
Abstract
As the majority of patients with basal-like breast carcinoma present with invasive, metastatic disease that do not respond to available therapies, it is essential to identify new therapeutic targets that impact invasion and metastasis. Protease-activated receptor 1 (PAR1), a G-protein coupled receptor has been shown to act as an oncogene, but underlying mechanisms are not well understood. Here, we show that ectopic expression of functionally active PAR1 in MCF-7 cells induced a hormone-refractory, invasive phenotype representative of advanced basal-like breast carcinoma that readily formed metastatic lesions in lungs of mice. PAR1 was found to globally upregulate mesenchymal markers, including vimentin, a direct target of PAR1, and downregulate the epithelial markers including E-cadherin, as well as estrogen receptor. In contrast, non-signaling PAR1 mutant receptor did not lead to an invasive, hormone refractory phenotype. PAR1 expression increased spheroid formation and the level of stemness markers and self-renewal capacity in human breast cancer cells. We identified HMGA2 (high mobility group A2) as an important regulator of PAR1-mediated invasion. Inhibition of PAR1 signaling suppresses HMGA2-driven invasion in breast cancer cells. HMGA2 gene and protein are highly expressed in metastatic breast cancer cells. Overall, our results show that PAR1/HMGA2 pathway may present a novel therapeutic target.
Similar content being viewed by others


Introduction
Protease-activated receptor 1 (PAR1) was originally discovered on platelets and serves as the prototype for this specialized protease-activated class of G-protein-coupled receptors.1 In addition to its well recognized roles in vascular biology, PAR1 has also been proposed to be involved in the invasive and metastatic processes of breast,2, 3 lung,4 pancreatic cancer5 and melanoma,6, 7, 8 and has been identified as an oncogene in the transformation of NIH3T3 mouse fibroblasts.9, 10 Bar-Shavit et al.2 demonstrated that high levels of PAR1 mRNA were found in infiltrating ductal carcinoma and very low amounts in normal and premalignant atypical intraductal hyperplasia. This observation was extended into a large study involving over 120 different breast patient samples. In another study, Korkola et al.11 found that PAR1 expression levels increased by an average of 4–10-fold in 106 invasive ductal and 17 invasive lobular tumors as compared with 6 normal breast specimens. Studies by our group and others showed that the invasive breast, lung and ovarian cancer cells express very high levels of functional PAR1,4, 12, 13 whereas minimally invasive MCF-7 cells have no PAR1.14 Recently, we also showed that blockade of PAR1 signaling with cell-penetrating pepducins greatly inhibits tumor growth in xenograft models4 and survival and metastasis to the lungs.15 The basal-like breast tumors16 are frequently triple negative (negative for estrogen receptor, progesterone receptor and HER2), and current chemotherapeutic regimens are unlikely to result in complete remission; thus, there is still a significant need to identify novel oncogenic targets that can enhance chemotherapeutic vulnerability to resistant disease. Here, we investigate the regulatory role of PAR1 in the basal-like breast cancer and identify the key transcription factor involved in tumor progression and metastasis.
The key molecular events leading to invasion is not well understood in basal-like breast cancer. The acquisition of invasive capacity by epithelial cells is observed in embryogenesis, wound-healing and metastasizing tumor cells.17 Epithelial cells form organized structures with cellular polarity, adjoined by specialized cellular junctions. In contrast, invading cancer cells depart from organized tissue structures, are highly motile and may morphologically resemble cells of mesenchymal origin. The mesenchymal phenotype is characterized by the loss of epithelial structural/adhesion proteins, such as cytokeratin, desmoplakin, zona occludens and claudin, replaced by their mesenchymal counterparts, vimentin, N-cadherin, OB-cadherin, integrin, fibronectin, laminin and smooth muscle actin.18 Vimentin, the intermediate filament family member is an important marker of mesenchymal phenotype.19 Identification of actionable targets that can regulate transition to an invasive/mesenchymal phenotype may reveal a new approach for the development of effective therapeutics.
Targeting master trasncriptional regulators that promote a mesenchymal phenotype and cellular invasion is an attractive approach to cancer therapy that can prevent metastasis and reduce therapeutic resistance. A seminal event in the acquisition of an invasive/mesenchymal phenotype is the loss of E-cadherin, a homotypic adherens junction molecule ubiquitously expressed in epithelial cells.18, 20 E-cadherin expression can be lost through a variety of genetic and epigenetic mechanisms, including the upregulation of classical transcriptional repressors such as SNAI1, SNAI2/SLUG and ZEB1.21, 22, 23 The high-mobility genes, principally HMGA-2 on chromosome 12, is implicated in the pathogenesis of soft tissue sarcomas, including liposarcoma24 and aggressive angiomyxoma.25 Recently the transcription factor HMGA2 (High Mobility Group AT-2 hook), a nuclear non-histone transcriptional regulator, was reported to be overexpressed in breast carcinomas of high histological grade.26 Importantly, the HMGA family of proteins have an important role in self-renewal, cellular proliferation and differentiation.27 HMGA proteins include HMGA1 (two splice varients HMGA1a and HMG1b) and highly related HMGA2 that bind AT-rich DNA chromatin and form multiprotein complexes known as ‘enhanceosomes’ that act on the promoter/enhancer region of genes that globally regulate gene transcription.28, 29
In this report, we show that overexpression of PAR1 in human breast cancer cell lines induces a basal-like phenotype though the non-histone chromatin-binding protein HMGA2. Furthermore, PAR1 ectopic expression leads to cancer stem cell marker expression—a hallmark of basal-like breast cancer.
Results
PAR1 overexpression transforms breast cancer cells into metastatic phenotype
Constitutive overexpression of PAR1 cells promotes anchorage and serum-independent growth, and has been strongly implicated in breast cancer invasion and metastasis. MCF-7 cells are thought to represent an early breast cancer phenotype because they are estrogen-sensitive and require transfection with oncogenes or invasogenic factors to become tumorigenic in nude mice.30 For this purpose, we have generated stable cell lines with ectopic expression of PAR1 in the PAR1-null human breast cancer cell line MCF-7 and tested their pro-invasive properties using three-dimensional (3D) matrigel growth. We evaluated the level of ectopic PAR1 expression in six randomly chosen stable MCF-7 clones using western blot analysis (Figure 1a). Furthermore, we made a stable cell line with a ‘signaling-dead’ PAR1 mutant that had a mutation in the third intracellular loop, R310E-PAR1. High levels of PAR1 expression that appear as highly glycosylated broad protein band31 were seen in clones N18, N26, N29, N55, R310E and MDA-MB-231 human breast cancer cell line with known expression of PAR115 (Figure 1a). These clones and parental cells were evaluated for 3D growth in matrigel to characterize their growth potential and invasiveness through basement membrane. MDA-MB-231 cells (Figure 1b) formed large branching stellate structures suggestive of highly invasive phenotype. In contrast, PAR1-null cell lines (clone N19 and N31) and R310E-PAR1 ‘dead’ receptor did not show any significant growth and formed a small number of round spheres. PAR1-expressing clones N18, N26, N29 and N55 all formed large branching stellate structures reminiscent of MDA-MB-231. These findings suggest that PAR1 expression promotes cancer cell growth and invasion. As an alternative to 3D growth in matrigel, invasive growth patterns in collagen type I were tested for MDA-MB-231, PAR1-MCF7/N55 and R310E-MCF7 cell lines. Collagen type I growth conditions revealed that MCF7 cells expressing functional PAR1 (N55) unlike mutant PAR1 (R310E) form infiltrative front similar to MDA-MB-231 and with a prominent stellate-appearance compared with matrigel (Figure 1c). To study the role of PAR1 in anchorage-independent growth, MCF7 cells expressing PAR1 and mutant R310E were grown in soft agar. Representative images of the wells with colonies developed inside soft agar on day 28 suggest that PAR1 significantly increased anchorage-independent colony formation (Supplementary Figure 1).

PAR1 promotes invasion and metastasis in breast cancer cells. (a) Series of MCF7-derived stable cell lines that either express PAR1 (N18, N26, N29, N55) or PAR1-null (N19 and N31) and R310E ‘dead PAR1’ were compared with MCF-7 parental and MDA-MB-231 cell lines for protein expression of PAR1 and β-actin using western blotting. Representative western blot of PAR1 and β-actin are from three independent experiments. (b and c) Representative 3D growth of breast cancer cell lines cultured in matrigel (b) and collagen type I (c) and photographed after 6 days with a light microscope and SPOT digital camera (Diagnostic Instruments) carried out in duplicate, two independent times (bar, 200 μm). (d and e) Metastatic potential of MCF7-PAR1/N55 (n=8), MDA-MB-231 (n=5), R310E-MCF7 (n=6) and MCF-7 (n=6) i.v. administered into NCR nu/nu mice. MCF7, MCF7-PAR1, R310E-PAR1 and MDA-MB-231 cells (2 × 106) introduced via the tail vein of female nude mice for 6 weeks. Representative sectioned, hematoxylin and eosin-stained lungs (bar, 100 μm) are shown 6 weeks after delivery of MCF7-PAR1, MD-MB-231, MCF7 and R310E-PAR1. T, tumor nodules.
Next, we tested whether the pro-invasive phenotype conferred by PAR1 in MCF-7 cells under in vitro conditions could be extended to an in vivo experimental metastasis tumor model. MCF7-PAR1/N55, the MCF-7 controls (PAR1-null) parental cell line and R310E ‘dead- PAR1’, and MDA-MB-231 cells (2 × 106, respectively) were injected via tail vein of female 6–7-week-old athymic (Harlan-Sprague-Dawley) nu/nu mice. After 6 weeks, mice were killed and the lungs were extracted for histologic evaluation of tumor growth. All mice that received MCF7-PAR1/N55 breast cancer cells developed extensive tumor nodules (8/8) in contrast to mice injected with MCF-7 cells (PAR1-null) or R310E (PAR1-dead) that had no detectable tumor formation (0/6 for both) after 6 weeks (Figures 1d and e; Supplementary Figure 2). Notably, the tumor burden was comparable in the N55- and MDA-MB-231-injected mice. The observations here suggest that ectopic PAR1 expression in MCF-7 cells is sufficient to confer growth and metastatic potential akin to those seen in MDA-MB-231, a cell line that models aggressive, hormone-independent breast cancer. Long-term goal is to determine whether PAR1 has role in spontaneous metastasis from primary tumors.
Overexpression of PAR1 regulates self-renewal
Given the striking ability of PAR1 to promote tumor growth and experimental metastasis, we then examined the ability of PAR1 to regulate stem-like self-renewal properties. We determined the ability of PAR1-MCF7 cells for self-renewal using serial colony-formation unit (CFU) analysis to differentiate between the highly proliferative progenitors vs potential stem cells. As shown in Figure 2a, each of the PAR1-expressing clones (N18, N26, N28 and N55) after initial seeding (P1) had 2–14-fold greater capacity (P<0.004, white bars) to form colonies larger then 30 cells, compared with cells without PAR1 expression (N19, N31) or without functional PAR1 (R310E). This was 9–69% capacity of CFU activity compared with the MDA-MB-231 cells. After 6 days, colonies were trypsinized and re-plated (P2) at a low density (equivalent to P1), which showed increase in their CFU activity for all the clones (P2, grey bars, P<0.0001). Upon the third serial passage, PAR1-expressing clones continued to enrich for CFUs (P3, P<0.000003) compared with PAR1-null (N19 and N31) and PAR1-‘dead’ (R310E) which exhibited no significant increase in capacity to form CFUs.

PAR1 regulates self-renewal. (a) PAR1-expressing cells are enriched for self-renewal activity. Serial CFUs from MCF7-PAR1-expressing clones 1-N18, 2-N26, 3-N29, 4-N55 and PAR1-null clones 5-N19, 6-N31 and PAR1-‘dead’ 7-R310E and 8-MDA-MB-231. Data are mean±s.d., n=3, representating three independent experiments. (b) Silencing of PAR1 expression in MCF7-PAR1/N55 cells with shPAR1. Surface expression of PAR1 was determined by flow cytometry. Pink and green traces: secondary antibody alone, blue and red traces: primary (SFLLRN-ab) plus secondary antibodies. (c) Serial CFUs for MCF7-PAR1/N55 and MCF7-PAR1 N55-shPAR1. Data are mean±s.d., n=3. (d) Quantitative analysis of tumorsphere formation efficacy of MCF7-PAR1/N55 and MCF7 cells. The sphere quantification and size (μm) were determined after 7 days. Data are mean±s.d., n=3. Statistical analysis using two-tailed Student’s t-test. *P<0.05, ***P<0.001.
To confirm that the increase in ability for self-renewal seen in PAR1-expressing clones is directly due to PAR1 expression, we tested the effect of silencing PAR1 with shPAR1 in the MCF7-PAR1/N55 clone. PAR1 surface expression was assessed by fluorescence-assisted cell sorting as shown in Figure 2b; there was 50% decrease in median surface expression of PAR1 relative to parental N55 control. The effect of reduced PAR1 surface expression was tested on the ability to form CFUs. As shown in Figure 2c, the shPAR1-derived cells formed 58% less CFUs as compared with the parental cell. This loss of PAR1 expression may be responsible for reduced capacity of these cells to self-renew and suggests that PAR1 may regulate self-renewal in breast cancer. N55-MCF7 cells were next tested for the capability to form tumorspheres. These cells formed threefold larger number of spheres (Figure 2d) that were also larger in size as compared with parental MCF7 cells, further supporting PAR1 role in both initiating and maintaining tumorspheres in the absence of cellular adhesion.
Increased pro-metastatic phenotype: switch in E-cadherin and vimentin expression
We have previously demonstrated that MCF-7 cells stably expressing PAR1 (MCF-7/PAR1) display significant increases in migration, invasion and tumorigenicity.12 Here, we correlate the invasive capacity of MCF-7/PAR1 cells with changes in cellular morphology. Consistent with the lack of motility, we observed that MCF-7 cells retain an epithelial morphology with clustered growth and significant cell-cell adhesion. In contrast, MCF-7/PAR1 cells obtained a spindle-shaped, fibroblast-like morphology with decreased cellular contacts (Figure 3a).

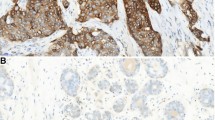
Expression of PAR1 correlates with vimentin expression in human breast cancer cell lines. (a) Immunofluorescence photomicrographs (40 × ) of MCF-7 and MCF-7/PAR1 cells stained for PAR1, phalloidin, E-cadherin or vimentin. Nuclear DNA was stained with 4',6-diamidino-2-phenylindole (blue). Representative data from multiple experiments are shown. (b) Total lysates of MCF-7, MCF7-PAR1-expressing clones 1-N18, 2-N26, 3-N29, 4-N55 and PAR1-null clones 5-N19, 6-N31 and PAR1-‘dead’ 7-R310E immunoblotted for vimentin and E-cadherin. β-actin was used as loading control. The protein size is indicated on the right in kilodalton (kDa). #PAR1 receptor is highly glycosylated and runs as a broad band. *Vimentin and its cleaved proteolytic fragments are detected as multiple bands. (c) NCI-60 panel of breast carcinoma cells were lysed and immunoblotted for PAR1, vimentin and E-cadherin. β-actin was used as the loading control. Representative data from multiple experiments are shown. (d) Representative immunohistochemistry images of vimentin in breast cancer tissue microarray samples (Car, carcinoma; H, high; M, medium; L, low; N, normal tissue; CA, carcinoma adjacent normal breast tissue). (e) Percentage of distribution according to immune staining of vimentin, scored for H, M and L expression, the score for negative expression (0%) and CA (100%).
PAR1-expressing MCF-7 cells are accompanied with increased formation of actin stress fibers as demonstrated with phalloidin staining (Figure 3a). To further characterize the phenotypic differences, we labeled the cells for E-cadherin (epithelial adherens junction protein) and vimentin (mesenchymal intermediate filament) and looked for changes associated with PAR1 expression. MCF-7 cells displayed a typical epithelial phenotype with E-cadherin localized at cell-cell junctions. We observed no expression of vimentin or PAR1. In association with PAR1 expression, we saw that MCF7/PAR1 cells gained vimentin, which was polymerized into filamentous cytoskeletal structures (Figure 3b). These cells further demonstrated a complete loss of E-cadherin expression both at the cell surface and in the cytoplasm. Consistent with the immunofluorescence results, western blot results demonstrated that PAR1 expression strongly correlates with the induction of vimentin and loss of E-cadherin (Figure 3b and Figure 5c). Full-sized vimentin gives a single-band of the expected size of ~56 kDa. However, several investigators have reported that vimentin can be detected as smaller sized degradation products due to proteolysis of the N-terminus, typically appearing as ~48 kDa products or smaller with stepwise removal of a 9 kDa units.32, 33 Proteolysis may be an important factor in regulating vimentin stability in these cells. MCF-7/PAR1 clones (Figure 3b and Figure 5c) express vimentin and cleaved proteolytic vimentin fragments, detected as multiple bands. Cells that lack PAR1 expression or express ‘dead’ PAR1 do not gain vimentin and instead, retain E-cadherin expression. In fact, among the NCI-60 panel of breast cancer cells, those expressing PAR1 (HS-578 T, Bt-549 and MDA-MB-231) are morphologically and immunophenotypically mesenchymal (vimentin (+), E-cadherin (−)) (Figure 3c). Conversely, PAR1-null cells (MCF-7 and T47D) are characteristically epithelioid (vimentin (−), E-caherin (+)).
We further examined vimentin expression in primary tumors from well- or moderately differentiated invasive ductal carcinoma using US Biomax (T088) breast cancer tissue array, a microarray panel that contained 16 panels of malignant tumor (Car), 4 normal tissue adjacent to tumor (CA) and 4 normal tissues. As shown in Figures 3d and e, vimentin expression increased along with the histologic grade of the tumor. In contrast, there was no expression detected in normal tissue. Most intriguingly, vimentin was detected in all cancer-adjacent normal tissue, although it is not clear whether these vimentin-positive cells are normal breast epithelial cells or desmoplastic stromal cells. Taken together, vimentin expression correlates with PAR1 expression and appears to indicate aggressive and invasive phenotypes.
PAR1 directly regulates vimentin expression
Given the significant upregulation of vimentin in primary breast cancer and breast cancer cell lines as seen above, we next characterized metastatic breast tumor to the lung (previously described in Figure 1d) for vimentin expression using immunohistochemistry. Consistent with previous data, vimentin expression was detected in metastatic breast tumor nodules formed by PAR1-expressing cells (N55 and MDA-MB-231; Figure 4a). Intense vimentin staining (a red color) was detected within all the breast tumor cells consistent with uniform robust vimentin expression detected in stably expressing PAR1 cell line (Supplementary Figure 3b). In contrast, MCF7 cells expressing either dead PAR1 or PAR1-null did not form tumors and had normal lung morphology.

PAR1 regulates vimentin expression. (a) Representative histology from sections of lung tissue from female nude mice tail vein injected with MCF7-PAR1 and MDA-MB-231 (2 × 106) after 6 weeks and immunohistochemically stained for vimentin. (b) MCF7 cells transiently transfected with PAR1 were stimulated with thrombin (1 nmol/l) or MMP-1 (1 nmol/l) for 0.5 to 2 h or 17β-estradiol (E2, 100 nmol/l) for 5 h. Expression level of vimentin and actin were determined using SYBR Green I-based quantitative PCR. The relative vimentin gene expression was normalized to the mean value of nonstimulated MCF7 cells transiently transfected with PAR1, and the value was set to 1. Individual points represent mean, and bars represent s.d. (c) MCF-7 cells were transiently transfected with PAR1 or vector (pcDEF3) control and 48 h after transfection, cells were immunostained for PAR1 (PE-red), vimentin (FITC-green) and 4',6-diamidino-2-phenylindole (blue) for nuclear DNA. All panels include MERGED image of three immunofluorescence photomicrographs with overlay of red+green+blue. MERGED stable MCF7-PAR1/N55 cells ( × 40, bar, 50 μm) are shown in the lower panel. (d) MCF-7/PAR1 cells were treated 1–5 days with vehicle (0.2% DMSO) or PZ128 (1 and 3 μM). Cells were lysed and immunoblotted for vimentin. β-actin was used as the loading control. Representative data from multiple experiments are shown.
The co-expression of PAR1 and vimentin is demonstrated in Figure 4c (bottom panel and Supplementary Figure 3b; vimentin-FITC (green) and PAR1-PE (red)). To directly show that PAR1 activation can induce vimentin in breast cancer cells, we transiently transfected MCF7 cells with PAR1 and activated with thrombin or matrix metalloprotease-1 (MMP1). Real-time reverse transcription–PCR (RT–PCR) analysis was used to quantify relative levels of vimentin mRNA. Treatment of PAR1 expressing MCF7 cells with 1 nmol/l thrombin or 1 nmol/l MMP1 caused a twofold induction of vimentin mRNA after 30 min with little effect on actin mRNA expression (Figure 4b). Interestingly, 17β-estradiol gave similar induction in vimentin mRNA at the 5-h time point.
Consistent with the RT–PCR results, transient expression of PAR1 resulted in co-expression of vimentin, as seen by immunofluorescence in Figure 4c (top panel and Supplementary Figure 3a). MCF-7 cells transfected with an empty vector (pcDEF3) lacked PAR1 and vimentin expression (Figure 4c, middle panel). Transient expression of PAR1 in MCF7 cells resulted in ~18% PAR1 transfection efficiency and ~25% of total cells expressed vimentin (Supplementary Figure 3a). On the basis of these data, majority (~77%) of PAR1 transfected cells expressed dual PAR1 and vimentin.
In a converse experiment, 5-day treatment of PAR1-expressing breast cancer cells with a PAR1 inhibitor, PZ128, almost completely downregulated vimentin in MCF-7/PAR1 cells, further supporting the regulatory role of PAR1 on vimentin (Figure 4d).
PAR1 induces epithelial to mesenchymal transition in the MCF-7 breast carcinoma cell line
Given the above changes in cellular phenotype and protein expression, we used the Human Genome U133A 2.0 Array (Affymetrix) containing an 18400 probe set representing 14 500 human transcripts to investigate global changes in gene transcription in response to PAR1 expression (Figure 5). Hierarchical gene clustering by euclidean distance revealed that, of all transcripts analyzed, vimentin experienced the greatest transcriptional induction (+3988-fold; P<1 × 10−3). Conversely, E-cadherin and Estrogen Receptor/Trefoil Factor demonstrated the greatest transcriptional reduction (−1383, −1681 and −3794-fold decrease, respectively; all P<1 × 10−3). Trefoil factor is a pro-angiogenic estrogen-responsive gene, and its downregulation reemphasizes the loss of estrogen receptor activity.34 The significant downregulation of estrogen receptor is in agreement with our recent finding that MCF-7/PAR1 cells are able to form tumors in the mammary fat pads of female nude mice without estrogen supplementation.

PAR1 induces mesenchymal transition. (a) Gene expression profile of MCF-7 and MCF-7/PAR1 cells, maintained in phenol red-free RPMI and 10% charcoal-stripped fetal bovine serum to prevent inadvertent estrogen receptor stimulation. Cells were quiesced overnight in serum-free media before RNA isolation. Purified total RNA was analyzed in triplicates using the Human Genome U133A 2.0 Array (Affymetrix) representing 14 500 human transcripts. Representative EMT-marker genes were selected after hierarchical clustering by euclidean distance. (b) Schematic summery of global change in gene transcripts upon PAR1 expression. (c) Western blot analysis of expression of PAR1, vimentin, integrin α6, laminin, estrogen receptor 1, E-cadherin, claudin, zona occludens, cytokeratin and β-actin as control in MCF7 and two independent MCF-PAR1/N26 and /N55 clones compared with highly aggressive MDA-MB-231. The protein size is indicated on the left in kilodalton (kDa).
Overall, we observed here a remarkable transcriptional shift highly indicative of a mesenchymal transition. There was an increase in expression of TGF-β family members that have been well established as a potent inducer of mesenchymal transition in mammary cells that involves acquisition of tumor stem-like properties.35 In MCF-7/PAR1 cells, the loss of epithelial junctional proteins (E-cadherin (adherens junction), claudin 3/zona occluden 3 (tight junction), desmoplakin (desmosome)) and epithelial intermediate filaments (cytokeratin 8/18/19) was accompanied by the gain of mesenchymal intermediate filament (vimentin), mesenchymal adhesion molecules (OB-cadherin, integrin α6) and stromal proteins (collagen, laminin, fibronectin) (Table 1). Protein levels of representative epithelial to mesenchymal transition (EMT) markers were also confirmed by western blot (Figure 5c). We analyzed the EMT status of two independent clones of MCF-7 cells stably expressing PAR1 (clones N26 and N55; N55 was used for the genome array). The mesenchymal state of both clones indicates that the observed shift in transcriptional events is specific to PAR1 expression and not the artifact of a fortuitous gene-insertion event. In fact, we see that both clones display significant similarities with MDA-MB-231, a highly invasive breast cancer cell line expressing high levels of endogenous PAR1. Taken together, we have demonstrated here that the ectopic expression of PAR1 in MCF-7 cells induces a hormone-refractory, mesenchymal phenotype representative of advanced stage metastatic breast carcinoma.
Regulation of PAR1 and HMGA2 expression
Lastly, microarray data revealed transcriptional regulators of the self-renewal and invasive program, such as HMGA2, and ZEB1 to be upregulated more than 200-fold, suggesting their potential functional relevance in PAR1-mediated tumor-invasive program (Supplementary Figure 4). The expression levels of other prominent EMT regulators, such as ZEB2/SIP1, SNAI1, TWIST1 and NFκB, did not significantly change (all microarray data discussed in this report has been deposited to NCBI gene expression and hybridization array data repository (GEO, www.ncbi.nlm.nih.gov/geo)). To correlate the expression of HMGA2 to PAR1, we analyzed both mRNA (qPCR) and protein (western blot) levels of HMGA2 levels in four invasive human breast cancer cell lines with varying levels of PAR1 expression: PAR1 high (MCF7-PAR1/N55, MDA-MB-231), intermediate (Hs578 T), low (BT-549) and negative (T47D and MCF7-310E (PAR1 ‘dead’)) (Figures 1a and 3c). Cell lines expressing high levels of PAR1 (MDA-MB-231, N55) demonstrated high HMGA2 expression, and conversely, cell lines with no PAR1 (MCF-7, T47D) had HMGA2 expression below the threshold for mRNA or protein detection (Figure 6a, mRNA; Figure 6b, protein). Cell lines with intermediate (HS-578 T) and low (BT-549) PAR1 expression do not have clear correlation. The HS-578T cell line reproducibly produced a potential HMGA2 band with higher mobility (~20 kDa) than the expected 18 kDa. Because HMGA2 is frequently disrupted or aberrantly expressed although most frequently in human neoplasis,36, 37 it remains feasible that this is a variant of HMGA2, however, this still needs to be confirmed. HMGA2 was not detectable by western blotting in BT-549.

Regulation of PAR1 and HMGA2 expression. (a) Quantitative RT–PCR analysis of HMGA2 mRNA in MCF7-PAR1 (N55), MDA-MB-231 (MDA), HS-578 T (Hs), Bt-549 (BT), T47D and MCF7. Values shown are fold-difference of RNA expression compared with MCF7 cells (arbitrarily assigned a value of 1). Error bars depict mean±s.e.m. from two independent experiments performed in triplicate. (b) Western blot analysis of HMGA2 and actin in the cell lines characterized in part a. *Hs cells detect an aberrant 20 kDa protein. (c) Invasion of MCF7-PAR1/N18 cells transfected with the ZEB1, Slug, HMGA2 and the control Luci siRNA, transwell invasion experiment through matrigel using NIH-3T3 conditioned media as the chemoattractant source in the bottom well. PZ128 (8 μM) was added as indicated to the lower well. For Luci control, the total number of cells was 557. Luci control was used to set invasion as 100% and every other treatment was normalized as compared with the Luci control. (d) MCF-7-PAR1/N18 cells were treated 3 days with vehicle (0.2% DMSO) or PZ128 (8 μM) as indicated. Cells were lysed and immunoblotted for HMGA2. β-actin was used as the loading control. Representative data are shown from two independent experiments. (e) Representative 3D growth of MCF-7-PAR1/N18 cell lines cultured in collagen type I and treated with vehicle (0.2% DMSO) or PZ128 (8 μm) as indicated and photographed after 6 days with a light microscope and SPOT digital camera (Diagnostic Instruments) carried out in duplicate, two independent times (bar, 100 μm). (f) ChIP assay to determine in vivo interaction between HMGA2 and the human PAR1 promoter using P1 to P5 primers (panel h, Supplementary Table 1). The immunopercipitated DNA from PAR1-MCF7/N55 cells was purified and amplified by PCR and run on 1.5% DNA agarose gel. Molecular weight markers are shown on right as base pair (bp). (g) ChIP assay to determine in vivo interaction between HMGA2 in hPAR1 promoter using high HMGA2-expressing PAR1-MCF7/N55 and low HMGA2-expressing R310E-MCF7 cell line. The immunoprecipitated DNA from the two cell lines was purified and amplified by PCR using P3 primers and run on 1.5% DNA agarose gel. The input DNA was used as a positive control for PCR amplification of GAPDH. (h) Schematic of the PAR1 promoter. A map of the 5’ flanking −2.9-kb sequence of the PAR1 gene and relative design of P1 to P5 primers. Potential AP-2 and Sp1 regulatory motifs have been previously described.8 Potential nucleotide binding P3 region (−1097 to −1674 bp) was identified. (i) Schematic of the proposed mechanism for PAR1 promoter by the transcriptional factor HMGA2. *P<0.05.
To address the functional role of HMGA2 and other prominent transcriptional regulators of the self-renewal and invasive programs such as ZEB1 and Slug, we silenced individual transcription factor gene expression using HMGA2-small interfering (siRNA), ZEB1-siRNA, Slug-siRNA or Luciferase-siRNA as control in MCF7-PAR1-expressing cells. As shown in Supplementary Figure 5, treatment of MCF7-PAR1 cells with individual siRNA caused a ~69%, 43% and 67% loss in Slug, HMGA2 and ZEB1 expression, respectively. To determine whether ZEB1, Slug or HMGA2 transcription factor may regulate invasion through collagen type I matrix, we set up a quantitative Boyden chamber invasion-like assay.12 We reproducibly observed reduction in invasion to be specific for the HMGA2 siRNA (P=0.017)-treated cells, whereas ZEB1 siRNA and Slug siRNA were unaffected (Figure 6c). Blockade of PAR1 with PZ128 on top of the HMGA2 siRNA treatment did not result in further decrease in invasion. Together these data suggest that PAR1 and HMGA2 affect invasion through a common signaling pathway.
To determine whether PAR1 can directly regulate HMGA2 protein expression, cells were treated for 3 days with PAR1 inhibitor, PZ128. There was a complete downregulation of HMGA2 protein, further supporting the link in the regulatory role of HMGA2 and PAR1 (Figure 6d). Furthermore, 3D growth in the presence of PZ128 completely blocked the formation of a migratory front and the stellate appearance (Figure 6e), consistent with the previous data that support a PAR1 mediated 3-D in vitro growth.
Given the strong correlation of high PAR1-expressing cell lines and HMGA2 expression, we then asked whether HMGA2 could be involved in the regulation of PAR1 gene expression. The interaction of HMGA2 protein with the PAR1 promoter was examined by the chromatin immunoprecipitation assay (ChIP). First, chromatin fragments were immunoprecipitated from MCF7-PAR1/N55 cells using anti-HMGA2 antibody (Ab). Upon isolation of IP-DNA, five sets of hPAR1 primers P1–P5 (spanning the PAR1 promoter) and input control GAPDH (sequence reported in Supplementary Table 1 and schematically shown in Figure 6h) were used for PCR amplification. Primers were designed to cover the −2.9 kb 5’ region of hPAR1 promoter (Figure 6h). As shown in Figure 6f, IP-HMGA2 Ab identified the P3 primer set (−1097 to −1674 bp) that amplified a 577-bp fragment of the PAR1 promoter as potential binding site for HMGA2. Because R310E-MCF7 (PAR1-dead) cells have minimal expression of HMGA2 and nonfunctional PAR1, these cells were used in the next series of comparative ChIP studies. IP-HMGA2 DNA from MCF7-PAR1/N55 (PAR1) and R310E-MCF7 (R310E) were isolated and a set of primers directed to P2 and P3 region of hPAR1 promoter were amplified. As expected, the PCR amplification for P3 in R310E cells was nearly undetectable, suggesting a weaker PAR1/HMGA2 association in the context of nonfunctional PAR1 (Figure 6g). P2 did not detect any specific binding from either IP-DNA prep. The input signal levels were confirmed using GAPDH to show that equal amounts of DNA were used. Together these data indicate that PAR1 promoter may bind HMGA2 transcription factor within a 577-bp fragment localized at −1097 to −1674 bp (Figure 6h).
Discussion
PAR1 is an oncogenic protein clinically associated with invasive breast carcinoma and characterized as a potent inducer of cancer cell migration, invasion, survival and metastasis.2, 12 Ectopic expression of PAR1 in mammary gland epithelia induces oncogenic transformation, however, the mechanistic basis for its tumorigenic function remains unclear. Here, we demonstrate that PAR1 expression results in a global change in gene transcription with loss of epithelial and gain of mesenchymal markers consistent with a shift to an invasive/mesenchymal phenotype. The in vivo ectopic expression of PAR1 promotes metastasis, suggesting that upregulated PAR1 expression may contribute to malignant transformation. The pathological manifestations of such transcriptional shifts were observed in our previous study in which MCF-7/PAR1 cells obtained hormone-independent in vivo tumor growth and ability to invade into surrounding tissue.12 We have shown that PAR1-mediated invasion is HMGA2-dependent in PAR1-expressing breast cancer cells. PAR1 was found to upregulate mesenchymal markers, including vimentin and downregulated the epithelial markers including E-cadherin resulting in all or none PAR1-dependent regulation of EMT. In contrast, non-signaling PAR1 mutant receptor did not lead to invasive transition. Blockade of PAR1 with intracellular pepducin inhibitor downregulated vimentin and HMGA2 transcription factor without changing E-cadherin expression. Furthermore, transient expression of PAR1 did not affect the loss of E-cadherin as observed with the stable PAR1 expression. Therefore, there may be two distinct programs that are regulated for the full EMT transition: (i) a gain of mesenchymal program that may be PAR1 regulated and (ii) a loss of epithelial program that is a secondary event not directly regulated by PAR1.
Inhibition of PAR1 signaling suppresses HMGA2 expression and PAR1/HMGA2 driven invasion in breast cancer. Also, based on the ChIP studies, PAR1 may be a direct downstream gene regulated via HMGA2 (Figure 6i), creating a feed-forward loop towards an invasive phenotype. In melanoma, PAR1 is negatively regulated by activator protein-2 (AP-2)8 and is in competition with a transcriptional activators such as Sp1 that regulate PAR1 expression. A gain of HMGA2 expression may have a profound effect on the regulation of AP-2/Sp1 binding to hPAR1 promoter region. Interestingly, in prostate cancer, a functional androgen-responsive element was identified upstream of a potential HMGA2-binding site,38 and in MCF7 breast cancer cells, estrogen-responsive elements were identified that regulate PAR1 expression.39 It remains to be determined whether HMGA2 is involved in these regulatory events. HMGA2 is a high-mobility-group non-histone chromosomal protein that possesses no intrinsic transcriptional activity but rather functions in orchestrating the assembly of structures through protein-DNA and protein-protein interactions.40 Recent reports identified HMGA2 as an important regulator of tumorigenesis and metastasis in lung cancer,41 melanoma,42 colon,43 ovarian cancer44 in addition to soft tissue sarcomas such as liposarcoma24 and deep (aggressive) myxoma25 of the female genital tract. Therefore, HMGA2 is becoming recognized as a key mediator in numerous mesenchymal and epithelial malignancies, and this report identifies HMGA2 as an important regulator in PAR1-dependent breast cancer progression.
Role of HMGA2 as it relates to PAR1 expression is not well understood. The ectopic expression of PAR1 in MCF-7 cells induces expression of TFG-β family members, most prominently TGF-β1 (Table 1). The TGF-β signaling pathway is a major inducer of the EMT. TGF-β has been previously identified45 as an important pathway that controls tumor invasiveness and metastasis via HMGA2. The dual role of PAR1/TFG-β and HMGA2 regulatory control warrants further investigation.
Basal-like breast cancer also called ‘triple-negative’ are associated with an aggressive phenotype and poor therapeutic response, and therefore still remains a challenge for clinical interventions. There is an urgent need to develop precise therapies and identify novel targets that impact metastatic spread to prevent higher recurrence in triple-negative breast cancer. After an initial response to chemotherapy, many triple-negative breast cancer patients have recurrence of drug-resistant metastatic disease. Recently, the US Food and Drug Administration approved vorapaxar SCH530348 (Zontivity), a novel PAR1 inhibitor to reduce the risk of myocardial infarction, stroke and cardiovascular death (TRACER; TRAP-2P: TIMI 50).46, 47, 48 PZ128 pepducin PAR1 inhibitor used in this study is presently in Phase I clinical trial.49 It remains to be determined whether PAR1 inhibitors hold great promise in the cancer field. Here, we show that PZ128 can regulate HMGA2 expression, further supporting the regulatory role of HMGA2 by PAR1. It is not clear whether vorapaxar inhibits PAR1 activity in a similar manner.
In summary, we describe here a novel approach taken by PAR1 to regulate EMT. The cooperative activity of PAR1 and HMGA2 ‘enhanceosomes’ to induce a fully mesenchymal phenotype suggests the utility of HMGA2-PAR1 as prognostic markers for metastatic behavior and therapeutic resistance in breast cancer. Furthermore, our results provide mechanistic rationale for the use of a PAR1 inhibitor for breast cancer therapy and HMGA2 as a potential biomarker for efficacy.
Materials and methods
Cell culture and generation of stable cell lines
Breast carcinoma cell lines MCF-7, MDA-MB-231, T47D, Bt-549 and HS-578 T were all obtained from the Developmental Therapeutics, National Cancer Institute/NIH (Frederick, MD, USA). Cells were cultured in NCI-recommended media. Briefly, MCF-7, MDA-MB-231, T47D, Bt-549 and HS-578 T cells were cultured in Dulbecco's modified Eagle's medium or RPMI 1640 media supplemented with 10% fetal bovine serum (Life Technologies/Invitrogen, Grand Island, NY, USA), 1% penicillin/streptomycin in 5% CO2 at 37 °C. Stably expressing PAR1 (MCF-7/PAR1) were generated in our laboratory as previously described.14
Reagents
N-palmitoylated peptide PZ128 also known as P1pal-7 was synthesized as described previously.13, 50 Activation of proMMP-1 with APMA was performed as previously described.12 Thrombin was obtained from Haematologic Technologies Inc (Essex Junction, VT, USA).
Extraction of RNA and real-time PCR analysis
Total RNA was extracted using the RNeasy kit (Qiagen, Valencia, CA, USA). cDNA was amplified as previously described51 and PCR was conducted using Q Syber Green Supermix, (Bio-Rad, Hercules, CA, USA) with specific primers for vimentin and actin, Vimenti-F: GACAATGCGTCTCTGGCACGT , Vimentin-R: TTCTTCTGCCTCCTGCAGGTTCTT ; β-actin-F: GGCTCTTCCAGCCTTCCTTCCT , β-actin-R : CACAGAGTACTTGCGCTCAGGAGG.
Small interfering RNA (siRNA)
RNA against HMGA2 SMARTpool siRNA (CCAGGAAGCAGCAGCAAGA) and firefly luciferase (CGTACGCGGAATACTTCGA) were synthesized by Dharmacon (Lafayette, CO, USA). MCF7/PAR1-N18 cells were transfected overnight with 0.2 μM siRNA using Oligofectamine (Invitrogen) in serum-free RPMI 1640 without antibiotics. Slug siRNA and ZEB1 siRNA were purchased premade from Thermo Scientific (Tewksbury, MA, USA). Cells were recovered for 24 h in 10% fetal bovine serum before use in assays.
Western blot analysis
Cells were washed in ice-cold phosphate-buffered saline and lysed in T-PER (tissue protein extraction reagent) supplemented with Halt protease and phosphatase inhibitor cocktail (all from Pierce/Life Technologies, Grand Island, NY, USA). Protein content was measured by Bradford Assay; 30–50 μg protein was resolved on 12% SDS–PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5% milk in TBST (0.1% Tween-20 in TBS) and incubated overnight with the following antibodies: PAR1-ATAP2, EGFR, estrogen receptor, Laminin (Santa Cruz, Dallas, TX, USA), Vimentin V9, E-cadherin, Integrin α6, Keratin 8/18, Claudin 3 (Abcam, Cambridge, MA, USA), Zona Occluden-3 (Chemicon, Billerica, MA, USA), SLUG, Zeb1 (Cell Signaling Technology, Danvers, MA, USA).
Immunofluorescence staining
Cells were cultured on eight-chamber polystyrene vessel tissue culture-treated glass slides (BD Falcon, Franklin Lakes, NJ, USA) and fixed with 4% formaldehyde, permeabilized with 0.1% Triton X-100 and blocked with 1% bovine serum albumin for 30 min. Cells were stained with PAR1-ATAP2, vimentin or E-cadherin and subsequently with Alexa Fluor 488 (Molecular Probes, Eugene, OR, USA). Slides were washed and mounted with ProLong Gold antifade reagent with 4',6-diamidino-2-phenylindole (Invitrogen) and visualized by fluorescence microscopy and image-captured by SPOT digital camera (Diagnostic Instruments, Sterling Heights, MI, USA).
Mouse models of lung metastasis
Experiments were conducted in full compliance with the Institutional Animal Care and Use Committee of Tufts Medical Center. MDA-MB-231/GFP (n=5), MCF7/PAR1-N55 (n=8), MCF7/R310E (n=6) or MCF7 (n=6) cells (2 × 106) were introduced via the tail vein of female NCR nu/nu mice (Taconic Farms, Hudson, NY, USA) mice.15 Lungs were collected and analyzed after 6 weeks for the presence of tumor nodules.
Lung histology
Paraffin-embedding, sectioning, hematoxylin and eosin staining and immunohistochemistry for vimentin were performed by the Tufts Medical Center Pathology department. Metastatic tumor nodules were counted throughout the entire lung section under a light microscope. Microscopy images were captured with a light microscope and SPOT digital camera.
Immunohistochemistry for tissue microarray analysis
Immunohistochemistry was performed by the Tufts Medical Center Pathology department using tissue microarray US Biomax (T088, Rockville, MD, USA). A tissue microslide were incubated with vimentin ab.52 Microscopy images were captured with a light microscope and SPOT digital camera.
Invasion assays
All invasion assays were performed using Boyden chambers with 8 μm pores (Costar, Tewksbury, MA, USA) and all cells were starved overnight in 1% fetal bovine serum. For invasion, 2.5 × 104 cells in 0.1% bovine serum albumin were placed in the top chamber and allowed to migrate towards NIH-3T3-conditioned media in the lower chamber. For invasion, type I rat tail collagen (BD Biosciences, San Jose, CA, USA) was set using Boyden membrane and cells were allowed to invade for 22 h. After 22 h, cells were removed from the upper chamber and membranes were stained using the Hema3 system; the invasion assays were quantified by counting the number of cells as previously described.53
3D cultured cells
Cells were seeded into eight-well plastic-chembered glass microscope slides containing growth factor-reduced Matrigel. Briefly, chambers were coated with matrigel and 5000 cells were seeded in a total volume of 100 ul of matrigel and covered with 100 ul of complete media. For Collagen type I studies, BD four-well chamber slide were coated with 100 μl of 1 mg/ml Corning (Lowell, MA, USA) collagen pH 7 and incubated at 37 oC for 30 min. Diluted cells (1000–10 000 per 1 ml) cell mixtures are added on top of collagen gel and put in 37 oC incubator. Fresh conditioned media is added every 3 days and photographed after 5–6 days of growth at 37 °C, 5% CO2.
Soft agar cellular growth
Soft agar experiments were performed in six-well cell-culture plates with 10 000 cells per well. Cells were seeded on pre-coated semi-solid agarose layer (0.8%), and the tumor cells were layered on top in 0.4% agarose. Following a 4-week period, the cells were stained with crystal violet blue, photographed and counted under microscope.
Colony-forming units
Five hundred cancer cells were seeded on non-adherent plates (Corning) in serum-free media. After 7 days, colonies greater than 30 cells were counted. Primary spheres were dissociated with trypsin and single cells passaged for second and third passage. In some experiments, single cells were embedded in Matrigel (BD Biosciences) as described.54 Cells were washed with 1 × phosphate-buffered saline and fixed with 100% MetOH for 10 min at room temperature. Crystal violet stain (Fluka AG 61135, 0.1% in 5% EtOH, Sigma-Aldrich, St Loise, MO, USA) was used to stain colonies prior to counting. Quantification of mammosphere and tumorsphere numbers was accomplished using a Multisizer 3 Coulter Counter (Beckman-Coulter, Brea, CA, USA) that provide number and size distribution of particles between 40 and 336 μm.
Tumorsphere formation assay
Using a total of 500 single cells/cm2, cells were plated on super-low adherence plates (Corning). The number of tumorspheres were counted using a Multisizer 3 Coulter Counter (Beckman Coulter).
Affymetrix microarray and data analysis
The microarray analysis was carried out at the Genomics Core of the Tufts Center for Neuroscience Research, Tufts University School of Medicine. MCF-7 and MCF-7/PAR1 cells were quiesced overnight in serum-free media and RNA was isolated using the RNEasy kit (Qiagen). Target RNAs from each cell line (MCF-7, MCF-7/PAR1; n=3) were fragmented and independently hybridized to Human Genome U133A 2.0 Array (Affymetrix, Santa Clara, CA, USA). Genechips were then washed and stained with Streptavidin R-phycoerythrin (Molecular Probes) and following washes, were scanned using the GeneChip scanner. Data analysis was conducted using the Bioconductor suite of programs. The 3'/5' RNA degradation plot was used to confirm the quality of the RNA samples hybridized to microarrays. Background correction, normalization and summarization of the raw probe intensities were carried out using the GCRMA protocol with default options. LIMMA module and topTable function was used to generate the list of differentially expressed genes. TIGR Multiexperiment Viewer (MeV) was used for statistical analysis and heatmap production. Expression data were deposited in the Gene Expression Omnibus (GEO).
ChIP and co-immunoprecipitation
PAR1-MCF7 (N55) and R310-PAR1-MCF7 cells were grown on 150 mm tissue culture plates overnight at 37 °C, 5% CO2. Cells were cross-linked with 1% formaldehyde in serum-free media, at room temperature for 15 min. Plates were washed twice with cold phosphate-buffered saline plus protease inhibitors (1 mM PMSF, 1 μg/ml pepstatin A and 1 μg/ml Aprotinin). Collected cells were treated with 1 ml Lysis buffer+protease inhibitors (ChIP kit, EMD Millipore, Billerica, MA, USA). Extracts were sonicated using the Misonix Sonicator 3000 (Newton, CT, USA) at high power until DNA fragments of 200–1000 bp were formed. The immunocomplexes were precipitated using Ab against HMGA2 (Cell Signaling Technology). Precipitated complexes were eluted and treated with Proteinase K (100 μg/ml) at 4 °C for 90 min. DNA fragments were purified by Qiagen Quick kit (Qiagen, Hilden, Germany) and quantified using a Nanodrop 2000 (Thermo Scientific). The DNA input and comparison between PAR1 expressing and ‘dead’ PAR1 cell line as control were used for PCR. The PCR primers of PAR1 promoter region and GAPDH control are listed in Supplementary Table 1. The experiment was carried out three independent times.
Statistical analysis
Cell culture experiments were carried out in minimum three replicates.
All quantified xenograft and in vitro assay results are presented as mean±s.d. or mean±s.e. as indicated. Statistical analysis were conducted using GraphPad Prism 5.0 software (San Diego, CA, USA). All statistical tests were two-sided. Student’s t-test for bar graph were performed. Statistical significance was defined as *P<0.05, **P<0.01 or ***P<0.001.
References
Vu T-KH, Hung DT, Wheaton VI, Coughlin SR . Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor action. Cell 1991; 64: 1057–1068.
Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y et al. Thrombin receptor overexpression in malignant and physiological invasion processes. Nat Med 1998; 4: 909–914.
Henrikson KP, Salazar SL, Fenton JW, Pentecost BT . Role of thrombin receptor in breast cancer invasiveness. Br J Cancer 1999; 79: 401–406.
Cisowski J, O'Callaghan K, Kuliopulos A, Yang J, Nguyen N, Deng Q et al. Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. Am J Pathol 2011; 179: 513–523.
Rudroff C, Seibold S, Kaufmann R, Zetina CC, Reise K, Schafer U et al. Expression of the thrombin receptor PAR-1 correlates with tumour cell differentiation of pancreatic adenocarcinoma in vitro. Clin Exp Metastasis 2002; 19: 181–189.
Nierodzik ML, Chen K, Takeshita K, Li JJ, Huang YQ, Feng XS et al. Protease-activated receptor 1 (PAR-1) is required and rate-limiting for thrombin-enhanced experimental pulmonary metastasis. Blood 1998; 92: 3694–3700.
Even-Ram SC, Maoz M, Pokroy E, Reich R, Katz B-Z, Gutwein P et al. Tumor cell invasion is promoted by activation of protease activated receptor-1 in cooperation with the alpha vbeta 5 integrin. J Biol Chem 2001; 276: 10952–10962.
Tellez C, Bar-Eli M . Role and regulation of the thrombin receptor (PAR-1) in human melanoma. Oncogene 2003; 22: 3130–3137.
Whitehead I, Kirk H, Kay R . Expression cloning of oncogenes by retroviral transfer of cDNA libraries. Mol Cell Biol 1995; 15: 704–710.
Martin CB, Mahon GM, Klinger MB, Kay RJ, Symons M, Der CJ et al. The thrombin receptor, PAR-1, causes transformation by activation of Rho-mediated signaling pathways. Oncogene 2001; 20: 1953–1963.
Korkola JE, DeVries S, Fridlyand J, Hwang ES, Estep ALH, Chen Y-Y et al. Differentiation of lobular versus ductal breast carcinomas by expression microarray analysis. Cancer Res 2003; 63: 7167–7175.
Boire A, Covic L, Agarwal A, Jacques S, Sharifi S, Kuliopulos A . PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005; 120: 303–313.
Agarwal A, Covic L, Sevigny LM, Kaneider NC, Lazarides K, Azabdaftari G et al. Targeting a metalloprotease-PAR1 signaling system with cell-penetrating pepducins inhibits angiogenesis, ascites, and progression of ovarian cancer. Mol Cancer Ther 2008; 7: 2746–2757.
Nguyen N, Kuliopulos A, Graham RA, Covic L . Tumor-derived Cyr61(CCN1) promotes stromal matrix metalloprotease-1 production and protease-activated receptor 1-dependent migration of breast cancer cells. Cancer Res 2006; 66: 2658–2665.
Yang E, Boire A, Agarwal A, Nguyen N, O'Callaghan K, Tu P et al. Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Res 2009; 69: 6223–6231.
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA et al. Molecular portraits of human breast tumours. Nature 2000; 406: 747–752.
Thiery JP, Sleeman JP . Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7: 131–142.
Slofstra SH, Bijlsma MF, Groot AP, Reitsma PH, Lindhout T, ten Cate H et al. Protease-activated receptor-4 inhibition protects from multiorgan failure in a murine model of systemic inflammation. Blood 2007; 110: 3176–3182.
Satelli A, Li S . Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci 2011; 68: 3033–3046.
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2000; 2: 737–744.
Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007; 26: 6979–6988.
Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A . The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 2003; 116: 499–511.
Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000; 2: 76–83.
Tallini G, Dal Cin P, Rhoden KJ, Chiapetta G, Manfioletti G, Giancotti V et al. Expression of HMGI-C and HMGI(Y) in ordinary lipoma and atypical lipomatous tumors: immunohistochemical reactivity correlates with karyotypic alterations. Am J Pathol 1997; 151: 37–43.
Medeiros F, Erickson-Johnson MR, Keeney GL, Clayton AC, Nascimento AG, Wang X et al. Frequency and characterization of HMGA2 and HMGA1 rearrangements in mesenchymal tumors of the lower genital tract. Genes Chromosomes Cancer 2007; 46: 981–990.
Peluso S, Chiappetta G . High-mobility group A (HMGA) proteins and breast cancer. Breast Care (Basel) 2010; 5: 81–85.
Pegoraro S, Ros G, Piazza S, Sommaggio R, Ciani Y, Rosato A et al. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013; 4: 1293–1308.
Reeves R . Nuclear functions of the HMG proteins. Biochim Biophys Acta 2010; 1799: 3–14.
Sgarra R, Zammitti S, Lo Sardo A, Maurizio E, Arnoldo L, Pegoraro S et al. HMGA molecular network: From transcriptional regulation to chromatin remodeling. Biochim Biophys Acta 2010; 1799: 37–47.
Chen JQ, Russo J . ERalpha-negative and triple negative breast cancer: molecular features and potential therapeutic approaches. Biochim Biophys Acta 2009; 1796: 162–175.
Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE . Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry 1999; 38: 4572–4585.
Nelson WJ, Traub P . Proteolysis of vimentin and desmin by the Ca2+-activated proteinase specific for these intermediate filament proteins. Mol Cell Biol 1983; 3: 1146–1156.
Lahat G, Zhu QS, Huang KL, Wang S, Bolshakov S, Liu J et al. Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS One 2010; 5: e10105.
Sun JM, Spencer VA, Li L, Yu Chen H, Yu J, Davie JR . Estrogen regulation of trefoil factor 1 expression by estrogen receptor alpha and Sp proteins. Exp Cell Res 2005; 302: 96–107.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–715.
Cleynen I, Brants JR, Peeters K, Deckers R, Debiec-Rychter M, Sciot R et al. HMGA2 regulates transcription of the Imp2 gene via an intronic regulatory element in cooperation with nuclear factor-kappaB. Mol Cancer Res 2007; 5: 363–372.
Tallini G, Dal Cin P . HMGI(Y) and HMGI-C dysregulation: a common occurrence in human tumors. Adv Anat Pathol 1999; 6: 237–246.
Salah Z, Maoz M, Cohen I, Pizov G, Pode D, Runge MS et al. Identification of a novel functional androgen response element within hPar1 promoter: implications to prostate cancer progression. FASEB J 2005; 19: 62–72.
Salah Z, Uziely B, Jaber M, Maoz M, Cohen I, Hamburger T et al. Regulation of human protease-activated receptor 1 (hPar1) gene expression in breast cancer by estrogen. FASEB J 2012; 26: 2031–2042.
Ferguson M, Henry PA, Currie RA . Histone deacetylase inhibition is associated with transcriptional repression of the Hmga2 gene. Nucleic Acids Res 2003; 31: 3123–3133.
Kumar MS, Armenteros-Monterroso E, East P, Chakravorty P, Matthews N, Winslow MM et al. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature 2014; 505: 212–217.
Sheen YS, Liao YH, Lin MH, Chu CY, Ho BY, Hsieh MC et al. IMP-3 promotes migration and invasion of melanoma cells by modulating the expression of HMGA2 and predicts poor prognosis in melanoma. J Invest Dermatol 2014; 135: 1065–1073.
Li Y, Zhao Z, Xu C, Zhou Z, Zhu Z, You T . HMGA2 induces transcription factor Slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett 2014; 355: 130–140.
Xi YN, Xin XY, Ye HM . Effects of HMGA2 on malignant degree, invasion, metastasis, proliferation and cellular morphology of ovarian cancer cells. Asian Pac J Trop Med 2014; 7: 289–292.
Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, Moustakas A . HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem 2008; 283: 33437–33446.
Becker RC, Moliterno DJ, Jennings LK, Pieper KS, Pei J, Niederman A et al. Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase II study. Lancet 2009; 373: 919–928.
Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med 2012; 366: 1404–1413.
Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med 2012; 366: 20–33.
Zhang P, Gruber A, Kasuda S, Kimmelstiel C, O'Callaghan K, Cox DH et al. Suppression of arterial thrombosis without affecting hemostatic parameters with a cell-penetrating PAR1 pepducin. Circulation 2012; 126: 83–91.
Covic L, Misra M, Badar J, Singh C, Kuliopulos A . Pepducin-based intervention of thrombin receptor signaling and systemic platelet activation. Nat Med 2002; 8: 1161–1165.
Hatziapostolou M, Polytarchou C, Panutsopulos D, Covic L, Tsichlis PN . Proteinase-activated receptor-1-triggered activation of tumor progression locus-2 promotes actin cytoskeleton reorganization and cell migration. Cancer Res 2008; 68: 1851–1861.
Covic L, Gresser AL, Kuliopulos A . Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 2000; 39: 5458–5467.
Kamath L, Meydani A, Foss F, Kuliopulos A . Signaling from protease-activated receptor-1 inhibits migration and invasion of breast cancer cells. Cancer Res 2001; 61: 5933–5940.
Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci USA 2010; 107: 21737–21742.
Acknowledgements
We thank Vishal Trivedi, Chris Parkin, Akiko Hata, Larry Feig and Anna Wronski for their expert advice and Ryan Stevenson for critically reading the manuscript. The microarray analysis was carried out in the Genomics Core of the Tufts Center for Neuroscience Research, Tufts University School of Medicine (supported by NNDS – P30 NS047243). This work was supported by NIH grants CA104406 (LC), Susan G. Komen BCTR0706763, Diane Connolly-Zaniboni Research Scholarship (LC) and a fellowship from Aid for Cancer Research, Boston, MA (EY) by NIH CA122992, HL64701 (to AK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
About this article

Cite this article
Yang, E., Cisowski, J., Nguyen, N. et al. Dysregulated protease activated receptor 1 (PAR1) promotes metastatic phenotype in breast cancer through HMGA2. Oncogene 35, 1529–1540 (2016). https://doi.org/10.1038/onc.2015.217
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.217
- Springer Nature Limited
This article is cited by
-
Notch-based gene signature for predicting the response to neoadjuvant chemotherapy in triple-negative breast cancer
Journal of Translational Medicine (2023)
-
HMGA2 regulates circular RNA ASPH to promote tumor growth in lung adenocarcinoma
Cell Death & Disease (2020)
-
MALT1 is a critical mediator of PAR1-driven NF-κB activation and metastasis in multiple tumor types
Oncogene (2019)
-
Overexpression of G protein-coupled receptor GPR87 promotes pancreatic cancer aggressiveness and activates NF-κB signaling pathway
Molecular Cancer (2017)
-
Protease-activated receptor-1 inhibits proliferation but enhances leukemia stem cell activity in acute myeloid leukemia
Oncogene (2017)