proliferation although at a slightly lower level than icODZ1 (Figure 8c). Consistent with our previous data, ROCK inhibitor reduced the proliferation promoted by icODZ1 and ODZ1 but had no effect on cells expressing ecODZ1 (Figure 8c). This result was confirmed by using a Rho inhibitor which significantly reduced ODZ1-mediated proliferation (Figure 8d). RhoA activation is crucial for the cell cycle G1-S progression through the regulation of CDKN/Cip family of CDK inhibitors. Our expression microarray data analysis showed that among members of this family CDKN1A (p21) and CDKN2C (p18) were downregulated in ODZ1-transfected cells (Figure 8e) which are both known to be modulated by RhoA.32 These results indicate that RhoA-ROCK axis plays a key role in mediating key activities of ODZ1, cell invasion and proliferation, in GSCs.
Discussion
We have focussed this work on ODZ1 gene, which is predominantly expressed in the developing brain. Two GSC samples with no or very little expression of ODZ1 were used as ODZ1-deficient GSCs in this work. GSCs grow in suspension forming neurospheres and in the presence of serum, they differentiate to proliferating intermediate precursors due, at least in part, to activation of the NFκB pathway24. Morphological changes are driven by cytoskeletal reorganization, which is a key feature of cell migration. We showed that ODZ1 promotes actin cytoskeletal remodelling, migration and invasion of GSCs as determined by using 2D and 3D in vitro systems, a xenograft model of chicken embryo and tumour specimens, in which ODZ1-expressing GSCs efficiently invade the surrounding environment. Previous work in Drosophila suggested that the ODZ1 homolog Ten-m is involved in remodelling of the postsynaptic cytoskeleton, physically linking the synaptic membrane to the cytoskeleton.16 Moreover, it has been described that chicken ODZ1 interacts with ponsin, which in turn binds to vinculin that could anchor the intracellular region of ODZ1 to the actin cytoskeleton.12 Consistently, we found that ODZ1 promoted long actin-filled protrusions and the localization of ponsin at projection tips in GSCs, which strengthen the role of ODZ1 as a regulator of cytoskeletal remodelling. Actin cytoskeleton has key roles in cell cycle progression33. In line with this, ODZ1 increases the proliferative capacity of GSCs. Invasiveness and proliferation are two hallmarks of GBM and strategies aimed at targeting pathways that control these processes, mainly in GSCs, are the focus of intensive research34,35,36. Rho GTPases are key players in controlling cytoskeleton dynamics, cell migration and cell division.18 Elevated levels of RhoA have been described in clinical samples of high-grade gliomas37. Interestingly, the intracellular fragment of ODZ1 (icODZ1) is able to induce the expression and activation of RhoA in GSCs. We found that icODZ1 is released following differentiation of GSCs by a proteolytic mechanism triggered by SPPL2a, a type II transmembrane protein-specific protease. SPPL2a is overexpressed in GBM compared with other brain tumours including oligodendrogliomas and low grade astrocytomas, as determined by searching the Rembrandt database. Consistent with the role of SPPL2a in ODZ1 processing, this protease has been shown to release the intracellular FasL domain, which translocates to the nucleus and regulates gene transcription38. Myc is a well- known oncoprotein that control the expression of a variety of genes involved in cell proliferation and differentiation39, and it has been described that Myc stabilization may be linked to the pathogenesis of GBM40. Our data strengthen the role of Myc in GBM by showing that icODZ1 binds to Myc and recruits this transcription factor to an E-box site in the promoter region of RhoA, which is in line with previous results where it is demonstrated that Myc induces RhoA transcription by recruiting a transcription complex to E-boxes within the RhoA promoter.30 By using different inhibition strategies, we demonstrated that RhoA signalling mediates the cytoskeletal remodelling and the increase in cell migration, invasion and proliferation promoted by ODZ1. In line with our data, high levels of RhoA activity have been associated with protrusion formation in different cancer models41,42. The relevance of Rho-ROCK pathway in promoting migration and growth of GBM cells has been mostly described by using established cell lines43,44. However, other models have strengthened this functional correlation. Although it has been described that invading GBM cells have low RhoA activity by using a rat GBM cell line45, many studies using primary human tumour cells support the key role of RhoA to provide an invasive phenotype to GBM cells46,47. Moreover, activation of RhoA in neural stem cells has also been shown to be key for cell migration and invasion48. Our data provide a novel ODZ1-RhoA-ROCK axis for therapeutic strategies against a pathway controlling key activities in GSCs. Blockade of RhoA expression or activation through its upstream regulator ODZ1 has the advantage that this protein is located at the cell membrane, which facilitates inhibition strategies with small molecules or antibodies. Additionally, contrary to other integral membrane proteins associated with GBM such as EGFR that is widely expressed in a number of adult tissues, or RhoA which is ubiquitously expressed across tissues, ODZ1 expression is mostly restricted to fetal brain, which could make ODZ1-targeted therapies more tumour-specific limiting their side effects.
In summary, our study shows for the first time that ODZ1, a protein that participates in the embryonic development of the brain, is also involved in cancer progression by promoting the growth and invasion capabilities of GSCs via a transcriptional pathway that induces the expression of RhoA and activation of downstream ROCK. Thus, these data provide a novel and promising target to develop therapeutic strategies aimed at blocking the two features that make GBM so aggressive and lethal.
Materials and methods
Primary cell cultures
Cells were maintained as neurospheres in serum-free DMEM/F12 medium (Invitrogen, Carlsbad, CA, USA) as previously described,24 and plated at a density of 3 × 106 live cells/60-mm plate. Neurospheres were dissociated every 4–5 days to facilitate cell growth. Cells were used between passages 10 and 20. To promote differentiation, neurospheres were cultured in the same medium but in the presence of 10 % FCS for 4 days. Tumour samples were obtained from patients after informed written consent had been given, as approved by the Research Ethics Board at the Valdecilla Hospital. Established GBM cell line RG1 (L0627), kindly provided by Dr Rosella Galli, was cultured as previously described.23 Cell proliferation and cell survival were evaluated with Alamar Blue bioassay (Life Technologies, Paisley, UK). Unless otherwise indicated, experiments were performed with undifferentiated GSCs. All cells used in this work were mycoplasma-free.
When indicated, cells were treated with 5 μM ROCK inhibitor H1152 (Tocris Bioscience, Bristol, UK), 1 μg/ml Rho Inhibitor I (Cytoskeleton, Inc., Denver, CO), 100 μM Temozolomide (Merck, Whitehouse Station, NJ, USA), 50 μM (Z-LL)2-Ketone (Calbiochem, MA, USA) or 10 μM L685,458 (Tocris Bioscience, Bristol, UK).
DNA copy number changes were evaluated using Affymetrix GeneChip 250-NspI/StyI SNP microarrays as previously described.17 DNA methylation was analysed by a pyrosequencing method49, which accurately measures the degree of methylation at several CpGs in close proximity with high quantitative resolution.
In vivo models
Tumour xenografts in chicken and mice50 were established as previously described. Stereotactically guided intracranial injections in athymic Nude-Foxn-1nu male mice, aged 8–9 weeks, were performed injecting 50.000 cells resuspended in 2 μl of culture medium. Survival of mice was analysed by the Kaplan–Meier method. No randomization was used. Images in chicken embryos are the integration of all Z-stacks taken along the dorsal-ventral plane at 15 μm intervals to cover the whole limb. The model of tumour xenografts in mice was reviewed and approved by the Research Ethics and Animal Welfare Committee at the Instituto de Salud Carlos III, Madrid. Magnetic resonance imaging analysis was performed in mice injected IP with Gd-DOTA (Dotarem, Bloomington, IN, USA) by using a 4.7 TBiospec BMT 47/40 spectrometer (Bruker, Billerica, MA, USA). No blinding experiments were done.
Immunofluorescence analysis
GSCs were grown on glass cover slips previously coated with 10 μg/ml fibronectin (Sigma-Aldrich, St Louis, MO, USA), or 10 μg/ml laminin (Sigma-Aldrich). Cells were incubated with antibodies against phospho-myosin light chain 2 (pMLC2) (#3671, Cell Signaling, Danvers, MA, USA), Ponsin (SC-25496, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Sox2 (MAB2018, R&D Systems, Minneapolis, MN, USA), GFAP (Z0334, DAKO, Glostrup, Denmark) or Tuj1 (T-8660, Sigma-Aldrich). Then, the cells were incubated with Texas red-conjugated or fluorescein isothiocyanate-conjugated goat anti-rabbit secondary antibodies (#111-035-144, Jackson ImmunoResearch, Cambridgeshire, UK). Actin cytoskeleton was stained with tetramethylrhodamine isothiocyanate- or fluorescein isothiocyanate-conjugated Phalloidin (Sigma-Aldrich) and nuclei visualized with 4',6-diamidino-2-phenylindole (Life Technologies). Confocal images were taken with a Ti-Eclipse microscope (Nikon, Tokyo, Japan). When indicated, fluorescence intensity was determined by measuring pixel intensity in a defined area (nucleus or the entire cell) using ImageJ software. Single plane confocal images were acquired with the same settings for each experiment.
Tissue microarray
Demographic, clinical and therapeutic data of patients included in the tissue microarray are presented in Supplementary Table S2. Replicate formalin-fixed paraffin-embedded tissue samples of representative tumour regions from 122 patients with GBM were collected for the preparation of four tissue microarrays. No samples were excluded from the analysis. Tissue microarrays were reviewed by an expert pathologist (NTV) blinded to all sample identifiers. A semiquantitative scoring system for the percentage of positive cells was used. Patients were divided into two groups of low (<35 %) and high (>35 %) number of ODZ1-positive cells. Differences in overall and disease-free survival between both groups were estimated by a Kaplan–Meier survival plot.
Immunohistochemical staining
Vibratome sections (200 μm thick) of mouse brains were paraffin-embedded and then consecutive semithin sections were processed and incubated with primary antibodies against Vimentin, GFAP and Ki67 (M7248, DAKO) followed by horseradish peroxidase-conjugated secondary antibodies (K4065, K5007, DAKO) and visualized by a chromogen or stained with hematoxilin-eosin. Slides containing tumour tissue from GBM patients or neurospheres from GSC cultures embedded in paraffin were stained with antibodies against the N-terminal region of ODZ1.
Expression analyses
To assess the expression of individual genes, a cDNA was generated and amplified using the following primers: CD133, Tuj1, GFAP, β-Actin (16),T-Cadherin (5′TTCTGTGCGTTCTCCTGTCC3′ and 5′TCTCAGAGCAACTAAGCCGC3′),N-Cadherin (5′GACAATGCCCCTCAAGTGTT3′ and 5′CCATTAAGCCGAGTGATGGT3′), Vimentin (5′TCGGCGGGACAGCAGG3′ and 5′GGTGGACGTAGTCACGTAGC3′), Snail (5′TAGCGAGTGGTTCTTCTGCG3′ and 5′AGGGCTGCTGGAAGGTAAAC3′), RhoA (5′ACCCGCCTTCGTCTCCGAGT3′ and 5′GGCAGCCATTGCTCAGGCAAC3′), Rac1 (5′AGGCCATCAAGTGTGTGGTG3′ and 5′CTTGTCCAGCTGTGTCCCAT3′), Cdc42 (5′GCCTATTACTCCAGAGACTGC3′ and 5′GTTCATAGCAGCACACACCTG3′), icODZ1 (5′ACTCAAGAGATGGAATTCTGTG3′ and 5′CTTAGTGCATGGTCAGGTG3′), ecODZ1 (5′ACAATGATGGACGGTGCCTT3′ and 5′GTGTCCCTCCCCTCTATGGT3′) ODZ1 exon 1 (5′GGACCAATTGTGAATCTGCC3′ and 5′CCTACAACCTCAGCTGGGC3′), ODZ1 exon 20 (5′GGGAAGGTTTTGCAGGC3′ and 5′CCACTGTGCTAGAGGCTGG3′), ODZ1 exon 32 (5′CTTGCAAGCCTGTCCTTTCC3′ and 5′CCCAGTGTTACCGATGAGC3′), Stag2 (5′CACGCCTGGCTAATTTTTGT3′ and 5′CAATACAGGGCAGGTGTGCT3′), S2P (5′GTTGCTGAGGACTCTCCTGC3′ and 5′AGCACACATCTGTGAGGCTG3′), SPPL2a (5′ATGAAGACAGGTGGGCTTGG3′ and 5′AGCTGCGAGTTCAACCATGA3′), SMARCA1 (5′CGTGGTCATAGAGGACGAGC3′ and 5’TAGGCGCTTTAGCAGCAAGT3’), BCORL1 (5′GCATGTGTGGCATCAACGAG3′ and 5′GGGATCGTCAGGCTTGTCTT3′), SH2D1A (5′TACCGAGTGTCCCAGACAGA3′ and 5′TCAGGATCTTCTCTTATCCCTGT3′). Quantitative real-time PCR was performed in a 7000 sequence detection system (Life Technologies).
Transfections, gene silencing and gene reporter assays
ODZ1-deficient GSCs were transfected with the entire ODZ1 cDNA, the extracellular (plus transmembrane) fragment (both from Origene, Rockville, MD, USA) or the intracellular fragment, that was PCR-amplified from ODZ1 cDNA and cloned into pCMV6. Stable transfectants were selected with 500 μg/ml geneticin (Life Technologies). icODZ1 was subcloned in a tetracycline-regulated retroviral vector.51 Cells were transiently transfected with this vector and the expression of ODZ1 was induced in the presence of 2 μg/ml doxycycline (Sigma-Aldrich).
GSCs were transfected with ODZ1-specific shRNAs (Thermo Fisher Scientific, Waltham, MA, USA) by using nucleofection or with ROCK1, ROCK2, S2P or SPPL2a siRNAs (Dharmacon’s SmartPools, Thermo Fisher Scientific) by Lipofectamine RNAiMax (Life Technologies). Knockdown of Myc was achieved by using specific shRNAs cloned in lentiviral pLKO (Sigma-Aldrich) and retroviral pRS52 vectors. For functional inactivation of Myc, a construct containing mutant c-Myc (pMLV-D106-143) that carries a deletion in the transactivation domain31 was used.
Cells were co-transfected with 1 μg of human RhoA promoter cloned into pGL3 vector30 and 50 ng of pRSV-β-galactosidase by nucleofection. When indicated, site-directed mutagenesis of E5 and E6 boxes, CA(C>A)(G>T)CG, of the RhoA promoter was conducted by using the QuickChange mutagenesis kit (Stratagene, La Jolla, CA, USA) as previously described53.
Expression microarray
Total RNA from empty vector- and ODZ1-transfected cells was extracted using the RNeasy mini kit (Qiagen, Valencia, CA, USA) and microarray gene expression analysis was performed with the Human Genome U133 Plus 2.0 array (Affymetrix, Santa Clara, CA, USA). The selection of those genes differentially expressed was performed using a criteria based on the fold-change value. Probe sets were selected as significant using a logFC cut-off of 1.5. The raw data have been deposited in a MIAME compliant database (GEO accession number, GSE65526).
Chromatin Immunoprecipitation (ChIP)
Purification of sonicated nuclear lysates and immunoprecipitation were performed as described. Precipitates using anti-Myc (SC-40, Santa Cruz Biotechnology) or irrelevant IgG were heated at 65 °C to reverse the cross-linking. Quantitative PCR was performed using primers (5′CGTGAAGAGTTGGCAGTTCG3′ and 5′ACGCCCTAAAAGCAAAACC3′) specific for a region (187 bp) of the RhoA promoter containing the functional cMyc binding sites,30 or primers (5′CAACCGACCAGTCACATCC3′ and 5′ATGTTGTTGAGGGCTTCCAG3′) that generate a fragment from exon 5 of SKP2, used as a negative control.
Electrophoretic mobility shift assay
Nuclear extracts (10 μg of total protein) were incubated with a 32p-labelled double-stranded DNA probe corresponding to E-boxes E5 (5′CCGCGCACGCGCACCTAA3′) and E6 (5′ATGCCCCACGCGGCTGCA3′). Samples were run on a 5 % non-denaturing polyacrylamide gel. Gels were dried and visualized by autoradiography.
Western blot, immunoprecipitation and pulldown assays
Total protein extracts were separated on 8 or 12 % polyacrilamide gels or 4–20 % gradient gels (Bio-Rad, Hercules, CA, USA) and transferred to nitrocellulose. Blots were incubated with antibodies against α-Tubulin (SC-23948, Santa Cruz Biotechnology), RhoA (#2117, Cell Signaling Technology, Danvers, MA, USA), ODZ1 (AF6324, R&D Systems) and N-Cadherin (C-2542, Sigma-Aldrich) followed by incubation with secondary anti-rabbit, anti-mouse or anti-sheep antibodies conjugated to horseradish peroxidase (SC-2004, SC2005, SC-2473, Santa Cruz Biotechnology). For protein immunoprecipitation, cells were treated as previously described.53 Cleared lysates were incubated with anti-cMyc antibodies and proteinA/G conjugated to agarose beads (Santa Cruz Biotechnology). Proteins were then electrophoresed, transferred to nitrocellulose membranes and incubated with anti-ODZ1 antibodies. For pulldown experiments, glutathione S-transferase-conjugated Rhotekin was expressed from pGEX-Rhotekin and assays were performed as described.17 Protein band quantification was carried out using ImageJ software.
Migration and invasion assays
For the wound-healing assay, cells were seeded into collagen I coated 24-well plates and grown to confluence. The cell monolayers were scratched with a pipette tip. Cell migration was expressed by the percentage of wound closure. For 3D assays, cells were included in collagen-based matrices and processed as previously described.17 The effect of protease inhibitors on the migration capacity of GSCs was analysed by using a modified Boyden chamber assay in 24-well plates (Transwell, Corning Incorporated, NY, USA). Cells were placed in the upper compartment and following 24 h of incubation, migratory cells in the lower face of the membrane were fixed and stained.
Statistical analysis
All statistics were calculated with the SPSS statistical package (version 13.0). Data are presented as mean±s.d. of at least three independent experiments. Differences between groups were tested for statistical significance using the unpaired 2-tailed Student’s t-test. The significance level was set at P<0.05.
Accession codes
References
Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M et al. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res 2010; 16: 2443–2449.
Giese A, Bjerkvig R, Berens ME, Westphal M . Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol 2003; 21: 1624–1636.
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006; 9: 157–173.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al. Identification of human brain tumour initiating cells. Nature 2004; 432: 396–401.
Yan K, Yang K, Rich JN . The evolving landscape of glioblastoma stem cells. Curr Opin Neurol 2013; 26: 701–707.
Roussel MF, Robinson GW . Role of MYC in medulloblastoma. Cold Spring Harb Perspect Med 2013; 3: a014308.
Kobayashi K, Hatano M, Otaki M, Ogasawara T, Tokuhisa T . Expression of a murine homologue of the inhibitor of apoptosis protein is related to cell proliferation. Proc Nat Acad Sci USA 1999; 96: 1457–1462.
Ambrosini G, Adida C, Altieri DC . A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997; 3: 917–921.
Teglund S, Toftgard R . Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta 2010; 1805: 181–208.
Tucker RP, Beckmann J, Leachman NT, Scholer J, Chiquet-Ehrismann R . Phylogenetic analysis of the teneurins: conserved features and premetazoan ancestry. Mol Biol Evol 2012; 29: 1019–1029.
Kenzelmann D, Chiquet-Ehrismann R, Leachman NT, Tucker RP . Teneurin-1 is expressed in interconnected regions of the developing brain and is processed in vivo. BMC Dev Biol 2008; 8: 30.
Nunes SM, Ferralli J, Choi K, Brown-Luedi M, Minet AD, Chiquet-Ehrismann R . The intracellular domain of teneurin-1 interacts with MBD1 and CAP/ponsin resulting in subcellular codistribution and translocation to the nuclear matrix. Exp Cell Res 2005; 305: 122–132.
Schoeler J, Ferralli J, Thiry S, Chiquet-Ehrismann R . The intracellular domain of teneurin-1 induces the activity of transcription factor MITF by binding to transcriptional repressor HINT1. J Biol Chem 2015; 290: 8154–8165.
Zhang W, Zang Z, Song Y, Yang H, Yin Q . Co-expression network analysis of differentially expressed genes associated with metastasis in prolactin pituitary tumors. Mol Med Rep 2014; 10: 113–118.
Huang Y, Prasad M, Lemon WJ, Hampel H, Wright FA, Kornacker K et al. Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc Nat Acad Sci USA 2001; 98: 15044–15049.
Mosca TJ, Hong W, Dani VS, Favaloro V, Luo L . Trans-synaptic teneurin signalling in neuromuscular synapse organization and target choice. Nature 2012; 484: 237–241.
Ruiz-Ontanon P, Orgaz JL, Aldaz B, Elosegui-Artola A, Martino J, Berciano MT et al. Cellular plasticity confers migratory and invasive advantages to a population of glioblastoma-initiating cells that infiltrate peritumoral tissue. Stem Cells 2013; 31: 1075–1085.
Jaffe AB, Hall A . Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 2005; 21: 247–269.
Ernst A, Hofmann S, Ahmadi R, Becker N, Korshunov A, Engel F et al. Genomic and expression profiling of glioblastoma stem cell-like spheroid cultures identifies novel tumor-relevant genes associated with survival. Clin Cancer Res 2009; 15: 6541–6550.
Jin X, Sohn YW, Yin J, Kim SH, Joshi K, Nam DH et al. Blockade of EGFR signaling promotes glioma stem-like cell invasiveness by abolishing ID3-mediated inhibition of p27(KIP1) and MMP3 expression. Cancer Lett 2013; 328: 235–242.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404.
Mazzoleni S, Politi LS, Pala M, Cominelli M, Franzin A, Sergi Sergi L et al. Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res 2010; 70: 7500–7513.
Nogueira L, Ruiz-Ontanon P, Vazquez-Barquero A, Lafarga M, Berciano MT, Aldaz B et al. Blockade of the NFkappaB pathway drives differentiating glioblastoma-initiating cells into senescence both in vitro and in vivo. Oncogene 2011; 30: 3537–3548.
Zhang M, Liu J, Cheng A, Deyoung SM, Chen X, Dold LH et al. CAP interacts with cytoskeletal proteins and regulates adhesion-mediated ERK activation and motility. EMBO J 2006; 25: 5284–5293.
Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 2012; 22: 21–35.
Heasman SJ, Ridley AJ . Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 2008; 9: 690–701.
Sahai E, Marshall CJ . RHO-GTPases and cancer. Nat Rev Cancer 2002; 2: 133–142.
Kamai T, Yamanishi T, Shirataki H, Takagi K, Asami H, Ito Y et al. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin Cancer Res 2004; 10: 4799–4805.
Chan CH, Lee SW, Li CF, Wang J, Yang WL, Wu CY et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat Cell Biol 2010; 12: 457–467.
Canelles M, Delgado MD, Hyland KM, Lerga A, Richard C, Dang CV et al. Max and inhibitory c-Myc mutants induce erythroid differentiation and resistance to apoptosis in human myeloid leukemia cells. Oncogene 1997; 14: 1315–1327.
Zhang S, Tang Q, Xu F, Xue Y, Zhen Z, Deng Y et al. RhoA regulates G1-S progression of gastric cancer cells by modulation of multiple INK4 family tumor suppressors. Mol Cancer Res 2009; 7: 570–580.
Lee K, Song K . Actin dysfunction activates ERK1/2 and delays entry into mitosis in mammalian cells. Cell Cycle 2007; 6: 1487–1495.
Ying Z, Li Y, Wu J, Zhu X, Yang Y, Tian H et al. Loss of miR-204 expression enhances glioma migration and stem cell-like phenotype. Cancer Res 2013; 73: 990–999.
Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol 2012; 189: 444–453.
Sherry MM, Reeves A, Wu JK, Cochran BH . STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009; 27: 2383–2392.
Yan B, Chour HH, Peh BK, Lim C, Salto-Tellez M . RhoA protein expression correlates positively with degree of malignancy in astrocytomas. Neurosci Lett 2006; 407: 124–126.
Kirkin V, Cahuzac N, Guardiola-Serrano F, Huault S, Luckerath K, Friedmann E et al. The Fas ligand intracellular domain is released by ADAM10 and SPPL2a cleavage in T-cells. Cell Death Differ 2007; 14: 1678–1687.
Shervington A, Cruickshanks N, Wright H, Atkinson-Dell R, Lea R, Roberts G et al. Glioma: what is the role of c-Myc, hsp90 and telomerase? Mol Cell Biochem 2006; 283: 1–9.
Shindo H, Tani E, Matsumuto T, Hashimoto T, Furuyama J . Stabilization of c-myc protein in human glioma cells. Acta Neuropathol 1993; 86: 345–352.
Timpson P, McGhee EJ, Morton JP, von Kriegsheim A, Schwarz JP, Karim SA et al. Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res 2011; 71: 747–757.
Jacquemet G, Green DM, Bridgewater RE, von Kriegsheim A, Humphries MJ, Norman JC et al. RCP-driven alpha5beta1 recycling suppresses Rac and promotes RhoA activity via the RacGAP1-IQGAP1 complex. J Cell Biol 2013; 202: 917–935.
Deng L, Li G, Li R, Liu Q, He Q, Zhang J . Rho-kinase inhibitor, fasudil, suppresses glioblastoma cell line progression in vitro and in vivo. Cancer Biol Ther 2010; 9: 875–884.
Zohrabian VM, Forzani B, Chau Z, Murali R, Jhanwar-Uniyal M . Rho/ROCK and MAPK signaling pathways are involved in glioblastoma cell migration and proliferation. Anticancer Res 2009; 29: 119–123.
Hirata E, Yukinaga H, Kamioka Y, Arakawa Y, Miyamoto S, Okada T et al. in vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family GTPases in glioblastoma cell invasion. J Cell Sci 2012; 125: 858–868.
Wong SY, Ulrich TA, Deleyrolle LP, MacKay JL, Lin JM, Martuscello RT et al. Constitutive activation of myosin-dependent contractility sensitizes glioma tumor-initiating cells to mechanical inputs and reduces tissue invasion. Cancer Res 2015; 75: 1113–1122.
Wang H, Han M, Whetsell W Jr, Wang J, Rich J, Hallahan D et al. Tax-interacting protein 1 coordinates the spatiotemporal activation of Rho GTPases and regulates the infiltrative growth of human glioblastoma. Oncogene 2014; 33: 1558–1569.
Danussi C, Akavia UD, Niola F, Jovic A, Lasorella A, Pe'er D et al. RHPN2 drives mesenchymal transformation in malignant glioma by triggering RhoA activation. Cancer Res 2013; 73: 5140–5150.
Tost J, Gut IG . DNA methylation analysis by pyrosequencing. Nat Protoc 2007; 2: 2265–2275.
Pozo N, Zahonero C, Fernandez P, Linares JM, Ayuso A, Hagiwara M et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J Clin Invest 2013; 123: 2475–2487.
Watsuji T, Okamoto Y, Emi N, Katsuoka Y, Hagiwara M . Controlled gene expression with a reverse tetracycline-regulated retroviral vector (RTRV) system. Biochem Biophys Res Commun 1997; 234: 769–773.
Bernard D, Pourtier-Manzanedo A, Gil J, Beach DH . Myc confers androgen-independent prostate cancer cell growth. J Clin Invest 2003; 112: 1724–1731.
Grande L, Bretones G, Rosa-Garrido M, Garrido-Martin EM, Hernandez T, Fraile S et al. Transcription factors Sp1 and p73 control the expression of the proapoptotic protein NOXA in the response of testicular embryonal carcinoma cells to cisplatin. J Biol Chem 2012; 287: 26495–26505.
Acknowledgements
This work was supported by the Instituto de Salud Carlos III (ISCIII), grants PI13/01760 (JLF-L), PI12/00775 (PS-G) and SAF2014-53526R (JL), and program Red Temática de Investigación Cooperativa en Cáncer (RTICC) grants RD12/0036/0022 (JLF-L), RD12/0036/0027 (PS-G), RD12/0036/0063 (JAM-C), RD12/0036/0003 (AP) and RD12-0036-0033 (JL), Instituto de Investigación Valdecilla (IDIVAL) grant APG/03 to JLF-L, and Cancer Research UK C33043/A12065 and Royal Society RG110591 to VS-M. We thank R. Galli for kindly providing RG1 cell line. We also thank M.E. Fernández-Valle for helping with magnetic resonance imaging experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Talamillo, A., Grande, L., Ruiz-Ontañon, P. et al. ODZ1 allows glioblastoma to sustain invasiveness through a Myc-dependent transcriptional upregulation of RhoA. Oncogene 36, 1733–1744 (2017). https://doi.org/10.1038/onc.2016.341
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.341
- Springer Nature Limited
This article is cited by
-
Downregulation of the Rho GTPase pathway abrogates resistance to ionizing radiation in wild-type p53 glioblastoma by suppressing DNA repair mechanisms
Cell Death & Disease (2023)
-
Molecular signature of excessive female aggression: study of stressed mice with genetic inactivation of neuronal serotonin synthesis
Journal of Neural Transmission (2023)
-
Glioblastoma invasion factor ODZ1 is induced by microenvironmental signals through activation of a Stat3-dependent transcriptional pathway
Scientific Reports (2021)
-
MEX3A contributes to development and progression of glioma through regulating cell proliferation and cell migration and targeting CCL2
Cell Death & Disease (2021)
-
Alternative splicing controls teneurin-latrophilin interaction and synapse specificity by a shape-shifting mechanism
Nature Communications (2020)