
Abstract
Protein arginine methyltransferase 5 (PRMT5) complexed with MEP50/WDR77 catalyzes arginine methylation on histones and other proteins. PRMT5-MEP50 activity is elevated in cancer cells and its expression is highly correlated with poor prognosis in many human tumors. We demonstrate that PRMT5-MEP50 is essential for transcriptional regulation promoting cancer cell invasive phenotypes in lung adenocarcinoma, lung squamous cell carcinoma and breast carcinoma cancer cells. RNA-Seq transcriptome analysis demonstrated that PRMT5 and MEP50 are required to maintain expression of metastasis and Epithelial-to-mesenchymal transition (EMT) markers and to potentiate an epigenetic mechanism of the TGFβ response. We show that PRMT5-MEP50 activity both positively and negatively regulates expression of a wide range of genes. Exogenous TGFβ promotes EMT in a unique pathway of PRMT5-MEP50 catalyzed histone mono- and dimethylation of chromatin at key metastasis suppressor and EMT genes, defining a new mechanism regulating cancer invasivity. PRMT5 methylation of histone H3R2me1 induced transcriptional activation by recruitment of WDR5 and concomitant H3K4 methylation at targeted genes. In parallel, PRMT5 methylation of histone H4R3me2s suppressed transcription at distinct genomic loci. Our decoding of histone methylarginine at key genes supports a critical role for complementary PRMT5-MEP50 transcriptional activation and repression in cancer invasion pathways and in response to TGFβ stimulation and therefore orients future chemotherapeutic opportunities.
Similar content being viewed by others


Introduction
Alterations in the histone code signaling of epigenetic information is highly correlated with cancer etiology and the Epithelial-to-Mesenchymal Transition (EMT) that leads to metastasis.1, 2, 3 Arginine methylation is a significant regulator of biological function4, 5 and of oncogenesis and tumor progression.5, 6, 7 PRMT1 is responsible for greater than 90% of asymmetric dimethylarginine (Rme2a) and PRMT5 is the main writer of symmetric dimethylarginine (Rme2s), while all PRMTs catalyze monomethylarginine (Rme1).8
Histone arginine methylation is an abundant post-translational modification (PTM). H4R3me2a and H3R2me2s have been correlated with transcriptional activation, while H4R3me2s, H3R2me2a and H3R8me2s repress transcription.9 Almost no information is known about the biological role and mechanism of action of histone monomethylarginine.10, 11
PRMT5 is always complexed with the WD-repeat protein MEP50 to bind and orient substrate to the catalytic site to preferentially produce monomethylarginine.12, 13, 14, 15 Elevated PRMT5 and MEP50 are found in many solid and blood cancers and often correlated with enhanced tumor growth and poor disease prognosis.4, 16, 17, 18, 19, 20 PRMT5 inhibition or loss disrupts cancer cell phenotypes21, 22, 23 and its direct interaction with proteins commonly misregulated or mutated in cancer indicates that PRMT5 plays a role in cancer as an oncogene.4
TGFβ signaling is frequently distorted during tumor progression and metastasis.24 This complex signaling pathway is convergent with EMT through altered expression and regulation of key transcription factors.25 Probing how PRMT5 activity may regulate tumorigenesis and cancer cell fate by histone methylation and transcriptional regulation is critical to molecular dissection of the pathway for biological understanding and better treatments in the clinic. Since PRMT5 expression is correlated with EMT18 and it catalyzes histone methylations widely distributed in chromatin, we speculated that the complex web of TGFβ signaling in cancer metastasis may be mediated by its activity.
Here, we demonstrate that PRMT5-MEP50 is a critical mediator of the TGFβ-response leading to invasivity and EMT via simultaneous histone H3R2 methylation-coupled transcriptional activation and H4R3 methylation-coupled transcriptional repression.
Results
PRMT5-MEP50 and histone methylarginine in cancers and cancer cells
To systematically identify the major human cancers correlated with altered PRMT5 and MEP50 expression, we probed The Cancer Genome Atlas (TCGA) and determined that lung cancers had the most significant positive alteration in expression (Figure 1a).4, 26, 27 Elevated PRMT5 and MEP50 are found in human lung adenocarcinoma and squamous cell carcinoma compared with corresponding normal tissue samples (Figure 1b and c and Supplementary Figure S1A). Expression of genes correlated with poor patient survival of lung cancers were correlated with elevated PRMT5-MEP50 expression.28 The Type I enzymes PRMT1, PRMT3, CARM1 and PRMT6 were increased in abundance in cancer cells compared with corresponding normal cells. The Type II enzyme PRMT5 and its cofactor MEP50 were also substantially increased in abundance in cancer cells, while the other putative type II enzymes PRMT7 and PRMT9 were not. (Figure 1d). Elevated PRMT5 and MEP50 was also observed in various lung cancer cell lines (Supplementary Figure S1B).

Aberrant PRMT5-MEP50 expression and histone methylation in lung cancer cells is a target for shRNA-mediated knockdown. (a) Dotplot of Wilcoxon rank-sum test P-values for PRMT5 and MEP50 expression in cancers with data collected from the TCGA. Upregulated expression (purple) and downregulated (green) are shown in dots scaled by–log(p). Corresponding cancer cell lines probed in this study are bolded. (b) Heatmap of gene expression of PRMT5 and MEP50 from TCGA lung cancer RNA-Seq data in normal and patients with lung adenocarcinoma (LUAD) or squamous cell carcinoma (LUSC), ranked by sample type (tumor or normal tissue) and then Shedden poor survival markers showing substantially increased expression of PRMT5 and MEP50 relative to normal tissue and poor survival markers. (c) Dot-plot showing distribution of TCGA individual sample data for LUAD or LUSC; mean FPKM expression RNA-Seq value shown by black bar; Comparison between tumor and normal tissue expression P<0.001 from Mann-Whitney Rank Sum Test. (d) Whole cell lysate immunoblots for PRMTs and MEP50 in the normal and corresponding cancer cell lines. GAPDH is a loading control. (e) Whole cell lysate and chromatin extracted from IMR90 and A549 probed with indicated antibodies. GAPDH and H3 are controls for lysate and chromatin immunoblots, respectively. (f) Immunoblots for PRMT5 or MEP50 from A549 cells expressing shRNA targeted against GFP as a control (GFPkd), against PRMT5 (PRMT5kd) or against MEP50 (MEP50kd). (g) Representative immunofluorescence images for PRMT5 (red) or MEP50 (red) and counterstained with DAPI (blue) from GFPkd, PRMT5kd and MEP50kd A549 cells demonstrating primarily cytoplasmic localization. Scale bar, 50 μm. (h) Blue-native gel immunoblots for PRMT5 or MEP50 from knockdown A549 cells as in E. Native Molecular mass markers are indicated. Recombinant human PRMT5-MEP50 complex (HsPRMT5-MEP50) or SDS/boiled HsPRMT5-MEP50 with SDS (lanes 1 and 2) are used for identification of intact complex (indicated) or free PRMT5 and MEP50, respectively.
We probed IMR90 lung fibroblasts and A549 extracts for PRMTs and their methylated substrates. A549 cells had elevated PRMT5 and MEP50 compared with IMR90 cells and also had increased H3R2me2s and H2A/H4 R3me2s, PRMT5-catalyzed histone PTMs previously shown to up- and down-regulate transcription, respectively (Figure 1e). We did not observe any difference in methylation of SmD3/SNRDP3 (methylated product recognized by the SYM10 antibody).
We used shRNA to independently knockdown PRMT5 and MEP50 in A549 lung adenocarcinoma cells (Figure 1f, roughly 70% protein loss; PRMT5kd, MEP50kd and control shRNA directed against eGFP, denoted GFPkd). We could not derive complete knockdown lines, consistent with an essential role for the complex in cell viability (Supplementary Figure S1C). PRMT5 knockdown caused loss of MEP50 expression while MEP50 knockdown caused PRMT5 loss, confirming the obligate pairing of these proteins.12, 13 Both proteins were found primarily in the cytoplasm (Figure 1g). Knockdown of PRMT5 did not result in free MEP50 nor did MEP50 knockdown result in free PRMT5 or other subcomplexes (Figure 1h).
PRMT5-MEP50 significantly alters the Transcriptome
Since PRMT5-MEP50 methylates histones, we employed RNA-Seq to probe the transcriptomes of the knockdown cell lines, in three biological replicates. We tested compensatory expression of other PRMTs by comparing FPKM of the PRMT genes from each replicate, validating the targeted knockdown of PRMT5 and MEP50 (Figure 2a). We measured the differentially expressed genes in these cells using the overlap of DESeq2 and edgeR output (Figure 2b, padj <0.01) and confirmed that the replicates of the PRMT5 and MEP50 knockdown RNA-Seq are highly correlated (DESeq2 clustering, Supplementary Figure S2A). 1604 genes were greater than 50% altered in expression (log2>|0.6|) for PRMT5kd while 1738 genes were similarly altered in the MEP50kd, with roughly equal distribution of up- and down-regulation. Most altered genes were within a 2-fold change of expression but with exceptionally high significance (volcano plot in Supplementary Figure S2B showing differentially expressed genes padj from 0.01 to 10−45).

RNA-Sequencing of PRMT5 and MEP50 knockdown cells revealed dramatic alterations of the cancer cell transcriptome. (a) FPKM (Fragments per kilobase of transcript per million mapped reads) of all 9 PRMTs (CARM1 is PRMT4) and MEP50 from the 3 RNA-Seq replicates for each of the control (GFPkd) and PRMT5kd and MEP50kd are shown on an arbitrary scale for each. PRMT5 and MEP50 reduced expression in each knockdown is indicated with the corresponding fold change from DESeq2 analysis. (b) Top: piechart showing up- (purple) and down- (green) regulated genes as calculated by both DESeq2 and edgeR algorithms with cutoffs as indicated. Bottom: Venn diagram showing the overlap in the total geneset of altered expression between PRMT5kd and MEP50kd. (c) Heatmap of the 955 genes altered in both knockdowns, showing PRMT5 and MEP50 up- (purple) and down- (green) regulated genes. The calculated Pearson correlation between the two knockdowns is shown. (d) Selected ConsensusPathDB output genes in protein complexes are shown, colored by up- (purple) and down- (green) regulation. Bolded genes are viewed in E. (e) IGV genome browser view of the RNA-Seq profile from a single replicate of the GFPkd, PRMT5kd and MEP50kd analysis. The fibrinogen locus (chr4: FGB, FGA and FGG) is shown as an example of downregulation upon knockdown (left panel, shaded green) and collagen 5α (chr9: COL5A1) is shown as an example of upregulation upon knockdown (right panel, shaded purple). (f) Example gene set enrichment analysis of the 955 genes altered in both knockdowns. Top panel: adhesion molecules known to be downregulated in lung cancer had a significant positive normalized enrichment score (NES) upon knockdown. Bottom panel: a gene set known to be upregulated in lung and breast cancers had a significant negative NES upon knockdown. (g) Dotplot of gene ontology terms enriched on both PRMT5 and MEP50 knockdown upregulated genes (top panel, purple) and downregulated genes (bottom panel, green) are shown in dots scaled by -log(p). (h) Dotplot of IPA diseases and functions enriched in both knockdowns are shown in dots scaled by–log(p). (i) Dotplot of IPA upstream pathways enriched in both knockdowns are shown in dots scaled by–log(p).
For the subsequent analyses we employed the 955 differentially expressed genes in both knockdowns (Figure 2b, bottom). We ranked the overlap between PRMT5kd and MEP50kd altered genes, further demonstrating the tight link between PRMT5 and MEP50 (Pearson correlation r=0.937, Figure 2c). To initially determine categories of regulated genes, we identified differentially regulated gene sets enriched in protein complexes with ConsensusPathDB29 (Figure 2d). Upregulated genes upon knockdown included groups involved in extracellular matrix formation and the TGFβ pathway, while downregulated genes included the cluster of fibrinogen genes recently shown to be involved in metastasis in A549 cells30 (FGB, FGA and FGG, RNA-Seq tracks shown Figure 2e). Collagen (col5a1) had the highest upregulation of any gene as shown in RNA-Seq tracks in Figure 2e. We therefore initially concluded that PRMT5-MEP50 had positive and negative roles in regulating transcription of extracellular matrix genes.
To further analyze this data set, we performed gene set enrichment analysis with candidate sets (Figure 2f and Supplementary Figure S2C). Genes involved in cellular adhesion, typically downregulated in lung cancer, were upregulated in the knockdowns (normalized enrichment score=+2.5), while genes known to be upregulated in kras mutated lung and breast cancers were downregulated in the knockdowns (NES=−1.69) (Figure 2f). These enrichments supported the hypothesis that PRMT5-MEP50 is necessary to maintain cancer cell identity.
We additionally probed the differentially regulated genes using Gorilla31 and REVIGO32 to consolidate and rank gene ontology enrichments. Highly significant upregulated GO terms upon PRMT5-MEP50 knockdown included cell adhesion, differentiation and extracellular matrix organization, while downregulated GO terms included cell-cell signaling, proliferation and metabolic processes (Figure 2g).
Finally, we used Ingenuity Pathway Analysis (IPA) to probe enriched pathways of the highly significant differentially expressed genes. IPA showed that cell migration and epithelial cancers were highly enriched terms (Figure 2h, individual knockdown IPA analysis in Supplementary Figure S2D). The TGFβ pathway was by far the most enriched upstream pathway, with a z-score>2 (Figure 2i and Supplementary Figure S2E).
PRMT5-MEP50 controls the proliferative and invasive phenotype of lung cancer cells
Since PRMT5-MEP50 alters transcription of cancer pathways, we probed a range of cancer phenotypes likely mediated by cell adhesion, migration, cancer and the TGFβ response pathways. First, we demonstrated that PRMT5 and MEP50 knockdowns have modest, but significant negative effects on proliferation after 6 days of culture (Figure 3a). Our subsequent assays measured phenotypes only within a 5 day window to minimize influence of altered proliferation.

PRMT5-MEP50 knockdown prevents cancer cell invasion. (a) Proliferation of A549 cells expressing shRNA targeted against GFP (red circle; GFPkd), PRMT5 (blue square; PRMT5kd) and MEP50 (orange triangle; MEP50kd). Plotted data are mean±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. Histogram (right): cell count on 6th day for each sample. (b) Colony formation assays of A549 cells expressing shRNAs are quantified for relative colony numbers (% of control). +Values are means±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. (c) Migration through 8 μm pores by GFPkd, PRMT5kd or MEP50kd A549 cells was measured. Left: representative crystal violet staining of migrated cells on the underside of the porous polycarbonate membrane under a phase-contrast microscope (20X). Right: quantification of the migrated cells. Values are mean±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. (d) Matrigel invasion through 8 μm pores by GFPkd, PRMT5kd or MEP50kd A549 cells was measured. Left: representative crystal violet staining of invaded cells on the underside of the porous polycarbonate membrane under a phase-contrast microscope (20 ×). Right: quantification of invaded cells. Values are mean±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. (e) Rate of wound-healing of GFPkd, PRMT5kd or MEP50kd A549 cells was measured over 24 h. Top: phase-contrast pictures (10 ×) of each cell line after scratching the confluent cells with the leading edge of cells indicated by a dashed yellow line. Bottom: quantification of the wound width (% 0hr wound width) after the scratch in indicated time points. Values are means±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. (f) 3D spheroid cell invasion assay of GFPkd, PRMT5kd or MEP50kd A549 cells. Cells were aggregated into spheroids and then induced to invade the invasion matrix for the indicated time courses. The flat area of the cell mass view was calculated at four time points to measure cell invasion rate. Top: representative spheroid images for individual A549 cell lines under a phase-contrast microscope (20 ×). Bottom: histogram depicting the Spheroid area (x106 μm2) measured with ImageJ 1.49. Values are means±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. (g) A549 cells were treated with 0, 10, 100, 500, 1, or 5 μM GS591 for 4 days and lysates or extracted histones were blotted for PRMT5, SYM10 (methylated SmD3), GAPDH (control), H4R3me2s and H3 as indicated. DB71 stain of extracted histones is also shown. (h) Invasivity of A549 cells treated with 500 nM GSK591 for 0 or 4 days were measured in a Matrigel assay. Top: Representative crystal violet staining of invaded cells on the underside of the porous polycarbonate membrane through a phase-contrast microscope (20 ×) are shown. Bottom: Quantification of the invaded cells. Values are mean±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. (i) Morphology (top micrographs) and viability (bottom plot) of A549 or IMR90 cells treated with 5 μM GSK591 for 4 days. Values are mean±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test.
To test anchorage-independent growth behavior, we performed a soft-agar colony formation assay in which we observed a dramatic and significant loss of colony formation in both PRMT5 and MEP50 knockdowns (Figure 3b and Supplementary Figure S3A), consistent with loss of cell autonomous behavior in the knockdowns. We observed pronounced and significant loss of migration (Figure 3c) and invasion through Matrigel (Figure 3d) in the PRMT5 and MEP50 knockdowns compared with the control, consistent with our hypothesis.
The dramatic loss of colony formation and invasivity prompted us to further examine the phenotypes of these knockdown cells. We employed a wound-healing assay and demonstrated a significantly reduced rate of closure in the knockdown cells compared with the control cells (Figure 3e, quantification in bottom panel). This reduced rate was not primarily due to reduced proliferation as the cell morphology and closure at 12 h, well below the A549 doubling time, was grossly distinct in the controls from the knockdown (Figure 3e, bottom).
To more robustly mimic an endogenous environment in the control and knockdown cells, we performed a 3D spheroid culture assay. Loss of either PRMT5 or MEP50 dramatically and significantly reduced the rate of growth and the spheroid volume of the cultured cells embedded in Matrigel (Figure 3f). In sum, these assays all pointed toward an important role for PRMT5 and MEP50 in invasivity and outgrowth.
A potent small molecule inhibitor of PRMT5 activity
PRMT5 and MEP50 are obligate partners and required for histone methylation, so the complementary knockdowns strongly supported the necessity of PRMT5 activity in those phenotypes. To provide additional support for this hypothesis we used GSK591/ EPZ015866, a recently developed potent and specific PRMT5 chemical probe.22, 33 Treatment of A549 cells with low concentrations of GSK591 lead to significant loss of PRMT5-catalyzed methylarginine on SmD3 (SYM10) and histone H4 (H4R3me2s) after 4 days of treatment (Figure 3g). 500 nM GSK591 caused complete ablation of histone and spliceosomal assembly factor methylation and abrogated cell migration and cell invasion of A549 cells (Figure 3h). Greater than 500 nM GSK591 killed A549 cells but not IMR90 lung fibroblasts, indicating that this compound has selectivity of cancerous vs normal cells (Figure 3i). GSK591 did not lead to significant loss of SYM10 and H4R3me2s after 4 days of treatment in the normal fibroblasts (Supplementary Figure S3B).
PRMT5-MEP50 mediates the EMT-response of TGFβ treatment of lung cancer cells
The phenotypic experiments and the RNA-Seq analysis all suggested that PRMT5-MEP50 may regulate the EMT. To test this hypothesis, we treated the control and knockdown cell lines with exogenous TGFβ, known to directly activate EMT in A549 cells34 and identified as the major upstream pathway in our IPA analysis. After 2 days of TGFβ treatment we observed non-contacting cells in the GFPkd control but adhering cells in the PRMT5 and MEP50 knockdowns (Figure 4a). TGFβ−treated control cells exhibited significantly increased migration through Matrigel while PRMT5 and MEP50 knockdown cells did not respond (Figure 4b).

PRMT5-MEP50 are novel regulators of the TGFβ response and invasion in metastatic lung and breast cancer cells (a) Representative cell morphology images of GFPkd, PRMT5kd or MEP50kd A549 cells. Cells were treated with (+) or without (−) 10 nM TGFβ1 for 2 days. Inserts show higher magnification views of selected cells. (b) Invasivity of GFPkd, PRMT5kd or MEP50kd A549 cells treated with (+) or without (−) 10 nM TGFβ1 for 2 days were measured in a Matrigel assay. Left: Representative crystal violet staining of invaded cells on the underside of the porous polycarbonate membrane through a phase-contrast microscope (20 ×) are shown. Right: Quantification of the invaded cells (+TGFβ, blue bars). Values are mean±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. (c) Top: Images of a 3D spheroid cell invasion assay of GFPkd, PRMT5kd or MEP50kd A549 cells treated with (+) or without (M) 10 nM TGFβ1 for 2 days are shown. Bottom: Quantification of spheroid area (× 106 μm2) measured with ImageJ 1.49 (+TGFβ, blue bars). Values are means±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. (d) Representative cell morphology images of A549 cells treated with (+) or without (−) 500 nM GSK591 for 4 days and (+) or without (−) 10 nM TGFβ1 for 2 days. Inserts show higher magnification views of selected cells. (e) Invasivity of A549 lung adenocarcinoma cells, (f). SK-MES-1 lung squamous cell carcinoma cells, and (g) HS578T breast carcinoma cells treated with (+) or without (−) 500 nM GSK591 for 4 days and (+) or without (−) 10 nM TGFβ1 for 2 days measured in a Matrigel assay. Top: Representative crystal violet staining of invaded cells on the underside of the porous polycarbonate membrane through a phase-contrast microscope (20 ×) are shown. Bottom: Quantification of the invaded cells (+TGFβ, blue bars). Values are mean±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test.
To further validate the role of PRMT5-MEP50 in mediating the TGFβ−stimulated EMT we turned again to the spheroid culture assay. 3D culture growth was substantially retarded by loss of both PRMT5 and MEP50, while the TGFβ−treated cells had significantly reduced spheroid area, consistent with a role for the complex in EMT (Figure 4c). GSK591 also blocked the TGFβ morphological and invasive response in A549 and H460 lung adenocarcinoma cells, SK-MES-1 lung squamous cell carcinoma cancer cells, and NCI H1334 large cell lung carcinoma cells, identical to the PRMT5 knockdown phenotype, and reduced invasion even in the absence of TGFβ (Figure 4d–f and Supplementary Figure S4). These data support a critical role for PRMT5 activity in invasive phenotypes of different types of lung cancer cells.
Invasive breast cancer cells also have upregulated PRMT5-MPE50 expression in human cancers (Figure 1a), so we used GSK591 to test the role of PRMT5 activity in HS578T triple-negative breast cancer cells’ phenotypes. TGFβ−treated control cells exhibited increased migration through Matrigel while GSK591-treated cells had significantly reduced response to TGFβ treatment (Figure 4g), consistent with similar PRMT5 function in invasive breast cancer and lung cancer.
Molecular mechanisms of the TGFβ-PRMT5-MEP50 axis
We hypothesized that histone methylation may be essential for the TGFβ−response mediated by PRMT5-MEP50. We treated the A549 knockdown lines with TGFβ for 2 days and immunoblotted these whole cell lysates (Figure 5a). In parental A549 cells, TGFβ-treatment dramatically increased the relative abundance of PRMT5 protein, but not PRMT7 or PRMT9, the other Type II arginine methyltransferases. TGFβ altered EMT protein markers with loss of E-cadherin and increased expression of Vimentin and Snail. In both the PRMT5 and MEP50 knockdown lines, we did not observe an alteration in the expression of EMT markers or changes in histone PTMs, consistent with the loss of invasive phenotypes in these cells. TGFβ caused much higher levels of H3R2me1 (of currently unknown function), H3R2me2s (correlated with gene activation) and H4R3me2s (correlated with gene repression) histone PTMs (Figure 5a, bottom blots).

TGFβ promotes increased PRMT5 expression and histone methylation concomitant with altered expression of EMT markers in lung and breast cancer cells. (a) Immunoblots for PRMTs, EMT markers, spliceosome factors, histone methylarginine PTMs, and controls as indicated, from A549 cells expressing shRNA targeted against GFP as a control (GFPkd), against PRMT5 (PRMT5kd) or against MEP50 (MEP50kd). These cells were treated with (+) or without (−) 10 nM of TGFβ1 for 2 days. (b) Immunoblots for the indicated proteins from A549 cells treated with (+) or without (−) 10 nM of TGFβ1 for 2 days and treated with (+) or without (−) 500 nM GSK591 for 4 days. (c) Immunoblots for PRMT5, MEP50, and histone methylarginine PTMs from H460 lung cancer cells and (d). HS578T breast cancer cells treated with (+) or without (−) 10 nM of TGFβ1 for 2 days and treated with (+) or without (−) 500 nM GSK591 for 4 days. (e) qRT-PCR of relative mRNA expression of A549 cells treated with (+, blue bars) or without (−) 10 nM TGFβ1 for 2 days.
To further test the requirement for PRMT5 activity in mediating the molecular consequences of EMT, we treated A549 and H460 lung cancer cells with TGFβ and GSK591 (Figure 5b and c compared with corresponding lanes in Figure 5a). These experiments showed that H3R2me1, H3R2me2s and H4R3me2s were all dependent on PRMT5 activity (Figure 5b bottom blots), as were the loss of expression of E-cadherin protein and gain of expression of Vimentin and Snail proteins, key players in EMT.
To test if the mechanism of PRMT5-dependent reduced invasivity of HS578T breast cancer cells was consistent between the different cancer cells, we treated these cells with TGFβ and GSK591 and measured histone methylation (Figure 5d). TGFβ treatment resulted in identical increases in H3R2me1, H3R2me2s and H4R3me2s and was completely abrogated upon GSK591 treatment, consistent with a similar TGFβ-PRMT5-MEP50 molecular axis in both cell types.
To determine if TGFβ treatment altered transcription of PRMT5 and MEP50, we performed qRT-PCR on treated A549 cells. We show that the PRMT5 transcript, but not the MEP50 transcript, was significantly increased upon treatment (Figure 5e), consistent with the observed increased protein.
EMT and cancer metastasis suppressor gene transcription is directly regulated by PRMT5-MEP50 histone H3 and H4 arginine methylation
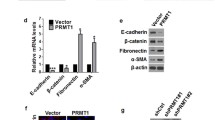
We established that invasive phenotypes in cancer cells were directly dependent on PRMT5-MEP50 enzymatic activity, likely through targeting of histones thereby altering transcriptional outcomes. To test this hypothesis, we designed methylarginine chromatin-immunoprecipitation (ChIP) studies at key genes around their promoter elements (−1 kb, TSS and +1 kb) in the presence or absence of TGFβ and in the PRMT5 and MEP50 knockdowns. We defined the genes to probe by performing qRT-PCR experiments on untreated or TGFβ-treated A549 cells with candidate ‘metastasis suppressor genes’ surmised from the literature.35, 36, 37 As shown in Figure 6a and Supplementary Figure S6A, these genes were significantly reduced in expression after TGFβ-treatment (left panel), while genes involved in activating EMT were significantly increased in expression (right panel).

PRMT5-MEP50 establishes a histone methylarginine code to downregulated metastasis suppressor genes and upregulate EMT activator genes (a) Relative mRNA levels of indicated genes in A549 cells (control, PRMT5kd and MEP50kd, as indicated on top) treated with 10 nM of TGFβ1 (blue bars) for 2 days were determined by qRT-PCR. β-Actin was used as an internal control. Values are means±s.e.m. of three independent experiments. *P<0.05 from two-tailed one-way ANOVA test. (b) qRT-PCR for the indicated genes from A549 cells expressing shRNA targeted against GFP as a control (GFPkd), against PRMT5 (PRMT5kd) or against MEP50 (MEP50kd). These cells were treated with (+, blue bars) or without (−) 10 nM of TGFβ1 for 2 days as indicated. Values are means±s.e.m. of three independent experiments. *P<0.05 from one-way ANOVA test. (c) Heatmap generated by ChIP-qPCR values to demonstrate the histone methylarginine code surrounding the candidate genes measured in (A). ChIP-qPCR enrichments from H3R2me1, H3R2me2s, H3R8me2s, H4R3me1, H4R3me2a, H4R3me2s and histone H3 from A549 on primers −1 kb, at the promoter, or +1 kb of the indicated genes are arrayed from blue (no enrichment) to yellow (maximal enrichment). ChIP-qPCR of TGFβ1-downregulated genes are arrayed on the top and TGFβ1-upregulated genes are on the bottom. (d) Selected ChIP-qPCR experiments of alternatively up-regulated (Snail1 and Vim) and down-regulated genes (CDH1 and SPDEF) from A549 cells (control, PRMT5kd and MEP50kd, as indicated on bottom) treated with 10 nM of TGFβ1 (blue bars) for 2 days are shown. The horizontal dotted line indicates the upper limit of the 95% confidence interval of the signal from no-antibody (NA) control ChIPs. Error bars, s.e.m. (n=3 independent cell passages). *P<0.05 from one-way ANOVA test.
To determine the role of PRMT5-MEP50 in the TGFβ-regulation of cancer metastasis and EMT genes, we probed expression of these genes in the PRMT5 and MEP50 knockdown cells. The majority of the TGFβ-transcriptional response, on both positively and negatively regulated genes, is lost in both knockdown lines compared with the control (Figure 6b). These observations strongly suggested that PRMT5 histone methylation may directly regulate transcription of these target genes.
We therefore systematically used ChIP to probe known PRMT5-catalyzed PTMs including H3R2me1, H3R2me2s, H3R8me2s, H4R3me1, H4R3me2s and the asymmetric mark H4R3me2a as a negative control for PRMT5 activity, on targeted genes (Figure 6c). TGFβ silenced genes were strikingly enriched in the H4R3me2s silencing mark in the control cells but not the knockdown cells (Figure 6c), while we observed enrichment of the previously unexplored H3R2me1 in TGFβ activated EMT genes (Figure 6c).
To highlight this novel independent up- and down-regulation of key metastasis markers by the same enzyme targeting distinct histone modifications, ChIP-qPCR for H3R2me1, H4R3me2s and H3 are shown for CDH1 and SPDEF (downregulated by TGFβ) and SNAIL1 and VIM (upregulated by TGFβ) (Figure 6d and Supplementary Figure S5B). These results demonstrate the simultaneous and independent up- and down-regulation of critical metastasis genes by PRMT5 activity towards H3 and H4, respectively.
TGFβ stimulates PRMT5-MEP50 H3R2me1 to recruit WDR5 and H3K4me3 methylation
Histone H3R2me2s enhances recruitment of the WDR5 protein, important for transcription through its associated MLL lysine methyltransferase to promote H3K4me3, while histone H3R2me2a blocks binding of the WDR5 protein and suppresses transcription.38 The structure of H3R2me2s bound to WDR5 suggested that H3R2me1 may also recruit WDR5 through a similar mechanism, so we hypothesized that H3R2me1 may promote H3K4me3 and stimulate transcription. Therefore, we employed OICR-9429, a new inhibitor of WDR5 interaction with the H3 tail,39 or depletion of WDR5 (Supplementary Figure S7 and 7A) to probe the molecular pathways downstream of H3R2 methylation.
Strikingly, both WDR5 inhibition or depletion partially abrogated the TGFβ stimulation of Snail and Vimentin expression (Figure 7b and c), consistent with our hypothesis that PRMT5-catalyzed H3R2me1 is responsible for their upregulation. To directly test this mechanism, we ChIPed H3R2me1 and H3K4me3 in the control and PRMT5kd A549 cells and probed Snail and Vimentin. These experiments further supported our hypothesis as the loss of PRMT5 activity significantly reduced H3K4me3 abundance on these genes (Figure 7d). Finally, we tested the direct involvement of WDR5 in this process by performing the ChIP in the presence of OICR-9429 or with depletion of WDR5. Consistent with our hypothesis, this treatment resulted in a significant loss of H3K4me3 after TGFβ treatment, with no change in H3R2me1 (Figure 7e and f).

Molecular mechanism of the simultaneous transcriptional activation and repression by the TGFβ-PRMT5-MEP50 axis through histone methylation. (a) Immunoblots for WDR5 and GAPDH from whole cell lysates of A549 treated with (+, blue bars) or without (−) TGFβ1 and with (+) or without (−) siRNA against WDR5. (b) qRT-PCR of relative expression of Snail and Vimentin in A549 cells treated with (+, blue bars) or without (−) TGFβ1 and with (+) or without (−) 1μM OICR-9429 for 2 days, a small molecule inhibitor of the WDR5:H3 interaction. (c) RT-qPCR as described in A except with A549 cells with (+, blue bars) or without (−) TGFβ1 and with (+) or without (−) expressing siRNA againstWDR5. (d) H3R2me1 and H3K4me3 ChIP-qPCR experiments of up-regulated genes (Snail1 and Vim) in GFPkd and PRMT5kd A549 cells treated with (+, blue bars) or without (−) TGFβ1. (e) H3R2me1 and H3K4me3 ChIP-qPCR experiments of up-regulated genes (Snail1 and Vim) in A549 treated with (+, blue bars) or without (−) TGFβ1 and with (+) or without (−) OICR-9429. (f) ChIP-qPCR as described in A except with A549 cells with (+, blue bars) or without (−) TGFβ1 and with (+) or without (−) expressing siRNA against WDR5. (g) Model depicting the TGFβ1-PRMT5-MEP50 histone code axis involved in lung cancer metastasis progression. TGFβ1 stimulates PRMT5 protein expression and activity towards H4R3me2s (left, histone tail exaggerated for illustration), downregulating expression of cancer metastasis suppressor genes, and towards H3R2me1 (right), stimulating expression of EMT activator genes through recruitment of WDR5 and MLL to methylate H3K4me3. The targeted genes are central to regulation of cellular movement, EMT, and metastasis. Knockdown of PRMT5 or MEP50 can prevent this axis. A novel, specific, and highly potent PRMT5 inhibitor GSK591 also inhibits TGFβ1 induced cancer phenotypes with limited effect on survival of normal lung cells.
Discussion
TGFβ, PRMT5-MEP50 and cancer phenotypes
A wealth of literature supports the essential biological functions of PRMT5-MEP50 activity targeting histones, splicesomal factors, p53, and many other targets in stem cells, germ cells, embryogenesis and cancer.4, 40 In cancer progression, TGFβ signaling and EMT are intimately connected pathways mediated by many events including transcriptional regulation.25, 41 In this manuscript we hypothesized that PRMT5-MEP50 may form a crucial intermediate in coupling signaling pathways and cancer progression to histone PTMs and transcriptional regulation.
Our extensive evidence now supports a model in which PRMT5-MEP50 histone methylation is responsible for mediating transcription of genes regulating cell adhesion, morphology, and invasivity and is essential for the TGFβ response, and therefore critical for cancer metastasis. PRMT5-MEP50 simultaneously targets histone H3 Arg2 for gene activation through the recruitment of WDR5 and histone H4 Arg3 for gene repression via currently unknown mechanisms. The bifurcation of histone methylation by the same enzyme to accomplish distinct and opposing outcomes on genes supporting the same biological pathways is an important and new function for a methyltransferase (Figure 7g), consistent with a recent study of PRMT5 activity in leukemia.42
PRMT5-MEP50 histone methylation and transcription
Symmetric dimethylation of histones H2A and H4 on R3 or histone H3 on R8, and asymmetric dimethylation of H3R2, repress transcription.4, 43 Conversely, we showed here that H3R2me1 and others have shown that H3R2me2s promote transcription by recruiting the WDR5 and the MLL methyltransferase that is responsible for transcriptional activation by H3K4 methylation.38, 44 Multiple PRMTs target similar residues with altered modification state (for example, H4 is monomethylated by PRMTs 1, 5, 7, asymmetric-dimethylated by PRMT1 and symmetric dimethylated by PRMT5). These distinct modification states appear to elicit distinct biological outcomes, likely through recruitment of unidentified effector molecules. H4R3me2s may bind the ADD domain of DNMT3a,45 although this conclusion remains in dispute.46 Recent evidence demonstrated H4R3me2s enrichment at G+C rich epigenomic loci in mouse ES cells,47 but this correlation did not provide a mechanism for regulating gene expression. We attempted many approaches to identify effector molecules for H4R3me2s without success (data not shown), perhaps indicating that this PTM functions through repulsion of other effectors.48
In vitro, PRMT5-MEP50 methylates H2A and H4, but not H3.12, 13, 49 H3 methylation may require additional factors present in the cell,50, 51 implying that subtle regulatory pathways and new complexes may alter the phenotypic outcome of PRMT5 activity. We did not observe any altered complex formation so future studies will examine transient interactions and cooperative function between pathways.
Our conclusions are important in that PRMT5-catalyzed histone methylation can simultaneously upregulate transcription through histone H3R2 methylation and repress transcription through H4R3 methylation, on independent genes in common biological pathways. This high degree of functional coordination through independent mechanisms by the same enzyme complex are unusual and may be a hallmark of PRMT function. Future investigations will determine how this epigenetic landscape is further coordinated among PRMTs.
Epigenetic targets, PRMT5-MEP50 methyltransferase activity and future clinical cancer therapies
As metastatic lung and breast cancers require new diagnostics as well as new drug candidates for personalized treatment, novel insight into epigenetic gene regulatory pathways is essential for the development of new clinical approaches. Recent observations have highlighted the strong potential for specific targeting of PRMT5-MEP50 in human cancers.52, 53 Here, we further established the molecular mechanisms by which PRMT5-MEP50 activity directly mediates cancer and invasive phenotypes through histone methylation, providing a compelling new pathway to target. We also provide strong evidence for the essential partnership between PRMT5 and MEP50 as both knockdowns elicit identical phenotypes to each other and to drug treatment.
Importantly, we demonstrated that the new, highly specific and potent PRMT5 inhibitor GSK591 blocks PRMT5 activity in cells and has selective killing activity towards cancer cells (A549) over normal IMR90 fibroblasts in culture. These observations are encouraging signs for future clinical development of this and related compounds and suggest that PRMT5 inhibition could be a novel therapeutic approach.
In conclusion, we demonstrate a new molecular pathway mediating metastasis in both invasive lung and breast cancer cells but not in non-cancerous IMR90 fibroblasts. PRMT5-MEP50 activity is now druggable by GSK591 and other molecules under active investigation.21, 22, 33, 54 Since PRMT5-MEP50 activity is upregulated in a range of human tumors with poor prognosis, it is clear that future studies leading to potential clinical applications are exciting and warranted. We anticipate that diagnostic tests probing expression of PRMT5-MEP50 combined with targeted inhibition of its enzymatic activity may lead to encouraging clinical outcomes preventing tumor metastasis.
Materials and methods
Cell culture and analysis
Normal cells (Primary human fetal lung fibroblast cells IMR90, human hepatocyte cells, human prostate epithelial cells, human mammary epithelial cells) and Lung Cancer cells (A549, H460, NCI H1334 and SK-MES-1 were cultured in RPMI with 10% FBS, MCF-7 human breast adenocarcinoma cells, LNCaP human prostate cancer cells, HepG2 human hepatocyte carcinoma cells and Hs578T human breast carcinoma cells were cultured as previously described.55 Cell line identities were confirmed by ATCC (A549 and HS578T; SK-MES-1 fresh vial from ATCC) or the Einstein Genomics Core (H460 and H1334). Cell lines were routinely tested for mycoplasma contamination.
Stable depletion of PRMT5, MEP50 or eGFP (as a control) in A549 was performed with retroviral-mediated expression of shRNA obtained from the TRC genome-wide shRNA collection. The targeting sequence for PRMT5 was 5′- GCCCAGTTTGAGATGCCTTAT-3′. The targeting sequence for MEP50 was 5′- GCAAAGTGAAGTCTTTGTCTT-3′. The targeting sequence for GFP was 5′-TACAACAGCCACAACGTCTAT-3′. The cells were selected and cultured under 1 μg/ml puromycin in RPMI with 10% FBS for 3 days. siRNA depletion of WDR5 in A549 was performed according to the manufacturer’s instructions with siRNA negative control (Ambion, 4390843) and WDR5-Silencer Select Pre-Designed & Validated siRNA (Ambion, 4392420 with siRNA ID s21862 and s21863).
Where indicated, cells were treated with the selective PRMT5 inhibitor GSK591 (Structural Genomics Consortium) or OICR-9429 (Cayman Chemical, 16095) a small molecular inhibitor of WDR5.
For cell proliferation curves, 5 × 104 cells were plated in 1 well of a 6-well plate. The cell numbers were counted up to 8 days with a hemocytometer. Immunofluorescence was essentially performed as previously described.56
TGFβ1 induced EMT (epithelial-mesenchymal transition) in A549 cells
Cells were starved 24 h in serum-free medium and plated into 6-well plates at a density of 104/well in medium supplemented with 1% FBS. Fresh recombinant human TGFβ1 (Sigma, T7039) was added at a final concentration of 10ng/ml each day for 2 days.
Cell phenotypic assays
Colony formation assay was essentially performed as previously described.57 Scratch wound healing assay was essentially performed as previously described.58 The wound width was measured by ImageJ 1.49. Cell migration and invasion assay were performed according to the manufacturer’s instruction (Corning). 3D tumor spheroid-based functional assay was performed with the commercial kit (Trevigen Inc., 3500-096-K) according to the manufacturer’s instruction. The area of the invasive structure of the spheroid was measured by ImageJ 1.49. Micrographs were captured by a Nikon Diaphot phase contrast microscope.
Immunoblots and acid-extraction of histones
Whole cell-lysis and acid-extraction of histones were performed as previously described.55 Immunoblots were performed on PVDF (Immobilon, Millipore), detected by ECL chemiluminescence (Lumigen, TMA-6) and imaged with an ImageQuant LAS4000 (GE). A list of antibodies used is presented in Supplementary Table 1.
Blue nativepage and immunoblotting
A549 protein samples were prepared with 4X Native PAGE Sample Buffer (Life Technologies). Purified human PRMT5 (Sinobiological) and PRMT5 in complex with MEP50 were prepared as described12 and used as controls. Proteins were separated on 4−16% Native PAGE Novex Bis-Tris Gels (Life Technologies).
ChIP and qRT-PCR
ChIP was performed as previously described.56 A list of antibodies and primers used for ChIP-qPCR are listed in Tables S1 and S2. RNA purification and qRT-PCR were performed as described previously.55 Primer sequences used in this study are listed in Table S3.
RNA-seq and TCGA analysis
RNA-seq was performed as described previously.55 Briefly, RNA-seq was performed on three biological replicates. Total RNA was extracted and purified with Rneasy mini kit (Qiagen, 74104). DNA was removed with DNA-free DNA Removal Kit (Thermo Fisher, AM1906). RNA quantitation and quality control was performed by Bioanalyzer 2100 (Agilent Technologies). Construction of stranded RNA-seq libraries from 0.1−1 μg of total RNA with depletion of ribosomal RNA (rRNA) was performed with KAPA stranded RNA-Seq kit with riboerase (HMR) following the manufacturer’s protocol (Kapa Biosystems, KR1151-v3.15). The barcoded libraries were sequenced on 2 HiSeq 2500 lanes (Illumina), 150nt paired-end Rapid-Run mode to produce approximately 30 million reads per replicate. Reads were trimmed and aligned to the human genome (hg19) with STAR. Gene counts for each replicate were determined with htseq-count, and differential expression between replicates was measured using DESeq259 and edgeR60 within the Chipster package.61 Overlaps were determined using vennt (http://drpowell.github.io/vennt/). FPKM (fragments per kilobase per million reads) were computed with cufflinks 2.2.1. Enriched functional annotations among genes were determined with ConsensusPathDB (www.consensuspathdb.org), GOrilla (http://cbl-gorilla.cs.technion.ac.il/), ReviGO (http://revigo.irb.hr/), and IPA (Qiagen). Gene Set Enrichment Analysis was performed with the GSEA tool (Broad Institute) and IGV (Broad Institute) was used as a genome browser. Dotplots and volcano plots were produced in SigmaPlot.
Data sets generated by TCGA Research Network (http://cancergenome.nih.gov/) were analyzed at the UCSC Cancer Genomics Brower (https://genome-cancer.ucsc.edu/). The Wanderer Website (http://gattaca.imppc.org:3838/wanderer/) was used to determine differential expression of PRMT5 or MEP50 in the tumor and normal samples and the Wilcoxon test P-values. SigmaPlot was used for dotplot generation and Mann-Whitney (Wilcoxon) rank sum tests.
Statistical analysis
All immunoblots were independently performed at least twice. All cell counting assay, colony formation assay, wound healing assay, migration and invasion assay, 3D tumor spheroid-based functional assay loci-specific ChIP and RT-qPCR experiments were repeated at least three times with independent biological samples. Results obtained are presented as means±s.e.m. One-way ANOVA (Analysis of variance) test was used to determine the significance of differences between samples indicated in figures. For RNA-seq analysis, Benjamini-Hochberg procedure was used in DESeq2 and edgeR differential expression analysis to calculate adjusted P-values (padj).
References
May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA . Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 2011; 13: 202.
Sarkar S, Horn G, Moulton K, Oza A, Byler S, Kokolus S et al. Cancer development, progression, and therapy: an epigenetic overview. Int J Mol Sci 2013; 14: 21087–21113.
Tam WL, Weinberg RA . The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med 2013; 19: 1438–1449.
Stopa N, Krebs JE, Shechter D . The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci 2015; 72: 2041–2059.
Yang Y, Bedford MT . Protein arginine methyltransferases and cancer. Nat Rev Cancer 2013; 13: 37–50.
Chi P, Allis CD, Wang GG . Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 2010; 10: 457–469.
Greenblatt SM, Liu F, Nimer SD . Arginine methyltransferases in normal and malignant hematopoiesis. Exp Hematol 2016; 44: 435–441.
Bedford MT, Clarke SG . Protein arginine methylation in mammals: who, what, and why. Molecular cell 2009; 33: 1–13.
Di Lorenzo A, Bedford MT . Histone arginine methylation. FEBS Lett 2011; 585: 2024–2031.
Dhar S, Vemulapalli V, Patananan AN, Huang GL, Di Lorenzo A, Richard S et al. Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Scientific reports 2013; 3: 1311.
Nicklay JJ, Shechter D, Chitta RK, Garcia BA, Shabanowitz J, Allis CD et al. Analysis of histones in Xenopus laevis. II. mass spectrometry reveals an index of cell type-specific modifications on H3 and H4. J Biol Chem 2009; 284: 1075–1085.
Burgos ES, Wilczek C, Onikubo T, Bonanno JB, Jansong J, Reimer U et al. Histone H2A and H4 N-Terminal Tails are Positioned by the MEP50 WD-Repeat Protein for Efficient Methylation by the PRMT5 Arginine Methyltransferase. J Biol Chem 2015; 290: 9674–9689.
Ho M-C, Wilczek C, Bonanno JB, Xing L, Seznec J, Matsui T et al. Structure of the Arginine Methyltransferase PRMT5-MEP50 Reveals a Mechanism for Substrate Specificity. PLoS ONE 2013; 8: e57008.
Wang M, Xu RM, Thompson PR . Substrate specificity, processivity, and kinetic mechanism of protein arginine methyltransferase 5. Biochemistry 2013; 52: 5430–5440.
Wang M, Fuhrmann J, Thompson PR . Protein arginine methyltransferase 5 catalyzes substrate dimethylation in a distributive fashion. Biochemistry 2014; 53: 7884–7892.
Bao X, Zhao S, Liu T, Liu Y, Liu Y, Yang X . Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J Histochem Cytochem 2013; 61: 206–217.
Chung J, Karkhanis V, Tae S, Yan F, Smith P, Ayers LW et al. Protein arginine methyltransferase 5 (PRMT5) inhibition induces lymphoma cell death through reactivation of the retinoblastoma tumor suppressor pathway and polycomb repressor complex 2 (PRC2) silencing. ⋄J. Biol. Chem 2013; 288: 35534–35547.
Ibrahim R, Matsubara D, Osman W, Morikawa T, Goto A, Morita S et al. Expression of PRMT5 in lung adenocarcinoma and its significance in epithelial-mesenchymal transition. Human pathology 2014; 45: 1397–1405.
Li Y, Chitnis N, Nakagawa H, Kita Y, Natsugoe S, Yang Y et al. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer discovery 2015; 5: 288–303.
Scoumanne A, Zhang J, Chen X . PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res 2009; 37: 4965–4976.
Alinari L, Mahasenan KV, Yan F, Karkhanis V, Chung JH, Smith EM et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015; 125: 2530–2543.
Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 2015; 11: 432–437.
Yan F, Alinari L, Lustberg ME, Katherine Martin L, Cordero-Nieves HM, Banasavadi-Siddegowda Y et al. Genetic Validation of the Protein Arginine Methyltransferase PRMT5 as a Candidate Therapeutic Target in Glioblastoma. Cancer Res 2014; 74: 1752–1765.
Ikushima H, Miyazono K . TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer 2010; 10: 415–424.
Lamouille S, Xu J, Derynck R . Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15: 178–196.
Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D et al. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Scientific reports 2013; 3: 2652.
Diez-Villanueva A, Mallona I, Peinado MA . Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin 2015; 8: 22.
Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med 2008; 14: 822–827.
Kamburov A, Stelzl U, Lehrach H, Herwig R . The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Research 2013; 41: D793–D800.
Wang H, Meyer CA, Fei T, Wang G, Zhang F, Liu XS . A systematic approach identifies FOXA1 as a key factor in the loss of epithelial traits during the epithelial-to-mesenchymal transition in lung cancer. BMC Genomics 2013; 14: 1–9.
Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z . GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 2009; 10: 48.
Supek F, Bosnjak M, Skunca N, Smuc T . REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 2011; 6: e21800.
Duncan KW, Rioux N, Boriack-Sjodin PA, Munchhof MJ, Reiter LA, Majer CR et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Medicinal Chemistry Letters 2015.
Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z . TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respiratory research 2005; 6: 56.
Liu J, Hu G, Chen D, Gong AY, Soori GS, Dobleman TJ et al. Suppression of SCARA5 by Snail1 is essential for EMT-associated cell migration of A549 cells. Oncogenesis 2013; 2: e73.
Stafford LJ, Vaidya KS, Welch DR . Metastasis suppressors genes in cancer. Int J Biochem Cell Biol 2008; 40: 874–891.
Yan J, Yang Q, Huang Q . Metastasis suppressor genes. Histology and histopathology 2013; 28: 285–292.
Migliori V, Muller J, Phalke S, Low D, Bezzi M, Mok WC et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol 2012; 19: 136–144.
Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat. Chem. Biol. 2015; 11: 571–578.
Koh C, Bezzi M, Guccione E . The Where and the How of PRMT5. Curr Mol Bio Rep 2015; 1: 19–28.
Yang H, Zhan L, Yang T, Wang L, Li C, Zhao J et al. Ski prevents TGF-beta-induced EMT and cell invasion by repressing SMAD-dependent signaling in non-small cell lung cancer. Oncology reports 2015; 34: 87–94.
Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 2015; 30: 789–799.
Guccione E, Bassi C, Casadio F, Martinato F, Cesaroni M, Schuchlautz H et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 2007; 449: 933–937.
Yuan C-C, Matthews Adam GW, Jin Y, Chen Chang F, Chapman Brad A, Ohsumi Toshiro K et al. Histone H3R2 Symmetric Dimethylation and Histone H3K4 Trimethylation Are Tightly Correlated in Eukaryotic Genomes. Cell Reports 2012; 1: 83–90.
Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol 2009; 16: 304–311.
Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, Shirakawa M . Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3 A ATRX-DNMT3-DNMT3L domain. EMBO reports 2009; 10: 1235–1241.
Girardot M, Hirasawa R, Kacem S, Fritsch L, Pontis J, Kota SK et al. PRMT5-mediated histone H4 arginine-3 symmetrical dimethylation marks chromatin at G+C-rich regions of the mouse genome. Nucleic Acids Research 2013; 42: 235–248.
Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X et al. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev 2012; 26: 2749–2762.
Wilczek C, Chitta R, Woo E, Shabanowitz J, Chait BT, Hunt DF et al. Protein Arginine Methyltransferase Prmt5-Mep50 Methylates Histones H2A and H4 and the Histone Chaperone Nucleoplasmin in Xenopus laevis Eggs. J. Biol. Chem. 2011; 286: 42221–42231.
Dacwag CS, Bedford MT, Sif S, Imbalzano AN . Distinct protein arginine methyltransferases promote ATP-dependent chromatin remodeling function at different stages of skeletal muscle differentiation. Mol Cell Biol 2009; 29: 1909–1921.
Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S . Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 2004; 24: 9630–9645.
Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016; 351: 1214–1218.
Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016; 351: 1208–1213.
Smil D, Eram MS, Li F, Kennedy S, Szewczyk MM, Brown PJ et al. Discovery of a Dual PRMT5-PRMT7 Inhibitor. ACS Med Chem Lett 2015; 6: 408–412.
Chen H, Ruiz PD, Novikov L, Casill AD, Park JW, Gamble MJ . MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat Struct Mol Biol 2014; 21: 981–989.
Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ . MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. cell 2015; 59: 719–731.
Kakuguchi W, Kitamura T, Kuroshima T, Ishikawa M, Kitagawa Y, Totsuka Y et al. HuR knockdown changes the oncogenic potential of oral cancer cells. Molecular cancer research: MCR 2010; 8: 520–528.
Wu YC, Tang SJ, Sun GH, Sun KH . CXCR7 mediates TGFbeta1-promoted EMT and tumor-initiating features in lung cancer. Oncogene 2015; 35: 2123–2132.
Love MI, Huber W, Anders S . Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15: 550.
Robinson MD, McCarthy DJ, Smyth GK . edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26: 139–140.
Kallio MA, Tuimala JT, Hupponen T, Klemela P, Gentile M, Scheinin I et al. Chipster: user-friendly analysis software for microarray and other high-throughput data. BMC Genomics 2011; 12: 507.
Acknowledgements
We are grateful to C. Arrowsmith, GlaxoSmithKline and the Structural Genomics Consortium for the gift of GSK591, C. Wilczek, R. Mazur and P. Bailey for preliminary knockdown experiments, E. Burgos for recombinant PRMT5-MEP50, S. Maqbool, R. Dubin, and B. Ye at the Einstein Center for Epigenomics for assistance with RNA-Seq and initial analysis, and members of the Shechter lab for comments. This work was supported by startup funds from the Albert Einstein College of Medicine, The Alexandrine and Alexander Sinsheimer Fund, The American Cancer Society – Robbie Sue Mudd Kidney Cancer Research Scholar Grant (124891-RSG-13-396-01-DMC) and NIH R01GM108646-01A1 (all to D.S.).
RNA-seq data have been deposited in the Gene Expression Omnibus database under accession code GSE80182.
Author contributions
HC planned and conducted all experiments and wrote the manuscript, BL performed the native gel analysis, VG assisted with bioinformatics analysis, DS planned experiments, performed bioinformatics analysis, wrote the manuscript, and supervised all work.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Chen, H., Lorton, B., Gupta, V. et al. A TGFβ-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 36, 373–386 (2017). https://doi.org/10.1038/onc.2016.205
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.205
- Springer Nature Limited
This article is cited by
-
Fibroblast-specific PRMT5 deficiency suppresses cardiac fibrosis and left ventricular dysfunction in male mice
Nature Communications (2024)
-
Epigenetic regulation of hybrid epithelial-mesenchymal cell states in cancer
Oncogene (2023)
-
Combination treatment of T1-44, a PRMT5 inhibitor with Vactosertib, an inhibitor of TGF-β signaling, inhibits invasion and prolongs survival in a mouse model of pancreatic tumors
Cell Death & Disease (2023)
-
The circSPON2/miR-331-3p axis regulates PRMT5, an epigenetic regulator of CAMK2N1 transcription and prostate cancer progression
Molecular Cancer (2022)
-
MYBL1 induces transcriptional activation of ANGPT2 to promote tumor angiogenesis and confer sorafenib resistance in human hepatocellular carcinoma
Cell Death & Disease (2022)