
Abstract
Small cell lung cancer (SCLC) is an aggressive malignancy characterized by early metastasis, rapid development of resistance to chemotherapy and genetic instability. This study profiles DNA methylation in SCLC, patient-derived xenografts (PDX) and cell lines at single-nucleotide resolution. DNA methylation patterns of primary samples are distinct from those of cell lines, whereas PDX maintain a pattern closely consistent with primary samples. Clustering of DNA methylation and gene expression of primary SCLC revealed distinct disease subtypes among histologically indistinguishable primary patient samples with similar genetic alterations. SCLC is notable for dense clustering of high-level methylation in discrete promoter CpG islands, in a pattern clearly distinct from other lung cancers and strongly correlated with high expression of the E2F target and histone methyltransferase gene EZH2. Pharmacologic inhibition of EZH2 in a SCLC PDX markedly inhibited tumor growth.
Similar content being viewed by others


Introduction
Despite an encouraging decline in lung cancer cases in the United States due, in large part, to a decreasing prevalence of tobacco use, lung cancer remains the number one cancer killer and is expected to remain so for some time.1 Worldwide tobacco use trends point to an increase in lung cancer cases and deaths, particularly in underdeveloped and developing countries.1, 2 A subset of lung cancers representing 10–15% of cases is classified as small cell lung cancer (SCLC),1, 3 typified by rapid proliferation, a high rate of early metastasis and extreme lethality; 2/3 of SCLC patients present with metastatic disease and have a 5-year survival rate of <1%.4 The primary standard of care for patients with advanced SCLC has remained the same for over 30 years: a combination of the cytotoxic compounds cisplatin and etoposide.5, 6
Two recent comprehensive genomic analyses have identified potential genetic drivers of SCLC.7, 8 One interesting outcome from these studies was the identification of hotspot mutations in a number of known chromatin modifiers including the histone acetyl transferase genes CREBBP and EP300, as well as epigenetic readers such as those encoded by MLL and MLL2. The effects of these alterations on site-specific DNA methylation and target gene expression have not been defined. A large-scale effort by The Clinical Lung Cancer Genome Project (CLCGP) to develop a genomics-based classification of human lung tumors has proven insightful in correlating specific genetic lesions with lung cancer subtypes and clinical outcomes.9 Although comprehensive in their genetic analysis, these several studies did not explore the role that epigenetics may have in SCLC etiology and classification.
A primary mechanism of epigenetic regulation of gene expression is DNA methylation. Hypermethylation of CpG islands associated with regulatory elements controlling gene expression can drive secondary recruitment of histone modifications, together leading to stable gene silencing. Several studies have demonstrated that key pathways can be altered in cancers by distinct mechanisms, including mutation, deletion and DNA methylation.10, 11, 12, 13 A clearer understanding of the patterns of DNA methylation in SCLC may yield cancer-specific biomarkers and help to identify clinically actionable drug targets and pathways through integrated genetic and epigenetic analysis.
Kalari et al.14 performed a study of DNA methylation patterns in 18 primary SCLC tumors and five SCLC cell lines using a target-specific CpG island promoter array. This study identified DNA methylation peaks in 73 genes specific to SCLC with a significant representation of neuroendocrine-specifying transcription factor genes including NEUROD1, HAND1, ZNF423, REST, as well as polycomb genes such as the HOXD cluster, but was not able to reliably differentiate tumor from normal lung. This study was further limited by relatively low resolving power of the technique used,15 and by the lack of complementary genetic analysis of these samples. The present study sought to substantially extend our understanding of genome-wide DNA methylation in SCLC at single base resolution by performing Illumina Human Methylation 450k analysis on a set of 47 extensively characterized SCLC samples, including 34 fresh-frozen primary SCLC tumors with available exome mutation, copy number and RNA-seq data as well as six distinct primary patient-derived xenografts and seven cell lines (Supplementary Table S1).7 Twenty-four of the primary SCLC tumors had matched normal lung control DNA available for analysis.
Using these complementary data sets, we show that SCLC primary xenografts are epigenetically more similar to primary SCLC than are cell lines, identify differentially methylated regions and individual CpG positions that are correlated with gene expression and define epigenetically distinct SCLC subtypes among primary patient samples that may have important therapeutic and diagnostic implications. SCLC is a disease that is characterized by extreme plasticity and cloning capacity consistent with a high level of stemness.16 We identified EZH2, a member of the PRC2 complex that promotes trimethylation of histone H3 lysine 27 (H3K27me3) and self-renewal as being both highly expressed in SCLC and strongly correlated with overall promoter methylation among many tumor types. Pharmacologic inhibition of EZH2 in LX92, a SCLC PDX, inhibited growth ex vivo and in vivo, providing preclinical evidence for the utility of this class of epigenetic therapy in SCLC.
Results
DNA methylation patterns in models of SCLC
Regions of DNA hypomethylation with high variation in both DNA methylation and gene expression have been reported in many human cancers.17 Consistent with these reports, we observed that DNA methylation levels were widely disparate in primary SCLC samples, PDX tumors and SCLC cell lines compared with normal lung (Supplementary Figure 1A). To estimate DNA methylation variability within each group, we examined the across-sample standard deviations in DNA methylation β-values for each CpG measured. The proportion of highly variable CpGs is low in normal lung and higher in every other group, consistent with high methylation instability in primary samples that is also present in both PDX and cell lines (Figure 1a).

DNA methylation patterns in normal lung, primary SCLC, PDX and cell lines. (a) Violin plot of CpG β-value standard deviations for each sample type demonstrate that tissue-specific methylation in primary normal samples is consistent between patients, whereas cancer samples can have a long tail of high variation. (b) Principal component analysis demonstrates clustering of 4/6 PDX with primary SCLC. Cell lines can be distinguished from all other samples using just two principal components. (c) Hierarchical clustering of Spearman correlation coefficients between all samples. (d) Concordance at the top (CAT) plot demonstrates that concordance with primary SCLC is consistent for PDX, whereas cell lines rapidly lose concordance as the size of the ranked gene list considered increases. (e and f) PDX have 98% concordance of differentially expressed genes (q-value cutoff 1 × 106), whereas cell lines exhibit epigenetic changes associated with long-term culture.
We hypothesized that DNA methylation patterns could discriminate between normal lung, primary SCLC, PDX and cell lines. Principal component analysis demonstrated that tissue-specific DNA methylation in lung was consistent between samples in contrast to DNA methylation in cancer samples, which was more variable (Figure 1b). Four of six PDX clustered with primary SCLC samples, whereas 7/7 cell lines fell outside of this cluster. All primary SCLC and PDX could be differentiated from cell lines by just two principal components, which account for almost half of the total variance (Supplementary Figures 1B and C). To further characterize differences among primary SCLC, PDX and cell lines, we performed unsupervised hierarchical clustering on inter-sample Spearman correlation coefficients and found that all primary normal samples clustered together, whereas primary SCLC also clustered together along with three classic subtype SCLC PDX, LX44, LX47 and LX48 (Figure 1c). All seven SCLC cell lines as well as three variant subtypes SCLC PDX, LX22, LX33 and LX36, clustered together. Variant subtype PDX may have clustered with SCLC cell lines due to similarity to the variant cell lines H446 and H82; we have previously reported that these cell lines and PDX share a common neurogenic differentiation program.18, 19 This finding is consistent with earlier reports suggesting that PDX are more similar to primary SCLC based on gene expression than are SCLC cell lines.20
We next compared β-values of primary SCLC, PDX and cell lines with normal lung and plotted the concordance at the top (CAT) of the ranked probe list obtained from primary SCLC with those obtained from either PDX or cell lines. Although both PDX and cell lines were >80% concordant with primary SCLC at the top of the ranked probe lists, cell line concordance dropped off rapidly with increasing probe list size, whereas PDX maintained high concordance with primary SCLC (Figure 1d).21 Using a significance cutoff on the basis of the concordance at the top plot (q<1 × 10−6) to select significant CpG sites as opposed to a rank order approach, we confirmed that PDX maintained 98% concordance with primary SCLC, whereas cell lines appear to accumulate divergent DNA methylation over time as a result of epigenetic drift and adaptation to cell culture (Figures 1e and f). Of note, several of these PDX models are of unknown high passage, yet still maintain high concordance with primary tumors. These data suggest that the irreversible changes in gene expression caused by long-term culture ex vivo may be driven in part by epigenetic dysregulation not observed when tumors are continuously passaged exclusively in mice.
Differential methylation between SCLC and normal lung
Of the differentially methylated CpG sites (Supplementary Table S2, Supplementary Figure 2A), the majority of CpGs were hypomethylated in SCLC relative to normal lung; however, the most significant methylation events were predominantly hypermethylated in SCLC (Figure 2a). Concordance of both PDX and cell lines with primary SCLC was strongly correlated with the fraction of differentially methylated CpGs that were methylated compared with normal lung (Supplementary Figure 2B). Probes associated with CpG island-containing promoters on the Illumina 450k platform are concentrated within 500 bp of the transcription start site (TSS). Significantly hypermethylated CpGs tend to follow a similar distribution within 500 bp of the TSS, whereas significantly hypomethylated sites are distributed over a wider range upstream of the TSS (Figure 2b), consistent with reports of general promoter hypomethylation accompanied by cancer-specific hypermethylation proximal to the TSS in bisulfite sequencing data.22

Characteristics of differential methylation between SCLC and normal lung. (a) CpG methylation vs normal lung in each group decreases as a function of the size of the ranked gene list considered increases, indicating that the majority of significant differences are methylation events. (b) CpGs hypermethylated in SCLC are more likely to be located immediately upstream of the TSS or within the first exon than hypomethylated CpGs, which are distributed more evenly throughout the promoter region (P<0.0001, two-tailed Wilcoxon rank-sum test). (c) CpG methylation is predominantly negatively correlated with gene expression, comprising 73% of differentially methylated CpGs. (d) Hypomethylated CpGs that are associated with gene expression are significantly more likely to be found in gene bodies than are hypomethylated CpGs associated with silencing (P=0.016, two-tailed Wilcoxon rank-sum test). (e) Methylation in the first exon of BCL2 is strongly correlated with gene expression silencing. BCL2 methylation may be one mechanism by which some SCLC tumors lack expression of BCL2 (P<0.0001, Spearman correlation test). (f) BCL2 methylation in SCLC PDX is strongly associated with silencing of Bcl-2 protein expression in variant subtype models as measured by western blot.
To characterize the significance of promoter methylation on gene expression, the Spearman rank correlation between the β-value at each CpG for every sample and the expression of the gene associated with that promoter was calculated. DNA methylation events that are strongly correlated with alterations in gene expression were calculated among samples where both Illumina 450k and RNA-seq data were available (Supplementary Table S3). Average fold change in gene expression is plotted vs differential β-value and summarized in Figure 2c. Among significantly hypomethylated CpGs, a distinct bimodal distribution is observed in those associated with high gene expression in contrast to those with apparent silencing, suggesting that demethylation in the gene body is associated with actively transcribed genes. Hypomethylated CpGs associated with high gene expression were more likely to be observed downstream of the TSS in expressed genes than hypomethyated CpGs associated with silenced genes, which predominantly cluster immediately upstream of the TSS (Figure 2d).
Four hundred and ninety-four ranges comprising 4033 unique CpGs were identified as significantly differentially methylated using a ‘bump hunting’ approach (Supplementary Table S5).23 The number of probes on the Illumina 450k array limits this general approach to interrogating 27% of probes in 12 502 clusters; however, it is useful for finding regions with consistent tumor-specific differences in local methylation. When comparing the regions identified with those reported in Kalari et al., 32/65 (49%) significant ranges were identified in our data set, while 462 additional unique ranges were detected (Supplementary Figures 2C and D).
One of the most differentially methylated genes in small cell lung cancer in comparison with normal lung that is also strongly correlated with gene expression is the oncogene BCL2 (Figure 2e; Supplementary Table S4). This gene is methylated and silenced in normal lung, but shows substantial variability among primary samples and PDX. We have previously reported that BCL2 message and its protein product Bcl-2 are more frequently highly expressed in classic subtype than in variant subtype SCLC PDX. We therefore compared methylation levels of BCL2 methylation with Bcl-2 protein expression across three PDX of each subtype and found that Bcl-2 was only detectable in classic subtype PDX, associated with minimal DNA methylation (Figure 2f). Upregulated BCL2 expression in classic subtype SCLC may be driven primarily by epigenetic mechanisms.24
We also found methylation and inactivation of two tumor suppressor genes, TCF21 (Supplementary Figure 2E), also identified by Kalari et al., and RB1.25 RB1 loss of function is typically by a combination of copy loss and mutation. In cases where these mutations are not fully inactivating, silencing by promoter methylation may have a role in further suppressing RB1 expression (Supplementary Figures 2F and G).
DNA methylation profiling defines distinct subtypes of SCLC
We hypothesized that disease subtypes characterized by differential DNA methylation and gene expression could be present among the primary SCLC samples in this study. We applied k-means consensus clustering to DNA methylation and gene expression data among primary SCLC samples. Three distinct groups are observed at both the DNA methylation and gene expression levels (Figures 3a and b). We refer to the three principal methylation clusters identified here as SCLC M1, M2 and SQ-P subgroups and the gene expression clusters as SCLC E1, E2 and SQ-P. Interestingly, extensive model tuning and silhouette analysis indicated the presence of a single, highly stable cluster closely related to a primitive phenotype gene expression cluster previously identified among lung cancers histologically identified as squamous cell lung cancer. This approach has also been applied to classification of other lung tumor types.9, 26 The SQ-P tumors in our tumor set originated from multiple cancer centers and were verified as SCLC by a thoracic pathologist. Other clustering solutions and model tuning approaches were explored, as described in detail in Materials and Methods; however, values of k>3 significantly decrease the quality of clustering of samples outside of the SQ-P cluster, whereas SQ-P cluster membership remains stable up to more than five groups (Supplementary Figure 3). Notably, 6/6 gene expression samples in the SQ-P cluster were concordant with the corresponding DNA methylation cluster, confirming that the methylation patterns observed are clearly tied to gene expression (Figure 3c).

Identification of SCLC subtypes by consensus clustering. (a) Consensus clustering of DNA methylation reveals three distinct groups of SCLC DNA methylation. We designate two groups here as SC-M1 and SC-M2, while a third group closely resembles primitive squamous cell tumors previously designated as SQ-P. (b) Consensus clustering of RNA-seq gene expression also revealed a stable cluster consistent with primitive squamous cell tumors as well as two larger clusters with a less distinct consensus. (c) Six out of six tumors in the gene expression SQ-P group belong to the identical methylation cluster. The other subtypes are less clearly distinguished exclusively by gene expression.
We examined DNA methylation patterns among the CpG sites with the highest variance and found that the cluster identified as SQ-P had what appeared to be overall lower methylation compared with the SCLC M1 and M2 subgroups (Figure 4a). We explored the possibility that CpG probe intensities could be affected in the SQ-P subset by tumor purity, a hypothesis that our data did not support. Increased methylation in the SCLC M1 and M2 subtypes can be visualized genome-wide in the density plot shown in Figure 4b and the mean promoter methylation scatter plot in Figure 4c. Consistent hypermethylation is present in M1 and M2 subtypes both genome-wide at single-nucleotide resolution as well as when considering average methylation of promoters. Dilution of the SQ-P subtype by admixture with normal lung tissue or immune infiltrate could be expected to increase the methylation of some promoters while decreasing others; however, DNA methylation was consistently increased in SCLC M1 and M2 subtypes relative to SQ-P. We plotted the number of methylated promoters in each subtype and found that the number of methylated promoters was significantly lower in the SQ-P subtype compared with all other subtypes (Figure 4d). We further identified which CpG sites were significantly differentially methylated between the SQ-P subtype and SCLC M1 and M2, and among this subset of sites found that the SCLC M1 and M2 subtypes were significantly more frequently methylated (Figure 4e). Significantly differentially methylated CpG sites were also significantly more likely to be found in CpG islands in SCLC M1 and M2 than in the SQ-P subtype (Figure 4f), whereas the SQ-P displayed a nonsignificant trend toward increased methylation in LINE repeats (P=0.075) that may reflect lack of statistical power due to the relative low coverage of CpG probes in these regions (Figure 4g). Other groups have identified a CpG island methylator phenotype among various cancer histologies that is often associated with a more aggressive tumor and poorer prognosis.27, 28, 29, 30, 31 DNA promoter methylation levels in SCLC are among the highest of any tumor type when compared with samples found in The Cancer Genome Atlas (TCGA), suggesting that this increased methylation level is biologically significant within the spectrum of human cancer (Figure 4h).

SCLC promoter methylation is higher in the SC-M1 and SC-M2 subgroups than in the SQ-P primitive squamous-like subtype or the majority of tumors in TCGA. (a) Unsupervised clustering of the most highly variable CpGs was consistent with consensus clustering approaches. The SCLC subtypes M1 and M2 had consistently higher DNA methylation. (b) Genome-wide CpG methylation is consistently higher in SCLC M1+M2 than SQ-P. (c) Average DNA methylation within promoters is also lower in SQ-P. (d) The number of methylated promoters in each sample is significantly lower in the SQ-P subtype than in other SCLC subtypes (Wilcoxon rank-sum test) (e) Global CpG methylation levels are consistently lower in SQ-P than other subtypes of SCLC (binomial distribution test). (f and g) Of the significantly differentially methylated CpGs between SQ-P vs other SCLC, the frequency of methylated CpGs in CpG island-containing promoters is significantly lower in SQ-P (proportionality test), whereas there is a trend of increased methylation in repetitive elements in SQ-P that fails to reach statistical significance. (h) Average methylation in CpG island-containing promoters in SCLC is among the highest of any tumors in TCGA, including tumor types with defined genetic mechanisms for genome-wide hypermethylation.
Interestingly, the hallmark SCLC genetic lesions including TP53 mutation and RB1 inactivation are present in all subtypes (Figure 5a). We have previously reported that expression of two genes, NEUROD1 and ASCL1, can accurately predict classic vs variant subtype SCLC cell lines and PDX.18 Differential expression analysis between SCLC E1 and E2 gene expression subgroups demonstrated that these genes follow a similar pattern in primary SCLC samples (Figure 5b). NEUROD1 expression was high and ASCL1 was low in the E1 subtype, whereas the converse was true in the SCLC E2 subtype (Figure 5; Supplementary Table S6).

Differentially expressed genes among SCLC subtypes. (a) Differentially expressed genes between the different subgroups indicate distinct differences in neuroendocrine differentiation. Despite the distinct epigenetic subtypes observed in this study, mutation and copy loss of RB1, a hallmark genetic lesion of SCLC, is nearly universal including within the group of primitive squamous-like tumors. (b) Among the top differentially expressed genes are ASCL1 and NEUROD1, which we have previously identified in patient-derived xenografts as important classifiers.
EZH2 expression is high in SCLC and correlates with high promoter methylation among cancers in TCGA
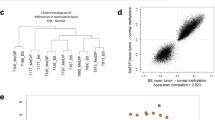
We also examined the genes that were highly expressed among all SCLC subtypes relative to normal lung for candidates that could account for the high levels of promoter methylation in SCLC. The chromatin modifier EZH2 was among the most significantly overexpressed genes in SCLC, expressed >12-fold higher than in normal lung (Figure 6a; Supplementary Table S7). EZH2, a histone methyltransferase component of the PRC2 complex, has been implicated in dysregulation of DNA methylation in cancer through its effects on histone methylation, particularly histone H3 lysine 27 trimethylation (H3K27me3).32 EZH2 is a known target of E2F, which is activated in SCLC by gene copy loss and loss of function mutations in RB1, the gene encoding the E2F repressor pRB.33

EZH2 is among the most significantly differentially expressed genes in SCLC compared with normal lung or other tumor types in TCGA. (a) EZH2 is the second most significantly differentially expressed gene when compared with normal lung control with >12-fold higher expression levels. (b) Median expression levels of EZH2 are significantly correlated with high levels of promoter methylation in SCLC and TCGA data sets (Spearman’s rho test of correlation). (c) EZH2 gene expression in SCLC is higher than any other TCGA tumor set and among the highest of any tumor type in the cell lines found in the Cancer Cell Line Encyclopedia (CCLE).
In addition to the high expression observed in SCLC compared with normal lung, we observed a significant correlation between median EZH2 gene expression and the number of methylated promoters in a given cancer type using data from TCGA (Figure 6b). Overall, EZH2 expression in SCLC is greater than or comparable to that of any tumor type represented in either TCGA for primary tumors or the Cancer Cell Line Encyclopedia for cell lines (Figure 6c). High-level EZH2 gene expression associated with increased methylation in CpG island-containing promoters is a common feature of SCLC.
We next asked whether EZH1 and EZH2 protein expression was detectable in a panel of 17 SCLC PDX by western blot (Figure 7a). Neither protein was universally expressed; however, EZH2 expression was detectable in 15/17 SCLC PDX (88%). We reasoned on the basis of these data that EZH2 could be of interest as a druggable target in SCLC. We therefore treated ex vivo cultures of the LX92 PDX with three EZH2 inhibitors: EPZ-5687, GSK343 and UNC1999 over a period of 7 days and measured cell viability by resazurin conversion over time (Figure 7b). All three compounds significantly inhibited viability ex vivo. To assess the extent of EZH2 inhibition, we performed western blots for EZH1, EZH2, H3K27me2 and H3K27me3 (Figure 7c). While all three inhibitors decreased levels of H3K27me3, only EPZ-5687 was able to profoundly decrease H3K27me2 levels, suggesting that this compound is a more potent inhibitor of EZH2 at the doses used.

Pharmacologic inhibition of EZH2 in SCLC inhibits growth in vitro and in vivo. (a) EZH1 and EZH2 are consistently expressed in a panel of SCLC PDX as measured by western blot (b) Ex vivo growth of the SCLC PDX LX92 is significantly inhibited by the EZH2 inhibitors EPZ-5687, GSK343 and UNC1999 as measured by resazurin conversion (two-way analysis of variance, adjusted for multiple comparisons by the method of Dunnet) (c) Western blot analysis of day 7 lysates from LX92 tumors treated with various EZH2 inhibitors. (d) In vivo growth of the SCLC PDX LX92 is significantly inhibited by the EZH2 inhibitor EPZ-6438. (e) Western blot analysis of end point LX92 tumor lysates treated with various EPZ-6438 treatment schedules. Tumors with growth inhibition greater than the median (TGI > median) for each treatment group were pooled separately from those with less robust response (TGI < median) to assess correlation between TGI and pharmacodynamic response. TGI, tumor growth inhibition.
EPZ-6438 is a more potent and bioavailable derivative of EPZ-5687 for in vivo use. We assessed the efficacy of EPZ-6438 in the LX92 SCLC PDX in vivo. EPZ-6438 was well tolerated (Supplementary Figure S4) and demonstrated remarkable efficacy at 100 mg/kg either QD or BID (Figure 7d). Tumors were collected on day 43 for pharmacodynamic analysis. Protein lysates from tumors were pooled based on treatment group and degree of tumor growth inhibition. EZH1 levels were decreased in all EPZ-5687 treatment groups. Interestingly, the best responding tumors in the BID group showed decreased levels of EZH2 and significant inhibition of both H3K27 di- and trimethylation, demonstrating that antitumor efficacy is consistent with target inhibition (Figure 7e).
Discussion
Our understanding of cancer genetics has grown exponentially over the last decade with increasing accessibility to high throughput sequencing technology and plummeting costs for data acquisition. It has been estimated that mutations in fewer than 200 genes drive cancer initiation and progression and that the majority of such oncogenic mutations have been identified.34 Many targeted therapies have been developed for these mutations; however, clinically actionable drug targets are disappointingly rare in SCLC. Targeting approaches on the basis of aberrant gene expression may be more tractable in SCLC. One focused proteomic study identified PARP1 and EZH2 as potential actionable targets.35 SCLC epigenetics has not been as comprehensively studied as tumor genetics, in part, due to the variety of DNA and histone modifications, chromatin-modifying enzymes, and the relatively scant tissue available from primary patient samples.
DNA methylation profiles of cells grown in culture tend to be distinct from those of primary tissues with both random and highly consistent changes in methylation being observed.36, 37 In contrast, we found that PDX maintain a DNA methylation pattern consistent with primary SCLC even after multiple passages. This supports our earlier gene expression findings and suggests that epigenetic dysregulation may account, in part, for the irreversible gene expression changes observed in cell lines and cell line xenografts.
Gene expression and DNA methylation data have begun to shed light on heterogeneity within lung cancer histologies.10, 26, 38 SCLC has been historically considered a relatively homogenous disease, supported by near-universal genetic alterations in TP53 and RB1 and a highly representative mouse model generated through conditional loss of these genes.39 Recent large-scale efforts have attempted to improve the classification of lung cancers on the basis of differential spectra of genetic alterations; primitive squamous and large-cell neuroendocrine tumors being among the histologies most difficult to accurately classify. We observed epigenetically distinct subgroups among histologically and genetically similar SCLC cases from multiple institutions and geographic cohorts that may represent clinically important populations. These subgroups have widely differing transcriptional profiles and can be delineated by differential expression of the neurogenic basic helix-loop-helix transcription factors ASCL1 and NEUROD1, suggesting that primary SCLC can be found along the same spectrum of differentiation found in cell lines and PDX.
Finally, we report striking overexpression of EZH2 in primary SCLC compared with normal lung as well as other primary tumors from TCGA and cell lines from the Cancer Cell Line Encyclopedia. EZH2 overexpression was correlated with increased promoter methylation across tumor types in TCGA. Various approaches to target EZH2 are being explored including inhibition of the catalytic domain40 and inhibition of protein–protein interactions to disrupt the PRC2 complex.41 We explored pharmacologic inhibition of EZH2 in SCLC and demonstrated that EPZ-6438 profoundly inhibited tumor growth in vivo in an SCLC PDX.
These data support further studies of the role that SCLC subtypes have in the clinical course of the disease. Given the exceptionally high level of promoter methylation observed in SCLC, new approaches to epigenetic targeting should be explored, inclusive of targeting EZH2.
Materials and methods
Primary SCLC samples
All clinical samples were extracted in a single batch using the Qiagen (Valencia, CA, USA) All-prep kit. All other samples were extracted from fresh-frozen pellets by the Hopkins Microarray and Deep Sequencing core facility. Aliquots of each sample were examined by agarose gel electrophoresis and spectrophotometry, and determined to be of good quality compared with a genomic DNA reference standard.
Methylation beadchip assay
Bisulfite conversion of DNA was carried out using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA) according to manufacturer’s instructions. Bisulfite-converted DNA was analyzed using Illumina’s (San Diego, CA, USA) Infinium Human Methylation 450 Beadchip Kit (WG-314-1001) according to the manufacturer’s instructions. Each beadchip probes 485 577 CpG loci in the human genome. Polymer-coated chips were image-processed in Illumina’s iScan scanner.
Data acquisition
Raw.idat files were imported using the Bioconductor (http://www.bioconductor.org) suite for R. Methylation levels, β, were represented according to the following equation:

Where M represents the signal intensity of the methylated probe and U represents the signal intensity of the unmethylated probe. Illumina recommends adding the constant 100 to the denominator to regularize β-values with very low values for both M and U. Probe dye bias was normalized using built-in control probes. Data points with a detection P-value <0.01 were dropped. Finally, probes from X and Y chromosomes were excluded, leaving 473 864 unique probes.
Cluster analysis
Principal component analysis was performed on quantile normalized data, excluding normal lung samples. Informative probes with a s.d.>0.2 were used for consensus clustering of Ilumina 450 k data. Different clustering algorithms and number of clusters were investigated extensively including k-means, PAM and hierarchical clustering approaches. The solution with the number of clusters reported here is based on cluster stability, silhouette width, the change in the area under the cumulative distribution function and biological significance of predicted subtypes. Scatter plots comparing methylation between two groups represent the average β-value at each probe for that group with color values indicating density after two-dimensional kernel density smoothing.
Differential methylation and gene expression
The difference in DNA methylation β-value was taken between each primary SCLC sample and its patient-matched normal lung. A linear model was fit to the remaining probes and P-values were calculated and adjusted by the method of Bonferroni. A q-value cutoff of 0.05 was used resulting in 1689 significantly differentially methylated CpG sites. Differentially methylated regions were identified as previously described.23 For gene expression analysis, RNA-seq counts were converted to log2 counts per million and correlated by the Spearman method to β-values of differentially methylated CpGs. Concordance at the top plots were generated by comparing the concordance between different contrasts at the top of the list of differentially methylated probes as a function of the size of the ranked probe list.
Patient-derived xenografts
PDX were propagated as previously described with the addition of a dissociation step using a gentleMACS automated dissociator and human tumor dissociation kit (Miltenyi, San Diego, CA, USA).20 LX lines were derived at Johns Hopkins University. Lu lines were obtained from Stem CentRx, Inc (San Francisco, CA, USA). PDX identity was confirmed by Short Tandem Repeat analysis using the PowerPlex 18 panel (Promega, Madison, WI, USA; DDC Medical is Fairfield, OH, USA). The LX92 PDX model was engrafted from fresh tumor tissue in 6–8-week-old female athymic mice (Hsd:Athymic-Nude:Foxn1nu). Briefly, 5 × 106 viable cells/mouse were injected with a 1:1 mix of Hank’s balanced salt solution and matrigel basement membrane (Corning, Corning, NY, USA) as a final volume of 100 μl subcutaneously into the right hind flank. Mice were randomized to treatment arms once tumors reached ~150 mm3. EPZ-6438 was purchased from ActiveBiochem and formulated in 0.5% NaCMC w/ 0.1% Tween-80 in water. Twice-daily (b.i.d.) dosing was done on a 1000 h/1600 h schedule, Monday through Friday. Tumor volumes and mice weights were measured twice weekly. Tumor tissues used for western blot were collected 2 h after the final dose.
Ex vivo cell culture
PDX were dissociated as above. Single-cell suspensions were filtered through a 70-μm mesh, washed twice with wash buffer (phosphate-buffered saline, 2% fetal bovine serum and 1 mM EDTA) and red blood cells were lysed with ACK buffer (Crystalgen Inc., Commack, NY, USA). Approximately 1 × 106 viable cells were seeded in 10 ml of DMEM:F12 supplemented with an EGM-2 growth factor kit (Lonza, Basel, Switzerland). Once cultures were established (~72 h post dissociation), 96-well plates were seeded with 5000 cells/well in 100 μl of ex vivo culture media and the following compounds were administered daily to a final concentration of 1μM: DMSO (vehicle), EPZ-5687, GSK343 or UNC1999 (Selleck Chemicals, Houston, TX, USA). Resazurin conversion of alamarBlue (Invitrogen, Carlsbad, NY, USA) was monitored on a Synergy Neo HTS plate reader (Biotek, Winooski, VT, USA).
Western blotting
Protein was extracted using RIPA buffer supplemented with Halt protease and phosphatase inhibitor cocktails (Pierce, Rockford, IL, USA). Extraction of nuclear proteins required three sequential 5-s pulses at 65% amplitude using a 120-W needle-tip ultrasonic processor (Fisher Scientific, Pittsburgh, PA, USA). The following antibodies were used for western blot according to the manufacturer’s recommendations: EZH1 (Thermo, Waltham, PA, USA; #PA1-41114), EZH2 (Cell Signaling, Danvers, MA, USA; #5246), β-actin (Cell Signaling; #4967), H3K27me2 (Cell Signaling; #9728), H3K27me3 (Cell Signaling; #9733) and total H3 (Cell Signaling; #4499).
Public data
RNA-seq, copy-number variation and mutation data are previously described and available at the European Genome-phenome Archive (EGAS00001000334).7 TCGA samples used for this study are listed in Supplementary Table S8.
References
Siegel R, Naishadham D, Jemal A . Cancer statistics 2012 CA Cancer J Clin 2012; 62: 10–29.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D . Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90.
Wistuba II, Gazdar AF, Minna JD . Molecular genetics of small cell lung carcinoma. Semin Oncol 2001; 28: 3–13.
Oze I, Hotta K, Kiura K, Ochi N, Takigawa N, Fujiwara Y et al. Twenty-seven years of phase III trials for patients with extensive disease small-cell lung cancer: disappointing results. PLoS One 2009; 4: e7835.
Madrigal PA, Manga GP, Palomero I, Gomez RG . VP16-213 combined with cis-platinum (CDDP) in the treatment of small cell carcinoma of the lung (SCLC). Cancer Chemother Pharmacol 1982; 7: 203–204.
Lara PN Jr., Natale R, Crowley J, Lenz HJ, Redman MW, Carleton JE et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol 2009; 27: 2530–2535.
Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 2012; 44: 1111–1116.
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012; 44: 1104–1110.
Clinical Lung Cancer Genome Project, Network Genomic Medicine. A genomics-based classification of human lung tumors. Sci Transl Med 2013; 5: 209ra153.
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489: 519–525.
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337.
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70.
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455: 1061–1068.
Kalari S, Jung M, Kernstine KH, Takahashi T, Pfeifer GP . The DNA methylation landscape of small cell lung cancer suggests a differentiation defect of neuroendocrine cells. Oncogene 2013; 32: 3559–3568.
Rauch T, Pfeifer GP . Methylated-CpG island recovery assay: a new technique for the rapid detection of methylated-CpG islands in cancer. Lab Invest 2005; 85: 1172–1180.
Hann CL, Rudin CM . Fast, hungry and unstable: finding the Achilles' heel of small-cell lung cancer. Trends Mol Med 2007; 13: 150–157.
Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 2011; 43: 768–775.
Poirier JT, Dobromilskaya I, Moriarty WF, Peacock CD, Hann CL, Rudin CM . Selective tropism of Seneca Valley virus for variant subtype small cell lung cancer. J Natl Cancer Instit 2013; 105: 1059–1065.
Carney DN, Gazdar AF, Bepler G, Guccion JG, Marangos PJ, Moody TW et al. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res 1985; 45: 2913–2923.
Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res 2009; 69: 3364–3373.
Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC et al. Multiple-laboratory comparison of microarray platforms. Nat Methods 2005; 2: 345–350.
Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 2012; 44: 40–46.
Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol 2012; 41: 200–209.
Gardner EE, Connis N, Poirier JT, Cope L, Dobromilskaya I, Gallia GL et al. Rapamycin rescues ABT-737 efficacy in small cell lung cancer. Cancer Res 2014; 74: 2846–2856.
Smith LT, Lin M, Brena RM, Lang JC, Schuller DE, Otterson GA et al. Epigenetic regulation of the tumor suppressor gene TCF21 on 6q23-q24 in lung and head and neck cancer. Proc Natl Acad Sci USA 2006; 103: 982–987.
Wilkerson MD, Yin X, Hoadley KA, Liu Y, Hayward MC, Cabanski CR et al. Lung squamous cell carcinoma mRNA expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clin Cancer Res 2010; 16: 4864–4875.
Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014; 506: 445–450.
Zouridis H, Deng N, Ivanova T, Zhu Y, Wong B, Huang D et al. Methylation subtypes and large-scale epigenetic alterations in gastric cancer. Sci Transl Med 2012; 4: 156ra40.
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010; 17: 510–522.
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38: 787–793.
Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropath 2012; 124: 547–560.
Chang CJ, Hung MC . The role of EZH2 in tumour progression. Brit J Cancer 2012; 106: 243–247.
Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K . EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 2003; 22: 5323–5335.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW . Cancer genome landscapes. Science 2013; 339: 1546–1558.
Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov 2012; 2: 798–811.
Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013; 500: 477–481.
Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 2013; 23: 555–567.
Wilkerson MD, Yin X, Walter V, Zhao N, Cabanski CR, Hayward MC et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS One 2012; 7: e36530.
Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A . Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003; 4: 181–189.
Wee ZN, Li Z, Lee PL, Lee ST, Lim YP, Yu Q . EZH2-mediated inactivation of IFN-gamma-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep 2014; 8: 204–216.
Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol 2013; 9: 643–650.
Acknowledgements
We thank Inna Kodos in the Sloan Kettering Antitumor Assessment Core for her technical expertise. We are grateful to all members of the Rudin and Hann labs for thoughtful discussions. SU2C, P30 CA008748 gave financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
CMR has been a paid consultant regarding cancer drug development for AbbVie, Aveo, Celgene, GlaxoSmithKline and Merck. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Poirier, J., Gardner, E., Connis, N. et al. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 34, 5869–5878 (2015). https://doi.org/10.1038/onc.2015.38
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.38
- Springer Nature Limited
This article is cited by
-
Expression of down-regulated ERV LTR elements associates with immune activation in human small-cell lung cancers
Mobile DNA (2023)
-
p53 restoration in small cell lung cancer identifies a latent cyclophilin-dependent necrosis mechanism
Nature Communications (2023)
-
Protein Expression of immune checkpoints STING and MHCII in small cell lung cancer
Cancer Immunology, Immunotherapy (2023)
-
Depletion of enhancer zeste homolog 2 (EZH2) directs transcription factors associated with T cell differentiation through epigenetic regulation of Yin Yang 1(YY1) in combating non-small cell lung cancer (NSCLC)
Medical Oncology (2023)
-
Novel Therapeutic Options for Small Cell Lung Cancer
Current Oncology Reports (2023)