
Abstract
The transcription factor nuclear factor kappa B (NF-κB) has been implicated in having a crucial role in the tumorigenesis of many types of human cancers. Although epidermal growth factor receptor (EGFR) can directly activate NF-κB, the mechanism by which EGFR induces NF-κB activation and the role of NF-κB in EGFR-associated tumor progression is still not fully defined. Herein, we found that mucosa-associated lymphoid tissue 1 (MALT1) is involved in EGFR-induced NF-κB activation in cancer cells, and that MALT1 deficiency impaired EGFR-induced NF-κB activation. MALT1 mainly functions as a scaffold protein by recruiting E3 ligase TRAF6 to IKK complex to activate NF-κB in response to EGF stimulation. Functionally, MALT1 inhibition shows significant defects in EGFR-associated tumor malignancy, including cell migration, metastasis and anchorage-independent growth. To further access a physiological role of MALT1-dependent NF-κB activation in EGFR-driven tumor progression, we generated triple-transgenic mouse model (tetO-EGFRL858R; CCSP-rtTA; Malt1−/−), in which mutant EGFR-driven lung cancer was developed in the absence of MALT1 expression. MALT1-deficient mice show significantly less lung tumor burden when compared with its heterozygous controls, suggesting that MALT1 is required for the progression of EGFR-induced lung cancer. Mechanistically, MALT1 deficiency abolished both NF-κB and STAT3 activation in vivo, which is a result of a defect of interleukin-6 production. In comparison, MALT1 deficiency does not affect tumor progression in a mouse model (LSL-K-rasG12D; CCSP-Cre; Malt1−/−) in which lung cancer is induced by expressing a K-ras mutant. Thus, our study has provided the cellular and genetic evidence that suggests MALT1-dependent NF-κB activation is important in EGFR-associated solid-tumor progression.
Similar content being viewed by others


Introduction
Nuclear factor kappa B (NF-κB) is a complex of transcription factors that regulates several important cellular functions, especially in the innate and adaptive immune responses.1 In tumor cells, NF-κB also contributes to several malignant phenotypes associated with cell proliferation, invasion, metabolism and survival.2 Although NF-κB is considered as a therapeutic target for cancer treatment, the upstream machinery leading to NF-κB activation varies among different types of tumor cells with different kinds of mutations or stimulations. Therefore, identifying a specific signaling component with therapeutic potential and high specificity in each tumor-promotive NF-κB pathway would be instrumental in improving therapeutic efficacy.
MALT1 (mucosa-associated lymphoid tissue lymphoma translocation gene 1) is an upstream signaling component of NF-κB with high therapeutic potential.3 MALT1 has been shown to be a key mediator in NF-κB activation in T and B lymphocytes, and is required for lymphocyte activation and survival through T-cell receptor (TCR) and B-cell receptor (BCR) signaling.4, 5 Activation of these receptors leads to the activation of protein kinase C, which phosphorylates the Caspase recruitment domain and membrane-associated guanylate kinase-like domain (CARMA) family proteins such as CARMA1 in lymphocytes.6, 7, 8, 9 Phosphorylation of the CARMA protein triggers a conformational change and further recruits MALT1 and B-cell lymphoma protein 10 (BCL10),8, 9 resulting in the assembly of the CARMA–BCL10–MALT1 (CBM) complex. The CBM complex activates the IκB kinase to trigger NF-κB activation.10 MALT1 is considered a critical component in constitutive NF-κB activation in certain types of lymphoma. In MALT lymphoma-associated with the genetic translocation t(11;18)(q21;21), a cIAP2 (cellular inhibitor of apoptosis 2)-MALT1 fusion protein gives constitutive NF-κB signals.11, 12, 13 In activated B-cell-like (ABC) diffuse large B-cell lymphoma (DLBCL), MALT1 also has a tumor-promotive role by bridging the constitutive BCR signaling to a dysregulated NF-κB activity.14
The therapeutic value of MALT1 is associated with the protein’s caspase-like domain, which contains an arginine-specific protease activity.15, 16 The protease activity of MALT1 facilitates optimal NF-κB and AP-1 activation by cleaving negative regulators, such as A20, CYLD and RelB.15, 17, 18 In MALT1 lymphoma, the protease activity of the cIAP2–MALT1 fusion protein cleaves and stabilizes NF-κB-inducing kinase, resulting in a constitutive NF-κB activity, enhanced adhesion and resistance to apoptosis.19 In addition, several independent studies of therapeutic applications of MALT1 protease inhibitors in ABC-DLBCL indicated that MALT1 inhibitors are selectively toxic to this type of lymphoma.20, 21, 22, 23 These results suggest that MALT1 inhibitors can potentially serve as therapeutic reagents for certain types of lymphoma. However, the tumor-promotive role of MALT1 has been confirmed only for the lymphoid system. As NF-κB contributes to tumor malignancy in a wide range of cell types, the oncogenic role and therapeutic potential of MALT1 need to be investigated in a nonhematopoietic system, such as solid tumors of epithelial origin. To date, no study has demonstrated a functional role of MALT1 in solid-tumor progression.
One of the most frequently mutated and overexpressed genes in solid tumors is the epidermal growth factor receptor (EGFR). EGFR overexpression and gain-of-function mutations are observed in nearly 30% of solid tumors, such as breast cancer, head-and-neck cancer and non-small-cell lung cancer (NSCLC).24 EGFR-mediated signaling contributes to many important malignant properties of tumors, such as cell growth, proliferation and metabolism. Recently, several groups including ours showed that in addition to its role in the phosphatidylinositide 3-kinase/Akt and MAPK/ERK (mitogen-activated protein kinase–extracellular signal-regulated kinase) pathways, EGFR-induced NF-κB activation has an essential role in malignant properties such as proliferation, survival, migration and metabolism.25, 26, 27 However, the exact molecular mechanism by which EGFR activates NF-κB remains unclear. In addition, a physiological role of NF-κB signaling in EGFR-associated tumor has not been demonstrated so far. In this study, we found that EGFR-driven NF-κB activation is mediated by MALT1, which recruits E3 liagase TRAF6 to IKK complex upon EGF stimulation. However, MALT1 protease activity is not required for EGFR-induced NF-κB activation. In addition, by using an EGFR-driven lung cancer mouse model, we further showed that EGFR-driven lung cancer progression requires MALT1-dependent NF-κB signaling, which transactivates STAT3 through interleukin-6 (IL-6) signaling. Thus, our data reveal that MALT1-dependent NF-κB activation is crucial for the development of EGFR-associated solid-tumor progression in preclinical models.
Results
MALT1 is required for EGF-induced NF-κB activation
Our previous results revealed that CARMA3, a MALT1-interacting protein, is involved in EGFR-mediated NF-κB signaling.26 To test whether MALT1 is involved in EGF-induced NF-κB activation, we knocked down MALT1 expression in A431 cells in which EGFR is highly expressed and examined NF-κB activation by gel shift assay upon EGF stimulation. We found that the suppression of MALT1 expression significantly impaired EGF- and PMA/Ionomycin-induced NF-κB activation, respectively, but not tumor necrosis factor-α (TNFα)-induced NF-κB activation, indicating that MALT1 is specifically involved in mediating EGF-induced NF-κB activation (Figure 1a). To further confirm the role of MALT1 in EGF-induced NF-κB activation in primary cells, we prepared MALT1-heterozygous (Malt1+/−) and -deficient (Malt1−/−) primary mouse embryonic fibroblasts (MEFs). Because EGFR expression in primary MEFs is low, we used PMA/Ionomycin to activate protein kinase C, a downstream component in EGFR signaling, which can induce NF-κB activation.28, 29 Consistently, PMA/Ionomycin-induced NF-κB was completely abolished in MALT1-deficient MEFs (Figure 1b). In addition, we found MALT1 suppression specifically abolished EGF-induced p65 nuclear localization (Figure 1c) upon EGF stimulation, but it had no impact on other pathways downstream of EGFR, as shown by the level of p-S6 Kinase, p-ERK and p-JNK (Figure 1d). These data collectively suggest that MALT1 is specifically involved in EGFR-mediated NF-κB activation.

MALT1 is selectively involved in EGFR-induced NF-κB activation. (a) A431 cells with a MALT1 knockdown (shMALT1) and control cells (shCtl) were stimulated with EGF (100 ng/ml), PMA and Ionomycin (50 ng/ml; 100 ng/ml) or TNFα (10 ng/ml) for indicated periods. NF-κB activation and Oct-1 (loading control) levels were determined by the gel shift assay. (b) MEFs from Malt1+/− and Malt1−/− embryos were isolated. Early-passage (P1) MEFs were stimulated with PMA and Ionomycin (50 ng/ml; 100 ng/ml) or TNFα (10 ng/ml) for indicated periods. NF-κB activation and Oct-1 levels were determined by the gel shift assay. (c) Nuclear lysates from shCtl and shMALT1 A431 cells were analyzed by immunoblotting using indicated antibodies. (d) shCtl and shMALT1 A431 cells were stimulated with EGF (100 ng/ml) for indicated periods. Cell lysates were analyzed by immunoblotting using indicated antibodies. L, long isoform; S, short isoform.
MALT1 functions as a scaffold protein by recruiting TRAF6 to IKK complex
It has been reported that MALT1 contains protease activity, which is required for optimal TCR- and BCR-induced NF-κB activity by inhibiting negative regulators in NF-κB pathway, such as A20 and RelB.15, 18 We therefore sought to determine whether MALT1 protease activity contributes to EGF-induced NF-κB activation. To this end, we reconstituted MALT1-silenced cells with either wild-type MALT1 or protease-deficient mutant MALT1 (MALT1C464A)18 in either MALT1-silenced A431 cells or MALT1-deficient MEFs (Supplementary Figure 1). We found there was not significant reduction of NF-κB in mutant MALT1-constituted MEFs compared with wild-type MALT1-reconstituted MEFs upon PMA/Ionomycin stimulation (Figure 2a), and both wild-type and the protease-deficient mutant of MALT1 rescued EGF-induced NF-κB in A431 cells (Figure 2b), suggesting that the protease activity of MALT1 is largely dispensable for EGFR-induced NF-κB activation. As an alternative approach, we examined EGF-induced NF-κB activation in the presence or absence of MALT1 specific inhibitor (z-VRPR-Fmk). Although MALT1 inhibitor completely blocked its protease activity as shown by cleaved BCL10 (Supplementary Figure 2a), it does not affect NF-κB activation in response to EGF stimulation (Supplementary Figure 2b). Taken together, these results demonstrate that MALT1 mainly functions as a scaffold protein and is selectively involved in the regulation of EGF-induced NF-κB activation.

MALT1 serves as a scaffold protein by recruiting TRAF6 to IKK complex. (a) MALT1-deficient MEFs (Malt1−/−), wild-type MALT1-reconstituted MEFs (Malt1−/−WT) and protease-deficient mutant-reconstituted MEFs (Malt1−/−C464A) were stimulated with PMA and Ionomycin (50 ng/ml; 100 ng/ml) for indicated periods, respectively. Nuclear lysates were isolated and subjected to gel shift analysis for NF-κB activation. (b) A431 cells with a MALT1 knockdown (shMALT1), wild-type MALT1-reconstituted cells (shMALT1WT) and protease-deficient mutant-reconstituted cells (shMALT1C464A) were stimulated with EGF (100 ng/ml) or TNFα (10 ng/ml) for indicated periods. Nuclear lysates were isolated and subjected to gel shift analysis for NF-κB activation. (c) MALT1-reconstituted cells were stimulated with EGF (100 ng/ml) for indicated time and MALT1-Flag was immunoprecipitated (IP) by anti-Flag conjugated beads. The IP samples and lysates were analyzed by immunoblotting using the indicated antibodies. (d) MALT1-reconstituted cells were stimulated with EGF (100 ng/ml) and TNFα (10 ng/ml), respectively. MALT1-Flag was immunoprecipitated (IP) by anti-Flag conjugated beads. The IP samples and lysates were analyzed by immunoblotting using the indicated antibodies. (e) Control (shCtl) or MALT1-silenced (shMALT1) A431 cells were either unstimulated or stimulated with EGF (100 ng/ml) for 15 min and IKKγ was immunoprecipitated (IP). The IP samples and lysates were analyzed by immunoblotting using the indicated antibodies. (f) A431 cells with a TRAF6 knockdown (shTRAF6) and control cells (shCtl) were stimulated with EGF (100 ng/ml), PMA and Ionomycin (50 ng/ml; 100 ng/ml) or TNFα (10 ng/ml) for indicated time points, respectively. NF-κB activation and Oct-1 (loading control) levels were determined by the gel shift assay.
To further delineate the molecular mechanism by which MALT1 activates NF-κB in response to EGF stimulation, we examined MALT1-interacting protein upon EGF stimulation. Upon EGF stimulation, MALT1 was inducibly associated with TRAF6, an E3 ligase that activates IKK complex30 (Figure 2c). Consistently, we also observed a notable, but weak, association between MALT1 and IKKα at a later time point upon EGF stimulation (Figure 2c), suggesting that MALT1 bridges TRAF6 to IKK complex in response to EGFR activation. The association of MALT1 and TRAF6 was specifically induced by EGF but not TNFα, indicating that MALT1 functions specifically downstream of EGFR (Figure 2d). In addition, MALT1 silencing abolished the association between IKKγ and TRAF6 upon EGF stimulation (Figure 2e), suggesting that MALT1 is functionally required for recruiting TRAF6 to IKK complex upon EGFR activation. To further confirm whether TRAF6 is functionally required for EGF-induced NF-κB activation, we knocked down TRAF6 expression by short hairpin RNA (Supplementary Figure 3) and examined NF-κB activation by gel shift assay. Consistently, TRAF6 inhibition significantly abolished EGF- and PMA/Ionomycin-induced NF-κB activation, respectively, suggesting that TRAF6 is also required for EGF-induced NF-κB activation (Figure 2f). Collectively, these results indicate that MALT1 serves as a scaffold protein by recruiting TRAF6 to activate IKK complex in response to EGFR stimulation.
MALT1 contributes to EGFR-associated tumor malignancy
On the basis of the observation that MATL1 is specifically required for EGF-induced NF-κB activation, we asked whether EGFR-MALT1-NF-κB signaling contributes to EGFR-associated tumor malignancy. To this end, we performed in vitro and in vivo assays in A431 cells, and a human lung cancer cell line HCC827 in which EGFR is mutated and constitutively activated. First, we found MALT1 suppression dramatically impaired cell migration and motility in both transwell migration assay (Figure 3a) and wound-healing assay (Figure 3b) in vitro. Consistently, we compared cell metastasis in vivo in a lung metastasis model and found that the number of lung metastatic spots was significantly reduced in MALT1-silenced cells compared with controls (Figure 3c). To access whether this effect is NF-κB dependent, we treated cells with IKK inhibitor, and found that IKK inhibition similarly blocked cell migration in vitro (Supplementary Figure 4). In addition, TRAF6-silenced cells showed a consistent defect of cell migration (Supplementary Figure 4), which indicates MALT1–TRAF6–IKK signaling controls cell migration. In addition, we found that treating MALT1 inhibitor does not affect cell migration in either A431 or HCC827 cell lines (Supplementary Figure 5), suggesting that MALT1 protease activity does not contribute to tumor migration. Taken together, these data suggest that MALT1-mediated NF-κB activity regulates cell migration in vitro and metastasis in vivo.

MALT1 contributes to EGFR-associated malignancy. (a) A431 and HCC827 cells with a MALT1 knockdown (shMALT1#1 and shMALT1#2) or a control knockdown (shCtl) were analyzed by transwell migration assays. Cells were fixed and stained 20 h after seeding (left panel). Cell numbers in five random fields were calculated and compared (right panel). (b) MALT1-silenced A431 cells and controls were analyzed by a wound-healing assay in the presence of EGF (1 ng/ml). The percentage of wound closure were calculated and analyzed. (c) MALT1-silenced A431 cells and controls were intravenously injected into SCID (severe combined immunodeficiency) mice. Four weeks after injection, mice were killed and the lungs were washed, fixed and stained with Bouin solution. Metastasis sites were visualized as white spots (left panel) and quantitated (right panel). (d) A431 (left) and HCC827 (right) cells with a MALT1 knockdown (shMALT1) and control cells (shCtl) were subjected to soft agar colony-formation analysis with or without EGF (2 ng/ml). The sizes of cell colonies were calculated and analyzed. ***P<0.001; ****P<0.0001.
Next we examined the role of MALT1 in cell proliferation. Although less profound to cell migration, we observed a notable suppression of cell proliferation in MALT1-silenced cells compared with controls as determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Supplementary Figure 6). By using the soft agar colony-formation assay to compare anchorage-independent growth rate, we found that MALT1 suppression significantly reduced the size of colonies formed in the agarose compared with controls (Figure 3d and Supplementary Figure 7). In addition, although control cells proliferated in response to EGF stimulation, MALT1-silenced cell showed no response to EGF in terms of proliferation (Figure 3d). Collectively, these data suggest that MALT1-dependent NF-κB activity is required for EGFR-dependent cell proliferation and survival in vitro.
MALT1 contributes to the progression of EGFR- but not K-ras-induced lung adenocarcinoma
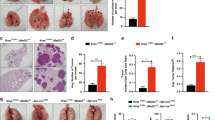
Next we sought to establish a physiological model to further investigate whether and how MALT1-mediated NF-κB activation affects EGFR-driven tumor progression in vivo. As EGFR mutation and overexpression have been frequently found in NSCLC, we crossed MALT1-knockout mice to a lung cancer mouse model (tetO-EGFRL858R; CCSP-rtTA; Malt1+/−or tetO-EGFRL858R; CCSP-rtTA; Malt1−/−), in which mutant EGFR-driven lung cancer will be developed in the presence or absence of MALT1 expression (Figure 4a). As MALT1 does not affect K-ras-associated NF-κB and tumor malignancy (data not shown), as a control, we also generated another mouse model in which lung tumor is driven by mutant K-ras expression with or without MALT1 expression (LSL-K-rasG12D; CCSP-Cre; MALT1+/− or LSL-K-rasG12D; CCSP-Cre; MALT1−/−; Figure 4b). After we successfully got EGFRL858R/CCSP-rtTA/Malt1+/−- and EGFRL858R/CCSP-rtTA/Malt1−/−-triple-transgenic mice, we found both Malt1 heterozygous and knockout mice express human EGFR mRNA in a similar level, indicating that MALT1 does not affect the expression and induction of mutant EGFR transgene (Figure 4c). To access lung tumor burden, we compared the ratio of lung weight with body weight. We found that Malt1-knockout mice showed significant lower ratio of lung to body weight compared with heterozygous controls (0.0350±0.001570, n=8 vs 0.02771±0.001672, n=7 P=0.0073; Figure 4d), suggesting a reduced lung tumor burden in Malt1-knockout mice. We reviewed the pathological slides of lung tissue and found that mice with homozygous Malt1 knockout showed a reduced level of cellularity and the frequency of atypical cells with irregular nuclei in lungs in comparison with Malt1 heterozygous controls, suggesting a less malignant phenotype in Malt1 homozygous knockout mice (Figure 4e). In addition, Malt1 heterozygous lungs also had less alveolar space and more frequent focal glandular neoplasms compared with Malt1-knockout littermates (Figure 4e). Collectively, these data suggest that Malt1 homozygous knockout mice had a reduced lung cancer burden and malignancy compared with its heterozygous controls.

MALT1 contributes to EGFR- but not K-ras-driven lung adenocarcinoma progression. (a) Graphic presentation of the generation of tetO-EGFRL858R; CCSP-rtTA; MALT1−/− triple-transgenic mice. (b) Graphic presentation of the generation of LSL-K-rasG12D; CCSP-Cre; MALT1−/− triple-transgenic mice. (c) Total RNA were extracted from the lungs of tetO-EGFRL858R; CCSP-rtTA; Malt1+/- mice (Het), tetO-EGFRL858R; CCSP-rtTA; Malt1−/− mice (KO), and HeLa cells, respectively. Reverse transcription PCR was performed using primers to amplify human EGFR and mouse LCa3, respectively. (d) The ratio of lung to body weight was compared between tetO-EGFRL858R; CCSP-rtTA; MALT1+/− (Het; n=8) and tetO-EGFRL858R; CCSP-rtTA; MALT1−/− (KO) mice (n=7). **P<0.01. (e) Hematoxylin and eosin (H&E) staining of the normal lung (NL), lung from tetO-EGFRL858R; CCSP-rTTA; MALT1+/− mice (Het) and lung from tetO-EGFRL858R; CCSP-rTTA; MALT1−/− mice (KO). (f) The ratio of lung to body weight was compared between LSL-K-rasG12D; CCSP-Cre; MALT1+/- (Het) (n=5) and LSL-K-rasG12D; CCSP-Cre; MALT1−/− (KO) mice (n=5). NS, no statistical significance. (g) The number of tumors on the lung in LSL-K-rasG12D; CCSP-Cre; MALT1+/− (Het) (n=3) and LSL-K-rasG12D; CCSP-Cre; MALT1−/− (KO) mice (n=3). NS, no statistical significance. (h) H&E staining of the lungs from LSL-K-rasG12D; CCSP-Cre; MALT1+/− mice (Het) and lung from LSL-K-rasG12D; CCSP-Cre; MALT1−/− mice (KO). Arrow shows tumor cells.
In comparison, such differences were not observed in mutant K-ras-induced lung cancer model in terms of the ratio of lung weigh to body weight (0.01573±0.001575, n=5 vs 0.01548±0.001560, n=5; Figure 4f), the number of visible tumors on the surface of lungs (Figure 4g) and the number and size of tumor showing by histology (Figure 4h). Therefore, these data indicate that MALT1 contributes to EGFR-induced but not K-ras-induced lung tumor progression in vivo.
MALT1 contributes to NF-κB and STAT3 activation in vivo
To further dissect how MALT1 deficiency inhibits EGFR-dependent tumor growth, we examined the activation of downstream pathways of EGFR. Consistent with our in vitro data, we found a lower NF-κB activity in Malt1-knockout tumor-bearing lungs compared with controls as showed by a reduced level of phosphorylated p65, whereas the level of phosphorylated-S6 ribosomal protein, phosphorylated AKT and phosphorylated ERK remained similar between Malt1 heterozygous and knockout mice (Figure 5). These results were consistent with our observation in vitro and suggest that MALT1 affects NF-κB activation in vivo.

MALT1 controls NF-κB and STAT3 activation in EGFR-driven lung cancer model. (a) Representative IHC staining of lung sections from tetO-EGFRL858R; CCSP-rtTA; MALT1+/− and tetO-EGFRL858R; CCSP-rtTA; MALT1−/− mice by using indicated antibodies. (b) The intensity × percentage of positive signals in a were compared for each staining. 10–15 random fields were chosen for each genotype, and were repeated three times using different mice (n=3) for each genotype. **P<0.001; ****P<0.0001.
It has been shown that STAT3 is also one of the downstream effectors of EGFR and is activated in EGFR-associated lung tumor in both mouse models and human samples.31 To our surprise, we found a significant defect of phosphorylated STAT3 level in Malt1-knockout mice compared with its heterozygous control, whereas the level of total STAT3 was comparable between Malt1 heterozygous and knockout mice (Figure 5). Thus, our data also suggest that MALT1 contributes to the EGFR-dependent activation of STAT3 in vivo.
MALT1 controls NF-κB-dependent IL-6 production in response to EGFR
It has been suggested that NF-κB regulates IL-6 production. We found both IL-6 neutralization and IKK inhibition significantly reduced P-STAT3 level in HCC827 cells, suggesting that both NF-κB and IL-6 are required for P-STAT3 activation in HCC827 cells (Supplementary Figure 8). In addition, MALT1-silenced HCC827 cells showed a lower level of P-STAT3 compared with control cells (Supplementary Figure 8). This result is consistent with a lower P-STAT3 level in the lung tumor from MALT1-deficient mice compared with control mice in vivo (Figure 5). To further determine whether MALT1 controls IL-6 production upon EGFR activation, we took A431 cells and examined IL-6 production upon EGF stimulation. We found that MALT1-silenced cells produced significantly less IL-6 compared with controls, whereas cells treated with MALT1 inhibitor produced similar amount IL-6 as control (Figure 6a). Consistently, MALT1-silenced HCC827, but not cells treated with MALT1 inhibitor, showed a similar defect in IL-6 production (Figure 6b). In our mouse model, we found that IL-6 mRNA level was much lower in the tumor-bearing lungs of Malt1-knockout mice compared with its heterozygous controls (Figure 6c). Taken together, these data indicate that MALT1 controls EGFR-driven IL-6 production in vitro and in vivo. In summary, our data suggest that MALT1 contributes to EGFR-associated lung cancer progression by coordinately controlling both NF-κB and STAT3 activation through IL-6-mediated cross talk between these two pathways (Figure 6d).

MALT1 is required for EGFR-induced IL-6 production. (a) Control A431 cells (shCtl), MALT1-knockdown cells (shMALT1) and control cells treated with MALT1 inhibitor z-VRPR-Fmk (75 μM) were stimulated with EGF (100 ng/ml), respectively. Total RNA was extracted for quantitative PCR analysis. The ratio of mRNA of human IL-6 to human HPRT1 were calculated and compared in the graph. *P<0.05. (b) Total RNA was extracted from control (shCtl) HCC827 cells, MALT1-knockdown cells (shMALT1) and control cells treated with MALT1 inhibitor z-VRPR-Fmk (75 μM), respectively. The ratio of mRNA of human IL-6 to human HPRT1 were calculated and compared in the graph. *P<0.05. (c) Total RNA was extracted from the lungs of tetO-EGFRL858R; CCSP-rtTA; MALT1+/− and tetO-EGFRL858R; CCSP-rtTA; MALT1−/− mice, respectively. The ratio of IL-6 to GAPDH was presented in the graph. *P<0.05. (d) The working model summarizing the finding in this study.
Discussion
NF-κB is a transcription factor that has been shown directly activated by EGFR. Although previously it has been shown that EGFR activates NF-κB in a CARMA3- and BCL10-dependent manner, the exact molecular mechanism by which EGFR activates NF-κB is still not clear. In this study, we have provided biochemical and genetic evidence suggesting that EGFR-induced NF-κB is mediated by MALT1. We found that MALT1 functions downstream of BCL10 and recruits E3 ligase TRAF6 to IKK complex upon EGFR stimulation. In lymphocytes, the activation of NF-κB requires two parallel signals to induce IKK ubiquitination and phosphorylation in response to TCR and BCR stimulation.32 A previous report suggests that EGFR activation induce IKK phosphorylation through protein kinase Cɛ.25 As we found that MALT1 deficiency does not alter the level of phosphorylated IKK in response to EGF stimulation (data not shown), we propose that MALT1–TRAF6 complex is responsible for IKK ubiquitination in response to EGF stimulation. This activation mechanism is similar to TCR- and BCR-induced NF-κB. However, unlike TCR- and BCR-induced NF-κB, the protease activity of MALT1 is not required for EGFR-induced NF-κB activation. This result is in part due to the lack of MALT1’s substrates in nonhematopoietic cells, such as A20 and RelB. It may be also due to the fact that EGFR does not activate NF-κB as strong as TCR or PMA plus Ionomycin does. However, even the cells express CYLD that is cleaved by MALT1 in response to TCR stimulation,17 we cannot detect proteolysis events upon EGF stimulation (data not shown). Therefore, our results suggest a differential requirement of MALT1 protease activity depending on different types of stimuli.
EGFR is having an oncogenic role in many types of human cancer. Although it has been well established that EGFR downstream effector pathways such as AKT and MAPK are having pivotal roles in cancer initiation and progression, less is known about how other signaling such as NF-κB contributes to EGFR-associated tumor progression. Here we show that MALT1 contributes to several important oncogenic phenotypes in EGFR-associated tumors. Our study found that MALT1 has an essential role in tumor migration and invasion in vitro, as well as in a lung metastasis model in vivo. We propose that such a defect is through the NF-κB-dependent gene expression based on the data that MALT1 deficiency specifically blocks NF-κB activation but not other signaling pathways downstream of EGFR. In line with this notion, inhibition of IKK kinase activity and TRAF6-silencing phenocopy MALT1 silencing in terms of cell migration, suggesting that MALT1 controls cell migration through NF-κB pathway. Indeed, our microarray data revealed that the expression of several matrix metalloproteinases is significantly downregulated in response to EGF stimulation in CARMA3-silenced cells (data not shown). This is consistent with the notion that NF-κB regulates the expression of many genes involved in migration and invasion.33 However, MALT1 may also contribute to migration and invasion in an NF-κB-independent manner. For example, a recent study showed that BCL10 regulates actin dynamics and polymerization.34 Therefore, it is possible that MALT1 also affects tumor cell migration and invasion by regulating these processes.
Although our previous study suggests that CBM complex-mediated NF-κB activation has an essential role for the EGFR-associated malignancy,26 this notion has not been tested in physiological tumor models. In this study, we used an EGFR-driven lung cancer mouse model to test how MALT1 is involved in the progression of EGFR-induced lung tumor progression. Our result shows that MALT1-dependent NF-κB contributes to the progression of EGFR-induced adenocarcinoma, but is not required for the tumor onset, which highly correlates with the in vitro data that MALT1 inhibition suppresses tumor growth but does not completely suppress tumor cells. Interestingly, although NF-κB activity has been shown to be important to K-ras-dependent lung cancer progression,35 MALT1 is dispensable for both the onset and progression of K-ras-induced lung cancer. This finding is consistent with the hypothesis that MALT1 is specifically involved in EGFR-induced NF-κB, but not K-ras-induced NF-κB that is likely mediated by TBK1.36 Therefore, our result has provided the genetic evidence supporting a rationale of targeting MALT1 or other components in NF-κB signaling in EGFR-associated lung cancer.
Another interesting finding in this study is that MALT1 deficiency abolishes the activation STAT3 in vivo. Previous studies suggest constitutively activated STAT3 is found in 50% of human NSCLC samples.31, 37 In addition, a blockage of STAT3 induces growth arrest of tumor, suggesting the functional importance of STAT3 in EGFR-associated lung cancer.31 However, how STAT3 is aberrantly activated in NSCLC is not clear. Here we provide genetic evidence suggesting that STAT3 activation in NSCLC is likely controlled by MALT1-dependent NF-κB activation. This result suggests that NF-κB not only controls cell proliferation and survival by itself, but also indirectly activates STAT3. Therefore, a blockage of the cross talk between NF-κB and STAT3 can be used as a potential therapeutic strategy for the treatment of EGFR-associated lung cancer. In the current study, we showed that IL-6 is one of the effectors that bridge NF-κB to STAT3 in response to EGFR activation. This result supports an idea to block IL-6 in EGFR-dependent lung cancer to interrupt the cross talk between NF-κB and STAT3. In fact, elevated IL-6 level has been connected to the survival of patients with advanced NSCLC.38 As anti-IL-6 and anti-IL-6 receptor antibodies are currently tested in several clinical studies including some types of cancers,39 targeting IL-6 in EGFR-dependent NSCLC is worth to be tested in preclinical models in the future.
Materials and methods
Antibodies and reagents
Phosphorylation-specific antibodies to ERK1/2 (9101) and IκBα (9246) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against phosphorylated EGFR (sc-12351), IκBα (sc-371), lamin B (sc-6216), ERK (sc-154), IKKγ (FL-419) and actin (sc-8432) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Monoclonal antibodies against the C terminus of MALT1 were generated in the Genentech (South San Francisco, CA, USA) central production facility. DNA-oligo probes for NF-κB (E3291) and Oct-1 (E3241) were purchased from Promega (Madison, WI, USA). Recombinant human EGF was purchased from Sigma-Aldrich (St Louis, MO, USA). PMA (16561-29-8) and ionomycin (56092-82-1) were purchased from Fisher Scientific (Pittsburgh, PA, USA). IL-6-neutralizing antibodies were purchased from Abcam, Cambridge, MA, USA (ab6672) and used at 1:400 dilution. TNFα was purchased from Thermo Fisher Scientific (Rockford, IL, USA). MALT1 protease inhibitor z-VRPR-Fmk was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY, USA).
Cell cultures
Human A431 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS). HCC827 cells were cultured with RPMI medium containing 10% FBS. Primary MEFs were isolated at E13.5 and cultured with DMEM–10% FBS. All cells were maintained at 37 °C with 5% CO2.
Immunoblotting and electrophoretic mobility shift assay
Immunoblotting was performed as descried elsewhere.26 For the electrophoretic mobility shift assay, two million cells were starved overnight in DMEM containing 0.5% serum and stimulated with reagents, and nuclear extracts were isolated. Nuclear extracts (5 μg) were incubated with 1 × 105 c.p.m. of 32P-labeled probes at room temperature for 15 min. The samples were separated on a native Tris-borate-EDTA polyacrylamide gel and analyzed by autoradiography.
Gene silencing and reconstitution
Lentivirus vectors were generated by cotransfection of HEK293T cells with plasmids encoding short hairpin RNA (target sequence for MALT1: 5′-CCTCACTACCAGTGGTTCAAA-3′; TRAF6: 5'-GCCACGGGAAATATGTAATATCT-3'): pCMV-VSV-G (Addgene #8454) and pCMV-dR8.2 (Addgene #8455). The complementary DNA of human MALT1 was amplified by using pcDNA3-MALT1 as a template and cloned into the pBabe-hygro (Addgene #1765) vector. The C464A mutation was introduced via site-directed mutagenesis. To reconstitute MALT1 in A431 cells with a MALT1 knockdown, six synonymous mutations in the short hairpin RNA-targeting sequence were introduced by PCR (5′-CCTCACTACCAGTGGTTCAAA-3′ to 5′-CCGCATTATCAATGGTTTAAG-3′), so that the short hairpin RNA targeted only endogenous MALT1. To produce retrovirus vectors for infecting MEF, pBabe-human MALT1 was cotransfected with pCL-Eco (Addgene #12371).
Colony formation, migration assay and wound-healing assays
Colony formation and migration assays were performed as described elsewhere.26 In brief, cells were mixed with agarose to a final concentration of 0.6% in complete medium, with or without EGF (1 ng/ml). Three weeks after culturing, the numbers and sizes of colonies were determined by inverted microscopy. Nine to ten fields were randomly selected and the size of each colony visualized was determined. For the wound-healing assay, confluent A431 cells were cultured in serum-free DMEM, and a uniform wound was made with a p200 pipette tip. Wounded monolayer cells were washed two or three times to remove detached cells. The initial size of the wound was determined by inverted microscopy immediately after the wash. After 16–20 h of incubation in serum-free medium, the size of the wound was analyzed again. Wound closure was calculated as the percentage of the initial wound area remaining.
Lung metastasis model
To establish a lung metastasis model, three million A431 cells were washed with phosphate-buffered saline, resuspended in 300 μl of serum-free DMEM, and intravenously injected into the tail veins of SCID mice. Three weeks after injection, mice were killed and the lungs were fixed by Bouin’s solution (Sigma-Aldrich, #HT10132), so that a metastasis site could be visualized as a white spot on a yellow background. Metastasis sites on each lung lobe were counted. Student’s t-test was used to determine statistical significance.
Mouse strains and models
To establish EGFR-induced lung cancer model, tetO-EGFRL858R mice,40 CCSP-rTTA mice41 and Malt1−/− mice5 were intercrossed to generate control mice (tetO-EGFRL858R; CCSP-rTTA; Malt1+/−) and experimental mice (tetO-EGFRL858R;CCSP-rTTA; Malt1−/−). After weaning, mice were administrated with doxycycline (Alfa Aesar, Ward Hill, MA, USA) containing water (2 mg/ml) for 2 months. For K-ras-induced lung caner model, we crossed LSL-K-rasG12D,42 CCSP-Cre43 and Malt1−/− mice to generate control mice (LSL-K-rasG12D; CCSP-Cre; Malt1+/−) and experimental mice (LSL-K-rasG12D;CCSP-Cre; Malt1−/−). These mouse strains were genotyped by PCR as previously described.40, 41, 42, 43 All animal experiments and procedures were conducted under the protocol and was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas MD Anderson Cancer Center.
Histology and Immunohistochemical staining
Mouse tissues were washed in PBS and fixed in 4% paraformaldehyde solution for overnight and embedded in paraffin. Paraffin sections were stained with hematoxylin and eosin by the histology core facility at the University of Texas MD Anderson Cancer Center and examined by a pathologist (MJY). For immunohistochemistry (IHC) staining, standard procedures were carried out according to the manual (DAKO, #K0673, USA). The stained sections were automatically processed by ACIS III system (DAKO, USA) and quantified based on 10–15 randomly chosen fields. Quantification of IHC staining was repeated three times by using at least three different mice per genotype. The following antibodies were used in IHC staining: P-S6 Ribosomal Protein (#4858, Cell Signaling), P-AKT (#3787, Cell Signaling), P-ERK1/2 (#4376, Cell Signaling), P-P65 (#3037, Cell Signaling), P-STAT3 (#9145, Cell Signaling) and STAT3 (#9139, Cell Signaling).
Real-time PCR
Total RNA was isolated using TRIzol RNA isolation reagent (Invitrogen, Grand Island, NY, USA) and reverse transcribed using SuperScriptIII (Invitrogen). Quantitative PCR was performed in triplicates using Power SYBR Green PCR Master Mix (Applied Biosystems, Grand Island, NY, USA).
Statistical analysis
GraphPad Prism software was used for all statistical analyses. The Student’s t-test (two-tailed t-test) was used to evaluate the difference of two groups of data.
References
Hayden MS, Ghosh S . NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Gene Dev 2012; 26: 203–234.
Karin M . NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 2009; 1: a000141.
Thome M . Multifunctional roles for MALT1 in T-cell activation. Nat Rev Immunol 2008; 8: 495–500.
Ruland J, Duncan GS, Wakeham A, Mak TW . Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity 2003; 19: 749–758.
Ruefli-Brasse AA, French DM, Dixit VM . Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science 2003; 302: 1581–1584.
Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L et al. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature 2000; 404: 402–407.
Wang D, You Y, Case SM, McAllister-Lucas LM, Wang L, DiStefano PS et al. A requirement for CARMA1 in TCR-induced NF-kappa B activation. Nat Immunol 2002; 3: 830–835.
Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B et al. Phosphorylation of CARMA1 plays a critical role in T Cell receptor-mediated NF-kappaB activation. Immunity 2005; 23: 575–585.
Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL et al. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity 2005; 23: 561–574.
Blonska M, Lin X . NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res 2011; 21: 55–70.
Akagi T, Motegi M, Tamura A, Suzuki R, Hosokawa Y, Suzuki H et al. A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene 1999; 18: 5785–5794.
Dierlamm J, Baens M, Wlodarska I, Stefanova-Ouzounova M, Hernandez JM, Hossfeld DK et al. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood 1999; 93: 3601–3609.
Morgan JA, Yin Y, Borowsky AD, Kuo F, Nourmand N, Koontz JI et al. Breakpoints of the t(11;18)(q21;q21) in mucosa-associated lymphoid tissue (MALT) lymphoma lie within or near the previously undescribed gene MALT1 in chromosome 18. Cancer Res 1999; 59: 6205–6213.
Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006; 441: 106–110.
Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol 2008; 9: 263–271.
Rebeaud F, Hailfinger S, Posevitz-Fejfar A, Tapernoux M, Moser R, Rueda D et al. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol 2008; 9: 272–281.
Staal J, Driege Y, Bekaert T, Demeyer A, Muyllaert D, Van Damme P et al. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J 2011; 30: 1742–1752.
Hailfinger S, Nogai H, Pelzer C, Jaworski M, Cabalzar K, Charton JE et al. Malt1-dependent RelB cleavage promotes canonical NF-kappaB activation in lymphocytes and lymphoma cell lines. Proc Natl Acad Sci USA 2011; 108: 14596–14601.
Rosebeck S, Madden L, Jin X, Gu S, Apel IJ, Appert A et al. Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical NF-kappaB activation. Science 2011; 331: 468–472.
Hailfinger S, Lenz G, Ngo V, Posvitz-Fejfar A, Rebeaud F, Guzzardi M et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci USA 2009; 106: 19946–19951.
Ferch U, Kloo B, Gewies A, Pfander V, Duwel M, Peschel C et al. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 2009; 206: 2313–2320.
Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell 2012; 22: 825–837.
Fontan L, Yang C, Kabaleeswaran V, Volpon L, Osborne MJ, Beltran E et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell 2012; 22: 812–824.
Sharma SV, Bell DW, Settleman J, Haber DA . Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7: 169–181.
Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang L et al. EGFR-induced and PKCepsilon monoubiquitylation-dependent NF-kappaB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell 2012; 48: 771–784.
Jiang T, Grabiner B, Zhu Y, Jiang C, Li H, You Y et al. CARMA3 is crucial for EGFR-Induced activation of NF-kappaB and tumor progression. Cancer Research 2011; 71: 2183–2192.
Pan D, Lin X . Epithelial growth factor receptor-activated nuclear factor kappaB signaling and its role in epithelial growth factor receptor-associated tumors. Cancer J 2013; 19: 461–467.
Stewart JR, O'Brian CA . Protein kinase C-{alpha} mediates epidermal growth factor receptor transactivation in human prostate cancer cells. Mol Cancer Ther 2005; 4: 726–732.
Fan QW, Cheng C, Knight ZA, Haas-Kogan D, Stokoe D, James CD et al. EGFR signals to mTOR through PKC and independently of Akt in glioma. Sci Signal 2009; 2: ra4.
Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ . The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell 2004; 14: 289–301.
Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest 2007; 117: 3846–3856.
Shambharkar PB, Blonska M, Pappu BP, Li H, You Y, Sakurai H et al. Phosphorylation and ubiquitination of the IkappaB kinase complex by two distinct signaling pathways. EMBO J 2007; 26: 1794–1805.
Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest 2004; 114: 569–581.
Marion S, Mazzolini J, Herit F, Bourdoncle P, Kambou-Pene N, Hailfinger S et al. The NF-kappaB signaling protein Bcl10 regulates actin dynamics by controlling AP1 and OCRL-bearing vesicles. Dev Cell 2012; 23: 954–967.
Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009; 462: 104–107.
Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009; 462: 108–112.
Haura EB, Zheng Z, Song L, Cantor A, Bepler G . Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin Cancer Res 2005; 11: 8288–8294.
Songur N, Kuru B, Kalkan F, Ozdilekcan C, Cakmak H, Hizel N . Serum interleukin-6 levels correlate with malnutrition and survival in patients with advanced non-small cell lung cancer. Tumori 2004; 90: 196–200.
Guo Y, Xu F, Lu T, Duan Z, Zhang Z . Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev 2012; 38: 904–910.
Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE . Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Gene Dev 2006; 20: 1496–1510.
Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Gen Dev 2001; 15: 3249–3262.
Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Gene Dev 2001; 15: 3243–3248.
Li H, Cho SN, Evans CM, Dickey BF, Jeong JW, DeMayo FJ . Cre-mediated recombination in mouse Clara cells. Genesis 2008; 46: 300–307.
Acknowledgements
We thank Dr Vishva Dixit (Genentech Corporation) for providing Malt1-deficient mice and Dr Francesco J DeMayo (Baylor College of Medicine) for providing CCSP-Cre mice and Dr Jeffrey Whitsett (Cincinnati Children’s Hospital Medical Center) for providing CCSP-tTA mice. This work is partially supported by grants, RP120316 from Cancer Prevention Research Institute of Texas (CPRIT) to XL, GM079451 and GM065899 from National Institutes of Health (NIH) to XL and R01 CA164346 (NCI/NIH), Developmental Research Awards in Leukemia SPORE CA100632 to MJY, and Center for Inflammation and Cancer, Center for Genetics and Genomics, IRG, Sister Institution Network fund of UT MD Anderson Cancer Center, and Cancer Prevention Research Institute of Texas to MJY.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Pan, D., Jiang, C., Ma, Z. et al. MALT1 is required for EGFR-induced NF-κB activation and contributes to EGFR-driven lung cancer progression. Oncogene 35, 919–928 (2016). https://doi.org/10.1038/onc.2015.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.146
- Springer Nature Limited
This article is cited by
-
In-depth quantitative proteomics analysis revealed C1GALT1 depletion in ECC-1 cells mimics an aggressive endometrial cancer phenotype observed in cancer patients with low C1GALT1 expression
Cellular Oncology (2023)
-
Notch1-ADAM8 positive feed-back loop regulates the degradation of chondrogenic extracellular matrix and osteoarthritis progression
Cell Communication and Signaling (2019)
-
TBKBP1 and TBK1 form a growth factor signalling axis mediating immunosuppression and tumourigenesis
Nature Cell Biology (2019)
-
CARD–BCL-10–MALT1 signalling in protective and pathological immunity
Nature Reviews Immunology (2019)
-
Hectd3 promotes pathogenic Th17 lineage through Stat3 activation and Malt1 signaling in neuroinflammation
Nature Communications (2019)