
Abstract
Polycyclic aromatic hydrocarbons (PAHs) are environmental pollutants, and many are potent carcinogens. Benzo[a]pyrene (B[a]P), one of the best-studied PAHs, is metabolized ultimately to the genotoxin anti-B[a]P-7,8-dihydrodiol-9,10-epoxide (BPDE). BPDE triggers stress responses linked to gene expression, cell death and survival. So far, the underlying mechanisms that initiate these signal transduction cascades are unknown. Here we show that BPDE-induced DNA damage is recognized by DNA damage sensor proteins to induce activation of the stress-activated protein kinase (SAPK) p38. Surprisingly, the classical DNA damage response, which involves the kinases ATM and ATR, is not involved in p38-SAPK activation by BPDE. Moreover, the induction of p38-SAPK phosphorylation also occurs in the absence of DNA strand breaks. Instead, increased phosphorylation of p38-SAPK requires the nucleotide excision repair (NER) and DNA damage sensor proteins XPC and mHR23B. Interestingly, other genotoxins such as cisplatin (CDDP), hydrogen peroxide and ultraviolet radiation also enhance XPC-dependent p38-SAPK phosphorylation. In contrast, anti-benzo[c]phenanthrene-3,4-dihydrodiol-1,2-epoxide, the DNA adducts of which are not properly recognized by NER, does not trigger p38-SAPK activation. As a downstream consequence, expression and secretion of the pro-inflammatory cytokine interleukin-6 is induced by BPDE and CDDP in vitro and by CDDP in the murine lung, and depends on XPC. In conclusion, we describe a novel pathway in which DNA damage recognition by NER proteins specifically leads to activation of p38-SAPK to promote inflammatory gene expression.
Similar content being viewed by others

Introduction
Chemical carcinogenesis is a multi-step process, involving procarcinogen activation to a genotoxin, induction of DNA damage and subsequent conversion into mutations, which eventually give rise to a tumor. Polycyclic aromatic hydrocarbons are prominent examples of environmental carcinogens. One of the most abundant and best-studied polycyclic aromatic hydrocarbon is the environmental pollutant benzo[a]pyrene (B[a]P), a constituent and contaminant of cigarette smoke, automobile exhaust, industrial waste and even food products, which is carcinogenic to rodents and humans.1 B[a]P binds to the intracellular aryl hydrocarbon receptor, thereby inducing its own metabolism by cytochrome P450s. Among several different metabolites that are formed initially, the B[a]P-7,8-epoxide is considered to be one of the most critical, as it is further metabolized to B[a]P-7,8-dihydrodiol by the microsomal epoxide hydrolase, which in turn is oxidized to the highly reactive electrophilic genotoxin and carcinogen anti-B[a]P-7,8-dihydrodiol-9,10-epoxide (BPDE).2, 3
During evolution, multiple surveillance and defense mechanisms have evolved to protect the cell from DNA damage. Specific signaling pathways operate to detect and repair different subtypes of lesions. In case the damage is only inadequately removed, expansion of damaged cells can be counteracted by the inhibition of proliferation, by triggering apoptosis or by the induction of senescence. Prominent examples of such cell cycle and survival regulators are transcription factors (for example, p53) and stress-activated protein kinases (SAPKs).4, 5, 6 The family of SAPKs is comprised of the c-Jun NH2-terminal kinases (JNKs) and the p38 kinases. Among the substrates of SAPKs are transcription factors such as c-Jun, which upon posttranslational modifications interact with co-activators or are relieved from repression to activate target gene expression.7, 8
Although many genotoxic stimuli activate SAPKs, the primary sensor(s) propagating the signal to SAPKs are still poorly defined. Both nuclear and cytoplasmic signal transducers have been identified.8 In the case of ultraviolet radiation (UVR), membrane-bound receptors are activated, presumably by intermediate reactive oxygen species, and are among the first targets to initiate signaling to SAPKs.8 However, nuclear targets have also been linked to genotoxin-induced SAPK activation. The most prominent signaling cascade that senses DNA damage and converts this into checkpoint activation is the ATM/ATR pathway.9, 10 The two kinases ATM (Ataxia-Telangiectasia mutated) and ATR (Ataxia-Telangiectasia and Rad3-related) orchestrate the DNA damage response (DDR) via multiple downstream effectors such as checkpoint kinase (Chk) 2 and 1, respectively. Interestingly, in response to double-strand-breaking agents, ATM/ATR seems to be involved in p38-SAPK activation.11
So far, only two studies have addressed immediate early signaling of reactive diol epoxides.12, 13 Activation of p38-SAPK and JNK by BPDE in murine epithelial cells was paralleled by an increased activity of the PI3K (phosphatidylinositol-3-kinase) as well as of the AKT kinase, which is a target of PI3K. The authors postulated a signaling chain in which BPDE first activates PI3K and subsequently AKT, which then via unknown intermediate steps triggers JNK but interestingly not p38-SAPK activation. Thus, the upstream regulatory events triggering p38-SAPK activity in response to reactive diol epoxides thus far remain unknown. A consequence of sustained p38-SAPK activation by BPDE is the induction of apoptosis in murine and human hepatoma cells as well as in murine fibroblasts, accompanied by cytochrome c release, PARP cleavage and upregulation of pro-apoptotic BCL2 family members.14 In parallel to p38-SAPK activation, BPDE triggers apoptosis that is dependent on the tumor suppressor protein p53 and the pro-apoptotic protein BAX in human colon carcinoma cells.15
In the present study, we investigated the mechanism of p38-SAPK activation in response to BPDE and other genotoxins. BPDE-induced DNA damage coincided with increased p38-SAPK phosphorylation, suggesting a critical role of the DDR in signal generation. However, the classical DDR components ATM/ATR were not involved in p38-SAPK activation by BPDE. Instead, the nucleotide excision repair (NER) and DNA damage recognition factors XPC (xeroderma pigmentosum, XP, complementation group C) and the homolog of the yeast protein RAD23 (HR23B) were required for this novel signaling chain. Furthermore, benzo[c]phenanthrene-3,4-dihydrodiol-1,2-epoxide (B[c]PhDE), whose DNA adducts are not properly recognized by NER,16 did not induce p38-SAPK activation. As intermediate signal transducers that link BPDE-mediated DNA damage recognition to p38-SAPK activation, we could identify the SAPK kinases MKK3 and 6, which act downstream of an unknown tyrosine kinase. As a consequence of enhanced p38-SAPK signaling in response to BPDE and cisplatin (CDDP) in fibroblasts, the pro-inflammatory cytokine interleukin-6 (IL-6) is induced. Interestingly, enhanced IL-6 expression and secretion was not only dependent on p38-SAPK but also required XPC. Finally, the instillation of CDDP suspensions into lungs of wild-type mice provoked IL-6 release, whereas this inflammatory response was greatly diminished in Xpc-knockout mice.
Results
Activation of p38-SAPK by BPDE does not require proliferation, de novo transcription and AKT signaling
In order to examine the underlying basic signaling cascade required for p38-SAPK activation by BPDE, we first tested the influence of cell proliferation. Genotoxic agents frequently disturb replication, which is the cause for initiating signaling cascades to cope with the damage, for example, by means of a cell cycle block.10, 17 In confluent murine fibroblasts, which in addition were serum starved overnight to inhibit proliferation (Supplementary Figure S1B), BPDE still activated p38-SAPK within minutes after exposure, comparably to logarithmically growing cultures (Figure 1a, upper panels). Hence, interference with replication was not a prerequisite to induce p38-SAPK phosphorylation. In contrast to the action on p38-SAPK, BPDE exerted no notable effect on the related extracellular signal-regulated kinases ERK1 and 2 (Figure 1a, lower panels). The observed p38-SAPK activation occurred also in the presence of the transcription inhibitor actinomycin D, demonstrating that an intermediate induced gene expression was not necessary for initiation of this signaling cascade (Figure 1b).

BPDE-induced p38-SAPK activation is independent of proliferation and de novo transcription but coincides with DNA damage. (a) Proliferation of cells is not a prerequisite for the activation of p38-SAPK by BPDE. Murine fibroblasts (NIH3T3) were either logarithmically growing (y=proliferation: yes) or grown to confluence and serum starved overnight to prevent proliferation (n=proliferation: no). Cultures were treated with BPDE (2 μM) or the solvent dimethyl sulfoxide (DMSO; 0.1%) for the indicated time. BPDE-induced phosphorylation of p38-SAPK and ERK was detected in whole-cell extracts by western blot analysis with phosphospecific antibodies. For loading, control membranes were reprobed with anti-p38 and anti-ERK2 antibodies, respectively. (b) BPDE-induced p38-SAPK activation is independent of de novo transcription. Confluent and serum-starved NIH3T3 cells were pretreated for 15 min with actinomycin D (5 μg/ml) to inhibit transcription. Subsequently, the cultures were incubated with BPDE (2 μM) or DMSO (0.1%) for the indicated time and phosphorylation of p38-SAPK was determined as described in a. (c) In contrast to BPDE, the nonreactive parent compound B[a]P does not induce p38-SAPK phosphorylation. NIH3T3 cells were treated for 60 min with DMSO (0.1%), benzo[a]pyrene (B[a]P, 2 μM) or BPDE (2 μM), respectively, and p38-SAPK phosphorylation was detected as described in a. (d) BPDE induces rapidly DNA adducts. NIH3T3 cells were treated for the indicated time with BPDE (2 μM). DNA adducts were determined with the 32P-postlabelling assay. (e, f) BPDE induces DNA single-strand breaks but not double-strand breaks. Cells were treated as described in d and induction of DNA strand breaks was measured by the alkaline comet or the neutral comet assay, respectively. For each time point, 100 cells were analyzed and intensity of the tail region was quantified by using the software VisComet 1.6.2.
As stated in the introduction, BPDE is the reactive metabolite of the parental nonreactive compound B[a]P. To investigate whether p38-SAPK activation is dependent on the chemical reactivity of BPDE, SAPK phosphorylation by B[a]P, which is not metabolized in fibroblasts, was examined. In contrast to BPDE, B[a]P did not cause phosphorylation of the SAPK p38 at equimolar concentrations (Figure 1c). Similarly, hydrolysis of BPDE to its inactive tetrol derivative also led to the loss of p38-SAPK activation (Supplementary Figure S1C; for formulas, see Supplementary Figure S1A). In conclusion, the reactive epoxide moiety is essential for p38-SAPK activation by BPDE.
As described for murine epithelial cells,12 we also observed induction of AKT phosphorylation by BPDE in mouse fibroblasts, the kinetics of which appeared similar to that of p38-SAPK activation (Supplementary Figure S1D). To examine whether in murine fibroblasts, like in epithelial cells, the PI3- and AKT kinase activities are also dispensable for BPDE-induced p38-SAPK activation, we investigated p38-SAPK phosphorylation in cells pretreated with the PI3 kinase inhibitor wortmannin. In the presence of increasing concentrations of wortmannin, BPDE-induced AKT phosphorylation was completely blocked, whereas BPDE-induced p38-SAPK phosphorylation remained unaffected (Supplementary Figure S1D).
BPDE-induced DNA damage coincides with p38-SAPK activation
Next we focused on nuclear events as possible initiators of p38-SAPK activation by BPDE and analyzed the emergence and kinetics of DNA damage. DNA adducts (as detected by the postlabeling assay, Figure 1d) and single-strand breaks (as detected with the alkaline comet assay; Figure 1e) were induced by BPDE within minutes after treatment. However, BPDE did not induce double-strand breaks, as determined by the neutral comet assay (Figure 1f). The emergence of DNA adducts and single-strand breaks correlated temporally with the activation of p38-SAPK (see Figure 1a). When comparing the kinetics of p38-SAPK activation with that of DNA damage induction, it appeared that kinase activation was already maximal after 10 min, whereas the level of DNA single-strand breaks further increased. Therefore, p38-SAPK activation seemed to precede DNA single-strand break formation and thus might be coupled to an early event during the generation of single-strand breaks.
Role of the DNA damage recognition proteins and signal transducers ATM, ATR and DNA-PK in p38-SAPK activation by BPDE and other genotoxins
The mammalian DDR is initiated by DNA damage sensors and the ATM and ATR kinases, which phosphorylate histone H2 AX and several other substrates. Accordingly, BPDE triggered phosphorylation of H2 AX in murine fibroblasts (Figure 2a). However, pretreatment of cells with ATM/ATR-kinase inhibitors did not prevent p38-SAPK activation (Figure 2a). A potent activator of ATM/ATR signaling is the topoisomerase II inhibitor doxorubicin, an anticancer drug that induces double-strand breaks (Supplementary Figure S1E). Although doxorubicin increased H2AX phosphorylation, it did not simultaneously activate p38-SAPK (Figure 2a). Similarly, doxorubicin activated the ATM/ATR pathway in human HeLa cells, as indicated by a rapid increase of Chk2 phosphorylation, whereas no parallel activation of p38-SAPK could be detected (Figure 2b). Thus, early ATM/ATR activation by doxorubicin does not lead to an immediate increase in p38-SAPK phosphorylation. However, at later time points after doxorubicin treatment, enhanced p38-SAPK phosphorylation became visible (Supplementary Figure S2A). Furthermore, late activation of p38-SAPK by doxorubicin was greatly reduced in human fibroblasts deficient for ATM (Supplementary Figure S2B). Hence, double-strand break-inducing genotoxins, in contrast to BPDE, trigger a delayed response that, as reported previously, couples ATM/ATR to p38-SAPK activation.11 In contrast to doxorubicin, however, BPDE-induced p38-SAPK phosphorylation was not impaired in ATM-deficient human fibroblasts (Figure 2c). Furthermore, simultaneous inhibition of ATM and ATR in human HeLa cells also did not prevent p38-SAPK activation by BPDE (Figure 2d).

The DNA damage sensors ATM and ATR are not involved in BPDE-induced p38-SAPK activation. (a) Chemical inhibition of ATM does not interfere with p38-SAPK activation by BPDE. NIH3T3 cells were pretreated for 30 min with dimethyl sulfoxide (DMSO; 0.1%) or the ATM/ATR-kinase inhibitor (KU55933 and ETP-46464, 10 μM each) and subsequently exposed for the indicated time to either BPDE (2 μM) or doxorubicin (10 μM). Extracts were analyzed as described in Figure 1a using phosphospecific antibodies to detect phosphorylated H2AX and p38-SAPK. (b) Early activation of ATM signaling by doxorubicin is not sufficient to trigger p38-SAPK phosphorylation. HeLa cells were treated for 60 min with BPDE or doxorubicin and phosphorylation of p38-SAPK and the ATM substrate Chk2 was analyzed as described above. (c) Induction of p38-SAPK phosphorylation by BPDE is independent of ATM. Human fibroblasts proficient or deficient for ATM were treated with DMSO (−) or BPDE (+) for 60 min and p38-SAPK phosphorylation was analyzed as described above. (d) Chemical inhibition of ATM and ATR does not prevent p38-SAPK activation by BPDE. HeLa cells were preincubated for 30 min with DMSO or the ATM/ATR-kinase inhibitor (KU55933 and ETP-46464, 2.5 μM each) and subsequently treated with DMSO (−; 0.1%) or 2 μM BPDE (+) for 30 min. p38-SAPK phosphorylation was analyzed as described above.
Thus, ATM/ATR signaling is dispensable for the activation of p38-SAPK activation by BPDE. The ATM/ATR target p53 that is activated by BPDE18 is also not involved in p38-SAPK activation, as there was no significant difference in p38-SAPK phosphorylation between wild-type and p53-knockout murine fibroblasts (Supplementary Figure S2C).
Another prominent signaling transducer of DNA damage is the DNA-dependent protein kinase DNA-PK.10 For example, DNA double-strand break-inducing compounds trigger activation of AKT and p53 via DNA-PK.19, 20 However, inhibition of DNA-PK did not abolish p38-SAPK activation by BPDE (Supplementary Figure S2D). Consistent results were obtained in murine DNA-PK-deficient severe combined immunodeficiency syndrome cells, in which BPDE still enhanced p38-SAPK phosphorylation (Supplementary Figure S2E).
Role of the DNA damage recognition proteins XPC and mHR23B in p38-SAPK activation by BPDE and other genotoxins
DNA adducts produced by BPDE are removed by NER, which can be divided into two subpathways—global genome NER (GG-NER) and transcription-coupled NER (TC-NER).21 GG-NER eliminates damage throughout the genome, whereas TC-NER specifically repairs the transcribed strand of active genes. The first step of DNA damage recognition by GG-NER involves the XPC and HR23B (homolog of the yeast protein RAD23) protein complex. Damage detection by TC-NER utilizes RNA polymerase II, stalled at transcription-blocking lesions, along with the CSA and CSB proteins. Subsequent to damage recognition, other repair proteins such as XPA, the XPD and XPB helicases of the TFIIH transcription initiation/repair complex, and nucleases are recruited to remove the lesion. Interestingly, in NIH3T3 mouse fibroblasts deficient for XPC, p38-SAPK activation was clearly reduced at early (Figure 3a) and later time points (Supplementary Figure S3A). In contrast, in CSB-deficient murine fibroblasts, no defect in signaling was detected (Figure 3b). Thus, the recognition of BPDE-generated DNA adducts by XPC seems to generate a signal for p38-SAPK activation.

The DNA damage recognition proteins XPC and mHR23B are essential for BPDE-induced p38-SAPK phosphorylation. (a) Activation of p38-SAPK is reduced in XPC-deficient immortalized murine fibroblasts. Wild-type (wt) and XPC-knockout (−/−) NIH3T3 cells were treated for 30 and 60 min with dimethyl sulfoxide (DMSO) or BPDE and analyzed for p38-SAPK phosphorylation as described in Figures 1a. (b) BPDE-induced p38-SAPK activation is not impaired in CSB-deficient cells. Wild-type and CSB-knockout NIH3T3 cells were treated and analyzed as described above. (c) Activation of p38-SAPK is also reduced in primary XPC-deficient MEF. Wild-type and XPC-knockout cells were treated and analyzed as described in a. (d) Depletion of XPC interferes with p38-SAPK activation by BPDE. Mouse hepatoma cells (Hepa-1) were transiently transfected with either control (ctr) siRNA or two different siRNA sequences (seq1 and 2) targeting XPC. Downregulation of XPC was confirmed by western blotting. Forty-eight hours after transfection, the cells were treated with DMSO or BPDE for 30 min and p38-SAPK phosphorylation was analyzed as described above. (e) Activation of p38-SAPK by H2O2, but not by hyperosmotic stress or anisomycin, depends on XPC. Cells were treated with 200 mM NaCl (30 min), 20 μg/ml anisomycin (20 min) or 50 μM H2O2 (30 and 60 min). Phosphorylation of p38-SAPK was analyzed as described in a. n.t., nontreated. (f) Induction of p38-SAPK phosphorylation by BPDE depends on mHR23A and B proteins. Wild-type and mHR23A/B double-knockout MEFs were treated for the indicated time with 0.1% DMSO (−) or 2 μM BPDE (+) and extracts were analyzed for p38-SAPK phosphorylation as described above.
As the described studies were performed in immortalized NIH3T3 cells, clonal differences between wild-type and Xpc-knockout cells could account for reduced p38-SAPK phosphorylation. Therefore, we next used primary mouse embryonic fibroblasts (MEFs) from wild-type (Xpc+/+) and Xpc-knockout (Xpc−/−) mice and tested for the activation of p38-SAPK after BPDE treatment. Again, p38-SAPK phosphorylation was drastically reduced in primary Xpc−/− MEFs (Figure 3c and Supplementary Figure S3B). Furthermore, knockdown of XPC in Hepa-1 C7 mouse hepatoma cells by small interfering RNA (siRNA) interfered with p38-SAPK activation by BPDE (Figure 3d). Thus, we could link BPDE-induced early p38-SAPK activation to the DNA damage recognition protein XPC.
Other genotoxins, like UVC, hydrogen peroxide (H2O2) and the anticancer drug CDDP, whose adducts are at least partially detected and repaired by NER21, also caused p38-SAPK activation (Figure 3e and Supplementary Figures S3C and D). Interestingly, p38-SAPK activation by these genotoxins was also reduced in NIH3T3 Xpc-knockout cells (Figure 3e and Supplementary Figures S3C and D). However, p38-SAPK phosphorylation by the well-known p38-SAPK activator and translational inhibitor anisomycin was not impaired in NIH3T3 Xpc-knockout cells (Figure 3e and Supplementary Figure S3C). Similarly, hyperosmotic shock triggered by incubation in 200 mM NaCl induced p38-SAPK phosphorylation in NIH3T3 Xpc-knockout cells as efficiently as in wild-type cells (Figure 3e and Supplementary Figure S3C). Taken together, the XPC protein, a sensor of bulky DNA lesions of various origins, is involved in the activation of p38-SAPK.
To obtain further evidence for a central role of DNA damage recognition by XPC in the initiation of p38-SAPK activation, we next investigated p38-SAPK activation by BPDE in mHR23A−/−/mHR23B−/− MEFs, using mHR23A+/−/mHR23B+/− MEFs as the corresponding wild-type control. In the absence of the mHR23A and B proteins (functionally redundant in GG-NER), XPC is unstable and rapidly degraded.22 Indeed, mHR23A−/−/mHR23B−/− MEFs phenocopy Xpc−/− MEFs for their lack of p38-SAPK activation after BPDE treatment (Figure 3f). This finding further highlights the role of XPC in p38-SAPK activation. Finally, the role of XPE in BPDE-induced signaling to p38-SAPK was addressed, as XPE acts upstream of XPC in lesion recognition. Indeed, patient-derived fibroblasts deficient for XPE also showed reduced phosphorylation of p38-SAPK in response to BPDE (Supplementary Figure S3E).
DNA strand breaks are not involved in p38-SAPK activation by BPDE
Loss of XPC prevents the subsequent steps in GG-NER, for example, recruitment of XPA and nucleases that introduce single-strand breaks in the 5' and 3' directions of the lesion. Therefore, the single-strand breaks induced by BPDE (Figure 1e) could be a consequence of the incisions during the repair process. Indeed, in NIH3T3 Xpc−/− fibroblasts, strand breaks in response to BPDE treatment could not be detected (Figure 4a). Absence of these strand breaks in NIH3T3 Xpc−/− fibroblasts could possibly explain the reduced phosphorylation of p38-SAPK. In fact, single- and double-strand breaks (which are further processed to single-stranded DNA) have been postulated as a key signal for the activation of the ATM/ATR pathway in the mammalian DDR.8 If so, NIH3T3 Xpa−/− cells (deficient in the XPA protein) should also show reduced p38-SAPK induction, as they also cannot recruit nucleases to the damaged DNA sites and hence cannot produce strand breaks. Indeed, Xpa−/− fibroblasts did not show increased levels of DNA strand breaks after exposure to BPDE, similar to Xpc−/− cells, which again demonstrated that single-strand breaks are the consequence of DNA repair (Figure 4b). Importantly, however, p38-SAPK activation in Xpa−/− cells was not disturbed (Figure 4c), and accordingly reduced DNA single-strand breaks were not the underlying cause for the reduced p38-SAPK induction observed in Xpc−/− fibroblasts.

Activation of p38-SAPK by BPDE is independent of DNA single-strand breaks. (a) XPC-dependent induction of DNA single-strand breaks by BPDE. XPC-deficient NIH3T3 cells and corresponding wild-type cells were treated for the indicated time with 2 μM BPDE. Induction of single-strand breaks was analyzed as described in Figures 1e. (b) XPA-dependent induction of DNA single-strand breaks by BPDE. Cells were treated and analyzed as described above. (c) Activation of p38-SAPK by BPDE is independent of XPA and occurs also in the absence of DNA single-strand breaks. Cells were treated and analyzed as described in Figures 1a. (d) B[c]PhDE adducts are not recognized by NER. NIH3T3 cells were treated for the indicated time with 2 μM BPDE or 2 μM B[c]PhDE. Induction of single-strand breaks was analyzed as described in a. (e, f) B[c]PhDE does not significantly induce phosphorylation of p38-SAPK. NIH3T3 (e) or HeLa (f) cells were treated and analyzed as described in c.
To further narrow down the specific factors of NER that contribute to p38-SAPK activation, we also investigated the role of the helicases XPB and XPD. In the absence of XPC, the recruitment of these helicases and thus DNA unwinding is also impaired, which might additionally explain impaired signaling to p38-SAPK. However, in patient-derived XPB- and XPD-deficient fibroblasts no obvious difference in p38 activation by BPDE was detected (Supplementary Figure S3F). Rather, the XPC protein seems to specifically induce a signaling cascade by translation of DNA adduct recognition into an intracellular signaling event, leading to p38-SAPK activation.
DNA damage recognition is a prerequisite for p38-SAPK activation by reactive epoxides
As reported here, cells completely lacking, or showing highly reduced levels of the DNA damage recognition GG-NER protein XPC (as in Xpc−/− and mHR23A−/−/mHR23B−/− MEFs, respectively), showed reduced p38-SAPK activation in response to BPDE. In order to support a key role of DNA damage recognition in p38-SAPK activation, we chose a chemical strategy as an alternative to the genetic approaches used above. B[c]PhDE (for formula, see Supplementary Figure S1A) is a highly carcinogenic and reactive polycyclic aromatic hydrocarbon metabolite that efficiently forms DNA adducts, which are not properly recognized by NER factors in human HeLa cells.16 In murine fibroblasts, addition of B[c]PhDE, in contrast to BPDE, only marginally induced single-strand breaks (Figure 4d), confirming that B[c]PhDE adducts are also not efficiently recognized by the NER machinery in rodent cells.16 More importantly, B[c]PhDE also did not induce p38-SAPK activation (Figure 4e). Similarly, B[c]PhDE treatment of human HeLa cells did not stimulate p38-SAPK phosphorylation either (Figure 4f). BcPhDE gives a much higher yield of DNA adducts compared with BPDE, as has been demonstrated by reacting both diol epoxides directly with isolated DNA.23, 24 Yet we cannot entirely rule out that B[c]PhDE in comparison with BPDE might induce lower levels of DNA adducts in cells. Clearly, more detailed studies on the type, amount and kinetics of the different adducts formed will be needed to validate our initial findings.
The upstream kinases MKK3/6 are essential for p38-SAPK activation by BPDE
Activation of p38-SAPK is usually mediated by the upstream kinases MKK3, MKK6 and SEK1.25 Indeed, BPDE treatment increased the phosphorylation of MKK3, MKK6 and SEK1 in NIH3T3 fibroblasts (Supplementary Figure S4A) and HeLa cells (Figure 5a) with similar kinetics compared with activation of p38-SAPK. Inactivation of MKK3/6 and SEK1 (Figure 5b) reduced phosphorylation of p38-SAPK in response to BPDE in HeLa cells. Activation of MKK3/6 and SEK1 by BPDE was also impaired in Xpc−/− cells (Figure 5c) as observed before for p38-SAPK and its substrate MK2 (Supplementary Figure S4B). For the identification of additional kinases further upstream of MKK3/6 and SEK1, we screened several different chemical inhibitors, of which the tyrosine kinase inhibitor PP1 clearly reduced MKK3/6, SEK1 and p38-SAPK phosphorylation induced by BPDE in NIH3T3 fibroblasts (Figure 5d) and HeLa cells (Supplementary Figure S4C). As some tyrosine kinases have previously been implicated in stress signaling to p38-SAPK, we selectively addressed the role of c-abl, c-src, yes and fyn by the use of knockout cells. However, in response to BPDE, no altered activation of p38-SAPK in the various kinase-deficient cells could be observed (Supplementary Figures S4D and E). In summary, DNA damage recognition by XPC initiates a signaling cascade involving an as yet unknown tyrosine kinase, which activates MKK3/6 and SEK1 to phosphorylate p38-SAPK.

The dual specificity kinases MKK3/6 and SEK as well as Src-family kinases are involved in BPDE dependent p38-SAPK activation. (a) MKK3/6 and SEK1 are activated in response to BPDE. HeLa cells were treated with BPDE (2 μM) for the indicated time points and extracts were analyzed by western blot for phosphorylation of MKK3/6, SEK1 and MKK-7 with specific antibodies. For loading, control membranes were reprobed with nonphosphospecific antibodies against the different kinases. (b) Downregulation of MKK3/6 and SEK1 inhibits BPDE dependent p38 activation. HeLa cells were transfected for 3 days with two sets of siRNAs (1 and 2) targeting two different sequences of MKK3/6 and SEK1, respectively. Subsequently, the cells were exposed to BPDE (2 μM) for 60 min. Cell extracts were analyzed by western blot for phosphorylation of p38 and for confirmation of MKK3/6 and SEK1 knockdown. (c) Activation of MKK3/6 and SEK1 after BPDE treatment is dependent on XPC. Wild-type (wt) and XPC-knockout (−/−) NIH3T3 cells were treated and analyzed as described in a. (d) Src-like kinases are involved in activation of p38-SAPK after BPDE treatment. NIH3T3 cells were pretreated for 15 min with dimethyl sulfoxide or the Src-family kinase inhibitor PP1 (10 μg/ml), followed by BPDE (2 μM) treatment for the indicated time points. Cell extracts were analyzed as described above.
BPDE and CDDP induce IL-6 expression and release in a p38-SAPK- and XPC-dependent manner
Finally, we explored the consequences of BPDE- and CDDP-mediated p38-SAPK activation. P38-SAPK regulates many different processes such as cell growth, survival, differentiation and gene expression.25
As activation of p38-SAPK by UV radiation initiates the G2/M checkpoint,26 we monitored progression through the cell cycle in the presence of BPDE. BPDE treatment of murine fibroblasts drastically reduced the percentage of cells entering S-phase (Supplementary Figure S5C). However, interference with cell cycle progression upon BPDE exposure was still observed in the presence of a p38-SAPK inhibitor and in p38 alpha-knockout fibroblasts (Supplementary Figure S5C). Increasing concentrations of BPDE not only inhibited proliferation but also triggered cell death in murine fibroblasts and HeLa cells, as indicated by decreased numbers of viable cells and a concomitant increase in apoptotic cells (Supplementary Figures S5A and B). Again, inhibition of p38-SAPK did not prevent the cytotoxic effects of BPDE (Supplementary Figures S5A and B).
p38-SAPK was initially identified as a target of the bacterial endotoxin LPS (lipopolysaccharide), and is now recognized as a central regulator of inflammation.27 As inflammation is critically linked to carcinogenesis,28, 29 we analyzed the effect of BPDE on inflammatory gene expression. Treatment of NIH3T3 fibroblasts with BPDE specifically increased the mRNA levels of IL-6, cyclooxygenase 2 and tumor necrosis factor alpha (Figure 6a and Supplementary Figures S5D and E). Induction of IL-6, cyclooxygenase 2 and, at least partially, tumor necrosis factor alpha mRNA levels was decreased by chemical inhibition of p38-SAPK (Figure 6a, left panel and Supplementary Figures S5D and E).

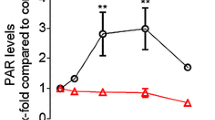
Activation of p38-SAPK by BPDE and CDDP treatment triggers IL-6 expression and release dependent on XPC. (a) Increase of IL-6 mRNA levels after treatment with BPDE and CDDP depends on p38-SAPK activation. NIH3T3 cells were pretreated with dimethyl sulfoxide or the p38-SAPK inhibitor SB203580 (25 μM) for 15 min and subsequently exposed to BPDE for the indicated time points (left panel) or CDDP (50 μM) for 60 min (right panel). IL-6 mRNA levels were analyzed by quantitative real-time PCR. Shown are the means±s.e.m. from three independent experiments analyzed in duplicates (n=6). (b, c) BPDE and CDDP induce IL-6 release dependent on p38-SAPK and XPC. NIH3T3 cells were either not transfected (−), transfected with control siRNA (ctr) or with different siRNAs targeting two different sequences (1 and 2) of p38-SAPK or XPC, respectively. After 3 days, cells were exposed to BPDE (b) or CDDP (c) for 24 h. IL-6 release into the cell culture medium was measured by enzyme-linked immunosorbent assay. The figure shows means±s.e.m. from at least three independent experiments. (d) Instillation of CDDP triggers release of IL-6 in the murine lung dependent on XPC. CDDP (15 μg) dissolved in 50 μl solvent (0.9% NaCl, indicated as ‘+’) or the solvent alone (indicated as ‘−’) were instilled into wild-type (each group: n=6) and XPC-knockout mice (solvent group: n=5; CDDP group: n=7). After 24 h, bronchoalveolar lavage was performed and the cell-free supernatant was used to determine the amount of IL-6 (left panel) and the total protein content as an indicator of vascular permeability (right panel).
In subsequent experiments, we focused our analysis on the regulation of IL-6 as a critical cytokine involved in transformation and carcinogenesis.30 Exposure to CDDP also increased IL-6 mRNA levels dependent on p38-SAPK activity (Figure 6a, right panel). As the final release of cytokines is a complex process that not only involves regulation at the mRNA level27, the cytokine levels in the medium of BPDE and CDDP exposed cells were determined. The amounts of the IL-6 protein were clearly increased upon BPDE and CDDP exposure (Figures 6b and c). Again, siRNA-mediated downregulation of p38-SAPK and XPC reduced induction of IL-6 secretion by BPDE and CDDP treatment (Figures 6b and c). Finally, as CDDP is a first-line drug to treat lung cancer, we analyzed its effects on IL-6 release in the murine lung. Indeed, instillation of CDDP suspensions increased IL-6 release in the lung after 24 h in wild-type mice (Figure 6d, left panel). Importantly, the release of IL-6 in response to CDDP exposure was clearly reduced in XPC-knockout mice (Figure 6d, right panel), thus demonstrating the relevance of XPC in promoting DNA damage-induced IL-6 release in vivo.
Discussion
The best-studied DDR to date is launched by the ATM/ATR pathway. Numerous downstream substrates, including p53 and checkpoint kinases, mount a cellular response critical for DNA repair, proliferation and cell survival (Supplementary Figure S6). Activation of p38-SAPK by genotoxic stimuli such as ionizing radiation, doxorubicin, camptothecin but also CDDP has been connected to ATM/ATR.11, 31 In this pathway, the primary event for signal initiation is possibly the generation of DNA double-strand breaks, which subsequently may also lead to single-stranded DNA. Recognition of DNA double-strand breaks or single-stranded DNA by ATM and ATR, respectively, then mounts an ill-defined response, culminating in p38-SAPK activation. The TAO (thousand and one amino acid) kinases interact with ATM and mediate, at least in part, activation of the p38-SAPK activating kinases MKK3 and 6.31
Here we describe a novel pathway in which DNA damage recognition by GG-NER signals to p38-SAPK via XPC. Compared with the 250 adducts/106 base pairs (bp) observed in our cell culture experiments, up to 34 adducts/108 bp are detected in the lung of smokers, corresponding to a 700-fold lower burden of DNA damage.32 Note, however, that fresh adducts are rapidly and efficiently repaired after acute exposure, whereas in the lung biopsies steady-state levels were measured. Thus, the levels of DNA adducts in our studies might still be within a range that is relevant for smoking-related diseases.33
Sensing of DNA adducts by the XPC protein involves binding to the DNA strand opposite to the lesion.34 Therefore, the pathway discovered here probably relies on the recognition of distorted DNA as a primary trigger to promote p38-SAPK activation in response to GG-NER-specific lesions. The intermediate factors that link XPC to p38-SAPK activation are still partly unknown. Clearly, the upstream kinases MKK3/6 and SEK1 are involved and are downstream of an as yet unknown tyrosine kinase activity. Future investigations based on the use of chemical inhibitors of known kinases, siRNA libraries or protein–protein interaction screens should help to identify more components of this novel signal transduction pathway. Indeed, a recent yeast two-hybrid screen revealed MAP3K5 (aka ASK-1), a known upstream activator of p38-SAPK, as a novel interaction partner of XPC35. Of note, we show that BPDE still triggers p38-SAPK activation in XPA- as well as XPB- and XPD-deficient cells. From these findings, we conclude that the first steps of lesion recognition in the GG-NER reaction are already coupled to p38-SAPK signaling.
We envisage a model in which XPC directly interacts with intermediary proteins to cross talk with p38-SAPK. XPC is also essential for p38-SAPK phosphorylation in response to H2O2 as well as CDDP, suggesting an even broader relevance of this pathway to other DNA-damaging agents. Interestingly, a regional interaction between p38-SAPK and XPC at sites of DNA damage has previously been observed, in which p38-SAPK promotes recruitment of XPC. In the context of our findings, this could indicate a potential positive feedback loop often observed in signaling cascades.36 Notably, XPC and p38-SAPK suppress the formation of lung tumors in mice.37, 38 Hence, the interaction of XPC and p38-SAPK in lung cancer and carcinogenesis in general needs to be further studied.
The genotoxins BPDE and CDDP increase IL-6 mRNA levels and subsequent release of IL-6 protein dependent on XPC and p38-SAPK. In support of our hypothesis that DNA lesion recognition by XPC is the primary event that initiates signaling culminating in the release of IL-6, fast removal of cyclobutane pyrimidine dimers in human keratinocytes by exogenously added photolyase (thus overruling slower repair of this lesion by NER) abrogates induction of IL-6 by UVR.39 IL-6 is involved in tumor cell proliferation and survival, angiogenesis, metastasis and inflammation.40 A self-maintaining loop of IL-6 signaling has been proposed to promote cellular transformation.41 Upregulation of IL-6 by BPDE has also been described recently in human lung fibroblasts, and IL-6 co-treatment enhanced transformation of human bronchial epithelial cells by BPDE.42 In our studies, murine fibroblasts also increased the expression and release of IL-6 in response to BPDE and CDDP, whereas in several other human or rodent cancer cell lines this was not observed (data not shown). Thus, fibroblasts might be the major source of IL-6 after DNA damage. Importantly, fibroblasts are a major component of the tumor microenvironment, and are more and more recognized as central players in cancer development.30, 43 Consistently, a protumorigenic role of IL-6 could be demonstrated in several tumor models.44, 45 Monoclonal antibodies targeting IL-6 are already used in the clinics and small-molecule inhibitors to inhibit IL-6 signaling are currently being investigated in clinical trials.40 Therefore, IL-6 is an attractive target for cancer therapy. Moreover, the impact of CDDP-induced IL-6 release in the lung, as observed by us, on the therapeutic outcome remains to be investigated. Interestingly, enhanced serum levels of IL-6 were observed in patients treated with CDDP and oxaliplatin, and coincided with myelosuppression and drug-induced fever, respectively.46, 47 IL-6 might also protect against CDDP-induced neuropathies and kidney injury.48, 49
In contrast to recent studies that have demonstrated activation of p38-SAPK in mouse breast tumors in response to CDDP,50 we failed to detect increased p38-SAPK phosphorylation in total homogenates from CDDP-treated lungs (data not shown). However, we note that instillated CDDP will only deposit in parts of the lung at the air–liquid interface, and hence more refined methods, such as immunofluorescence studies, are needed to monitor p38-SAPK phosphorylation in vivo. Clearly, identification of the responding cell type(s) such as macrophages, fibroblasts, lung epithelial or endothelial cells will be instrumental to better understand the kinetics and cellular context of CDDP-induced p38-SAPK signaling in the lung.
In conclusion, we have uncovered a novel signaling cascade in which DNA damage recognition via XPC couples to p38-SAPK activation to promote release of IL-6. The impact of IL-6 on chemical carcinogenesis but also cancer treatment by CDDP warrants further investigations.
Materials and methods
Materials
Anti-AKT1/2 (H-136), anti-ATR (N-19), anti-ERK2 (D-2), MKK-3/6 (H90), SEK-1 (H98), MKK-7 (H160), anti-p38 (C-20) and anti-XPC (H-300) antibodies were purchased from Santa Cruz Biotechnology (Heidelberg, Germany). Antibodies for detection of ATM (D2E2) and for detection of phosphorylated kinases (AKT, Chk1, Chk2, ERK, H2A.X, MKK-3/6, SEK-1, MKK-7, p38) were purchased from Cell Signaling Technology (Frankfurt am Main, Germany). Anti-Chk2 (clone 7) was purchased from Millipore (Schwalbach, Germany). Benzo[a]pyrene was purchased from the Biochemical Institute for Environmental Carcinogens, Grosshansdorf, Germany with a purity of >99% as determined by gas chromatography–mass spectrometry. Synthesis of anti-dihydrodiol epoxides of benzo[a]pyrene51, 52 and benzo[c]phenanthrene53, 54 was performed according to literature methods. CDDP, actinomycin D, anisomycin, wortmannin and doxorubicin were purchased from Sigma (München, Germany); ATM/ATR - (KU55933 and ETP-46464) and DNA-PK II (NU7026) Kinase inhibitors from Merck Millipore (Darmstadt, Germany); PP1 (src-kinase inhibitor) from Biomol (Hamburg, Germany) and SB203580 from Enzo Life Sciences (Loerrach, Germany).
Cell lines and cell culture
NIH3T3 wild-type-, CSB- (Csbm/m)) and XPA- (Xpa−/−) deficient MEFs (both kindly provided by B Epe, University Mainz, Germany), p38 alpha wild-type and knockout MEFs (kindly provided by A Nebreda, IRB Barcelona, Spain and described in Porras et al.,55 SYF cells (MEFs deficient for src, yes and fyn were purchased from ATCC), c-abl wild-type and knockout MEFs (kindly provided by A Koleske, Yale University, USA), p53-knockout and DNA-PK- (severe combined immunodeficiency syndrome) deficient cells (kindly provided by C Blattner, KIT, ITG, Germany) and corresponding wild-type MEFs as well as murine Hepa-1 C7 and human HeLa cells were cultured in Dulbecco’s modified Eagle’s Medium supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 mg/ml streptomycin. Xpc−/− and mHR23A−/−/mHR23B−/− MEFs and corresponding wild-type cells have been described22 and were cultured in a 1:1 mixture of Dulbecco’s modified Eagle’s Medium and F-10 supplemented with 10% fetal calf serum, penicillin and streptomycin (each 100 U/ml). Patient derived fibroblasts deficient for XPE, XPB and XPD have been previously described.56, 57, 58, 59, 60 Annotations of the human NER-deficient cell lines are XP-B (1), XP-B (2), XP-D (1), XP-D (2), XP-E (1) and XP-E (2) and refer to XPCS1BA, XP131MA, XPCS2, 96RD362, XP215HE and XP2RO, respectively. ctrl (1) and ctrl (2) are the proficient lines C5RO and C7RO, respectively. In the text and figures, the nomenclature for NIH3T3 wild-type (wt) and corresponding knockout (−/−) cells is indicated for the relevant gene, for example, Xpc as NIH3T3-XPC wt or −/−. When nonimmortalized wt MEFs and the corresponding knockout cells are studied, the abbreviation MEF wt or −/− is used. Transient depletion of selected gene products in human HeLa cells (Figure 5b), murine Hepa C7 (Figure 3d) and NIH3T3 (Figures 6b and c) cells by siRNA transfections is specified as siRNA (seq.) followed by the respective siRNA sequence (control or sequence 1 or 2 targeting either the MKK3/6, SEK1, XPC or p38 alpha mRNA, for details see below). Human ATM-proficient (GM00637) and ATM-deficient fibroblasts (GM05849) were purchased from Coriell Cell Repositories (Camden, NJ, USA) and cultured in minimum essential medium supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 mg/ml streptomycin.
Western blotting, mRNA extraction and real-time PCR analysis
Whole-cell extracts were separated by SDS–polyacrylamide gel electrophoresis and analyzed as previously described.15 Cell extracts derived from different cell lines were always loaded onto the same gel, blotted onto the same membrane and exposed to the same film during subsequent ECL detection to guarantee for identical exposure times. In a few cases, lanes that were irrelevant for the experiment were cut from the image and this is indicated by a black line. Reverse transcription-PCR was performed as published.18 Supplementary Table S1 shows the sequences of the used primers.
Enzyme-linked immunosorbent assay
Secretion of IL-6 in NIH3T3 wt cells was determined using an anti-mouse IL-6 OptEIA enzyme-linked immunosorbent assay kit according to the manufacturer's instruction (BD Biosciences, Heidelberg, Germany).
Single-cell gel electrophoresis (comet) assay
Induction of DNA strand breaks was assessed by using the comet assay as published.61 After electrophoresis, the glass slides were either neutralized (alkaline, 0.4 M Tris-HCl, pH 7.5) or directly washed with aqua bidest and were then incubated with ethanol for 5 min, dried at room temperature and examined under a fluorescence microscope. Images were quantified by the comet analysis software VisComet (Impuls Bildanalyse, Gilching, Germany).
Determination of DNA adducts
Specific DNA adducts from BPDE were determined with the 32P-post-labelling assay.62, 63, 64
siRNA procedures
Transfection of HeLa, Hepa C7 and NIH3T3 cells was performed using HiPerfect (Qiagen, Hilden, Germany) according to the manufacturer's instructions. siRNAs were either purchased from MWG-Biotech (Ebersberg, Germany; XPC, p38) or Qiagen (MKK3, MKK6 and SEK1; Supplementary Table S2). In brief, for transient transfection of siRNAs, 1 × 105 cells/well (24-well plate) or 1 × 104 cells/well (96-well plate) were seeded and transfected with 20 pM (24-well plate) or 5 pM (96-well plate) siRNA according to the manufacturer's instructions. After 48–72 h, cell extracts were prepared and subjected to western blot analysis.
Animals, bronchoalveolar lavage and analysis
All experiments were performed according to European and German statutory regulations (license number AZ 35-9185.81/G-63/13). C57BL/6 Xpc wild-type and knockout mice were kindly provided by E Friedberg, University of Texas, Southwestern Medical Center, Dallas, USA. The genotype of the mice was determined by PCR analysis of genomic DNA from tissue biopsies as described previously.65 For the experiments, 8–10-week-old male mice were used. Bronchoalveolar lavage and the subsequent analysis was performed as previously described.66, 67 The bronchoalveolar lavage fluid from lavages 1 and 2 were pooled, centrifuged (450 × g, 10 min at 4 °C) and the cell-free supernatants were used for measurements of cytokine expression. IL-6 expression was determined by enzyme-linked immunosorbent assay as described above. Total protein content was determined using the bicinchoninic acid Protein Assay (Thermo Scientific, Dreieich, Germany) according to the manufacturer’s instructions.
References
Straif K, Baan R, Grosse Y, Secretan B, El GF, Cogliano V . Carcinogenicity of polycyclic aromatic hydrocarbons. Lancet Oncol 2005; 6: 931–932.
Pelkonen O, Nebert DW . Metabolism of polycyclic aromatic hydrocarbons: etiologic role in carcinogenesis. Pharmacol Rev 1982; 34: 189–222.
Thakker DR, Yagi H, Levin W, Wood AW, Conney AH, Jerina DM . aromatic hydrocarbons: metabolic activation to ultimate carcinogens. In: Anders MW (ed.), Bioactivation of Foreign Compounds. Academic Press, Inc.: New York: NY, 1985. 177–242.
Toledo F, Wahl GM . Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 2006; 6: 909–923.
Weston CR, Davis RJ . The JNK signal transduction pathway. Curr Opin Cell Biol 2007; 19: 142–149.
Han J, Sun P . The pathways to tumor suppression via route p38. Trends Biochem Sci 2007; 32: 364–371.
Weiss C, Schneider S, Wagner EF, Zhang X, Seto E, Bohmann D . JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. EMBO J 2003; 22: 3686–3695.
Herrlich P, Karin M, Weiss C . Supreme EnLIGHTenment: damage recognition and signaling in the mammalian UV response. Mol Cell 2008; 29: 279–290.
Shiloh Y . ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3: 155–168.
Kastan MB, Bartek J . Cell-cycle checkpoints and cancer. Nature 2004; 432: 316–323.
Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB . p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38 MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007; 11: 175–189.
Li J, Tang MS, Liu B, Shi X, Huang C . A critical role of PI-3K/Akt/JNKs pathway in benzo[a]pyrene diol-epoxide (B[a]PDE)-induced AP-1 transactivation in mouse epidermal Cl41 cells. Oncogene 2004; 23: 3932–3944.
Li J, Chen H, Tang MS, Shi X, Amin S, Desai D et al. PI-3K and Akt are mediators of AP-1 induction by 5-MCDE in mouse epidermal Cl41 cells. J Cell Biol 2004; 165: 77–86.
Chen S, Nguyen N, Tamura K, Karin M, Tukey RH . The role of the Ah receptor and p38 in benzo[a]pyrene-7,8-dihydrodiol and benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide-induced apoptosis. J Biol Chem 2003; 278: 19526–19533.
Donauer J, Schreck I, Liebel U, Weiss C . Role and interaction of p53, BAX and the stress-activated protein kinases p38 and JNK in benzo(a)pyrene-diolepoxide induced apoptosis in human colon carcinoma cells. Arch Toxicol 2012; 86: 329–337.
Buterin T, Hess MT, Luneva N, Geacintov NE, Amin S, Kroth H et al. Unrepaired fjord region polycyclic aromatic hydrocarbon-DNA adducts in ras codon 61 mutational hot spots. Cancer Res 2000; 60: 1849–1856.
Bakkenist CJ, Kastan MB . Initiating cellular stress responses. Cell 2004; 118: 9–17.
Schreck I, Chudziak D, Schneider S, Seidel A, Platt KL, Oesch F et al. Influence of aryl hydrocarbon- (Ah) receptor and genotoxins on DNA repair gene expression and cell survival of mouse hepatoma cells. Toxicology 2009; 259: 91–96.
Bozulic L, Surucu B, Hynx D, Hemmings BA . PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 2008; 30: 203–213.
Boehme KA, Kulikov R, Blattner C . p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc Natl Acad Sci USA 2008; 105: 7785–7790.
Maillard O, Camenisch U, Blagoev KB, Naegeli H . Versatile protection from mutagenic DNA lesions conferred by bipartite recognition in nucleotide excision repair. Mutat Res 2008; 658: 271–286.
Ng JM, Vermeulen W, van der Horst GT, Bergink S, Sugasawa K, Vrieling H et al. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev 2003; 17: 1630–1645.
Sayer JM, Chadha A, Agarwal SK, Yeh HJC, Yagi H, Jerina DM . Covalent nucleoside adducts of benzo[a]pyrene 7,8-diol 9,10-epoxides: structural reinvestigation and characterization of a novel adenosine adduct on the ribose moiety. J Org Chem 1991; 56: 20–29.
Dipple A, Pigott MA, Agarwal SK, Yagi H, Sayer JM, Jerina DM . Optically active benzo[c]phenanthrene diol epoxides bind extensively to adenine in DNA. Nature 1987; 327: 535–536.
Wagner EF, Nebreda AR . Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009; 9: 537–549.
Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O et al. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001; 411: 102–107.
Gaestel M, Kotlyarov A, Kracht M . Targeting innate immunity protein kinase signalling in inflammation. Nat Rev Drug Discov 2009; 8: 480–499.
Mantovani A, Allavena P, Sica A, Balkwill F . Cancer-related inflammation. Nature 2008; 454: 436–444.
Coussens LM, Zitvogel L, Palucka AK . Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 2013; 339: 286–291.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Raman M, Earnest S, Zhang K, Zhao Y, Cobb MH . TAO kinases mediate activation of p38 in response to DNA damage. EMBO J 2007; 26: 2005–2014.
Phillips DH, Hewer A, Martin CN, Garner RC, King MM . Correlation of DNA adduct levels in human lung with cigarette smoking. Nature 1988; 336: 790–792.
Lloyd DR, Hanawalt PC . p53-dependent global genomic repair of benzo[a]pyrene-7,8-diol-9,10-epoxide adducts in human cells. Cancer Res 2000; 60: 517–521.
Maillard O, Solyom S, Naegeli H . An aromatic sensor with aversion to damaged strands confers versatility to DNA repair. PLoS Biol 2007; 5: e79.
Lubin A, Zhang L, Chen H, White VM, Gong F . A human XPC protein interactome—a resource. Int J Mol Sci 2014; 15: 141–158.
Zhao Q, Barakat BM, Qin S, Ray A, El-Mahdy MA, Wani G et al. The p38 mitogen-activated protein kinase augments nucleotide excision repair by mediating DDB2 degradation and chromatin relaxation. J Biol Chem 2008; 283: 32553–32561.
Hollander MC, Philburn RT, Patterson AD, Velasco-Miguel S, Friedberg EC, Linnoila RI et al. Deletion of XPC leads to lung tumors in mice and is associated with early events in human lung carcinogenesis. Proc Natl Acad Sci USA 2005; 102: 13200–13205.
Ventura JJ, Tenbaum S, Perdiguero E, Huth M, Guerra C, Barbacid M et al. p38alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat Genet 2007; 39: 750–758.
Petit-Frere C, Clingen PH, Grewe M, Krutmann J, Roza L, Arlett CF et al. Induction of interleukin-6 production by ultraviolet radiation in normal human epidermal keratinocytes and in a human keratinocyte cell line is mediated by DNA damage. J Invest Dermatol 1998; 111: 354–359.
Ara T, DeClerck YA . Interleukin-6 in bone metastasis and cancer progression. Eur J Cancer 2010; 46: 1223–1231.
Iliopoulos D, Hirsch HA, Struhl K . An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009; 139: 693–706.
Chen W, Xu X, Bai L, Padilla MT, Gott KM, Leng S et al. Low-dose gamma-irradiation inhibits IL-6 secretion from human lung fibroblasts that promotes bronchial epithelial cell transformation by cigarette-smoke carcinogen. Carcinogenesis 2012; 33: 1368–1374.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D . Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 2010; 17: 135–147.
Schafer ZT, Brugge JS . IL-6 involvement in epithelial cancers. J Clin Invest 2007; 117: 3660–3663.
Naugler WE, Karin M . The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med 2008; 14: 109–119.
Chen YM, Whang-Peng J, Liu JM, Kuo BI, Wang SY, Tsai CM et al. Serum cytokine level fluctuations in chemotherapy-induced myelosuppression. Jpn J Clin Oncol 1996; 26: 18–23.
Ulrich-Pur H, Penz M, Fiebiger WC, Schüll B, Kornek GV, Scheithauer W et al. Oxaliplatin-induced fever and release of IL-6. Oncology 2000; 59: 187–189.
Callizot N, Andriambeloson E, Glass J, Revel M, Ferro P, Cirillo R et al. Interleukin-6 protects against paclitaxel, cisplatin and vincristine-induced neuropathies without impairing chemotherapeutic activity. Cancer Chemother Pharmacol 2008; 62: 995–1007.
Mitazaki S, Kato N, Suto M, Hiraiwa K, Abe S . Interleukin-6 deficiency accelerates cisplatin-induced acute renal failure but not systemic injury. Toxicology 2009; 265: 115–121.
Pereira L, Igea A, Canovas B, Dolado I, Nebreda AR . Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol Med 2013; 5: 1759–1774.
Yagi H, Thakker DR, Hernandez O, Koreeda M, Jerina DM . Synthesis and reactions of the highly mutagenic 7,8-diol 9,10-epoxides of the carcinogen benzo[a]pyrene. J Am Chem Soc 1977; 99: 1604–1611.
McCaustland DJ, Engel JF . Metabolites of polycyclic aromatic hydrocarbons. II. Synthesis of 7,8-dihydrobenzo[a]pyrene-7,8-diol and 7,8-dihydrobenzo[k]pyrene-7,8-epoxide. Tetrahedron Lett 1975; 30: 2549–2552.
Sayer JM, Yagi H, Croisy-Delcey M, Jerina DM . Novel bay-region diol epoxides from benzo[c]phenanthrene. J Am Chem Soc 1981; 103: 4970–4872.
Kumar S . A new and concise synthesis of 3-hydroxybenzo[c]phenanthrene and 12-hydroxybenzo[g]chrysene, useful intermediates for the synthesis of fjord-Region diol epoxides of benzo[c]phenanthrene and benzo[g]chrysene. J Org Chem 1997; 62: 8535–8539.
Porras A, Zuluaga S, Black E, Valladares A, Alvarez AM, Ambrosino C et al. P38 alpha mitogen-activated protein kinase sensitizes cells to apoptosis induced by different stimuli. Mol Biol Cell 2004; 15: 922–933.
Vermeulen W, Scott RJ, Rodgers S, Müller HJ, Cole J, Arlett CF et al. Clinical heterogeneity within xeroderma pigmentosum associated with mutations in the DNA repair and transcription gene ERCC3. Am J Hum Genet 1994; 54: 191–200.
Graham JM Jr., Anyane-Yeboa K, Raams A, Appeldoorn E, Kleijer WJ, Garritsen VH et al. Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet 2001; 69: 291–300.
Keeney S, Eker AP, Brody T, Vermeulen W, Bootsma D, Hoeijmakers JH et al. Correction of the DNA repair defect in xeroderma pigmentosum group E by injection of a DNA damage-binding protein. Proc Natl Acad Sci USA 1994; 91: 4053–4056.
Oh KS, Khan SG, Jaspers NG, Raams A, Ueda T, Lehmann A et al. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum Mutat 2006; 27: 1092–1103.
Broughton BC, Thompson AF, Harcourt SA, Vermeulen W, Hoeijmakers JH, Botta E et al. Molecular and cellular analysis of the DNA repair defect in a patient in xeroderma pigmentosum complementation group D who has the clinical features of xeroderma pigmentosum and Cockayne syndrome. Am J Hum Genet 1995; 56: 167–174.
Gehrke H, Frühmesser A, Pelka J, Esselen M, Hecht LL, Blank H et al. In vitro toxicity of amorphous silica nanoparticles in human colon carcinoma cells. Nanotoxicology 2013; 7: 274–293.
Randerath K, Randerath E, Danna TF, van GL, Putman KL . A new sensitive 32P-postlabeling assay based on the specific enzymatic conversion of bulky DNA lesions to radiolabeled dinucleotides and nucleoside 5'-monophosphates. Carcinogenesis 1989; 10: 1231–1239.
Lagerqvist A, Håkansson D, Prochazka G, Lundin C, Dreij K, Segerbäck D et al. Both replication bypass fidelity and repair efficiency influence the yield of mutations per target dose in intact mammalian cells induced by benzo[a]pyrene-diol-epoxide and dibenzo[a,l]pyrene-diol-epoxide. DNA Repair (Amst) 2008; 7: 1202–1212.
Phillips DH, Castegnaro M . Standardization and validation of DNA adduct postlabelling methods: report of interlaboratory trials and production of recommended protocols. Mutagenesis 1999; 14: 301–315.
Cheo DL, Ruven HJ, Meira LB, Hammer RE, Burns DK, Tappe NJ et al. Characterization of defective nucleotide excision repair in XPC mutant mice. Mutat Res 1997; 374: 1–9.
Brown RH, Walters DM, Greenberg RS, Mitzner W . A method of endotracheal intubation and pulmonary functional assessment for repeated studies in mice. J Appl Physiol 1999; 87: 2362–2365.
Stoeger T, Reinhard C, Ritter B, Karg E, Schröppel A, Heyder J . Comparison of the inflammatory effects of five different particles in BALB/cJ miceIn: effects of air contaminants on the respiratory tract. In Heinrich U (ed.), Interpretations From Molecules To Meta Analysis. Fraunhofer IRB Verlag: Stuttgart, Germany, 2004. 75–84.
Acknowledgements
This work was supported by ECNIS (Environmental Cancer Risk, Nutrition and Individual Susceptibility), a network of excellence operating within the European Union 6th Framework Program, Priority 5: ‘Food Quality and Safety’ (Contract No 513943). We thank Dorit Mattern for technical assistance; Jonathan Sleeman for critical reading and editing, Selma Huber, Simone Maier, Tobias Stöger and Bärbel Ritter for supervision and performance of the animal experiments. Heinz Frank is greatly acknowledged for his contribution to the synthesis of PAHs and sadly passed away.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
About this article

Cite this article
Schreck, I., Grico, N., Hansjosten, I. et al. The nucleotide excision repair protein XPC is essential for bulky DNA adducts to promote interleukin-6 expression via the activation of p38-SAPK. Oncogene 35, 908–918 (2016). https://doi.org/10.1038/onc.2015.145
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.145
- Springer Nature Limited
This article is cited by
-
DNA damage mediated by UV radiation and relative repair mechanisms in mammals
Genome Instability & Disease (2022)
-
XPC is an RNA polymerase II cofactor recruiting ATAC to promoters by interacting with E2F1
Nature Communications (2018)
-
p38-MK2 signaling axis regulates RNA metabolism after UV-light-induced DNA damage
Nature Communications (2018)
-
ATM inhibition prevents interleukin-6 from contributing to the proliferation of glioblastoma cells after ionizing radiation
Journal of Neuro-Oncology (2018)
-
Dual roles of extracellular signal-regulated kinase (ERK) in quinoline compound BPIQ-induced apoptosis and anti-migration of human non-small cell lung cancer cells
Cancer Cell International (2017)