
Abstract
Both cell polarity and c-Jun N-terminal kinase (JNK) activity are essential to the maintenance of tissue homeostasis, and disruption of either is commonly seen in cancer progression. Despite the established connection between loss-of-cell polarity and JNK activation, much less is known about the molecular mechanism by which aberrant cell polarity induces JNK-mediated cell migration and tumor invasion. Here we show results from a genetic screen using an in vivo invasion model via knocking down cell polarity gene in Drosophila wing discs, and identify Rho1–Wnd signaling as an important molecular link that mediates loss-of-cell polarity-triggered JNK activation and cell invasion. We show that Wallenda (Wnd), a protein kinase of the mitogen-activated protein kinase kinase kinase family, by forming a complex with the GTPase Rho1, is both necessary and sufficient for Rho1-induced JNK-dependent cell invasion, MMP1 activation and epithelial–mesenchymal transition. Furthermore, Wnd promotes cell proliferation and tissue growth through wingless production when apoptosis is inhibited by p35. Finally, Wnd shows oncogenic cooperation with RasV12 to trigger tumor growth in eye discs and causes invasion into the ventral nerve cord. Together, our data not only provides a novel mechanistic insight on how cell polarity loss contributes to cell invasion, but also highlights the value of the Drosophila model system to explore human cancer biology.
Similar content being viewed by others


Introduction
Correct cell polarity maintenance is essential for tissue homeostasis and cell physiology, whereas dysregulation of cell polarity has been characterized as an important step and a hallmark of cancer progression.1 Consequently, an overwhelming amount of studies have focused on the connections between polarity regulation and cancer development.2, 3, 4, 5 In particular, studies on Drosophila have defined numerous signaling modulators acting to establish and maintain epithelial polarity, including the basolaterally localized Scribble complex, which is composed of Scribble (Scrib), discs large (Dlg) and lethal giant larva (Lgl).6 In fly, loss of any of these proteins can cooperate with oncogenic Ras to induce invasive tumors;7 whereas in mammal, homologs of these proteins show consistently aberrant expression in multiple cancers, including lung, breast, colon and prostate.8, 9, 10 Despite the well-established connection between dysregulation of cell polarity and cancer, the factors that act downstream of the Scrib complex to mediate cancer progression remain poorly understood.
During the past decade, Drosophila melanogaster has become an excellent model to investigate the genetic mechanism of cancer biology,11, 12 and several invasion and metastasis models have been established in larvae and adult flies.12, 13 For instance, in the developing wing epithelia, loss-of-cell polarity genes or C-terminal SRC kinase (Csk) along the anterior/posterior compartment boundary using patched-Gal4 driver produces an invasive migration phenotype,14, 15, 16, 17 which has been used to study cell invasion in vivo.11
The structure and function of Scribble complex members have been highly conserved from fly to human,18, 19 which enables us to study the downstream signaling and underlying mechanism in simpler organisms like fly. To identify additional factors downstream of the Scribble complex involved in cell invasion and cancer progression, we performed a genetic screen using the previously described loss of scrib-induced cell invasion model in Drosophila wing discs.14, 15 We found that Rho1, a small GTPase, and Wallenda (Wnd), a Ser/Thr protein kinase of the mitogen-activated protein kinase kinase kinase (MAPKKK) family, are crucial regulators of loss-of-cell polarity or ectopic Src42A-induced cell invasion. Our genetic analysis indicates that Rho1 acts upstream of Wnd to regulate c-Jun N-terminal kinase (JNK)-dependent aspects of cell invasion, including loss of adherence junction, activation of metastasis markers and epithelial–mesenchymal transition (EMT). Furthermore, our biochemical studies confirm a physical interaction between Rho1 and Wnd. Finally, we show that Wnd cooperates with oncogenic Ras to stimulate wingless (Wg) expression, cell proliferation and tumor invasion.
Results
Rho1 and Wnd are required for cell invasion
As previously reported, RNA interference (RNAi)-mediated downregulation of the cell polarity gene scrib, driven by ptc-Gal4, triggered a strong cell invasion phenotype toward the posterior part of wing discs (Figures 1a and b), along with upregulation of matrix metalloprotease1 (MMP1; Supplementary Figure 1A),14, 15 a protein that is essential for basement membrane degradation and a hallmark of EMT.20, 21 This invasion phenotype and MMP1 upregulation depends on JNK signaling,14, 15, 22 which is activated by multiple upstream factors including mitochondrial respiratory complexes, small GTPases, MAPKKK and MAPKK family members, and so on,23, 24, 25, 26, 27, 28 yet it has remained unknown which factors mediate JNK activation induced by the loss-of-cell polarity. To investigate the molecular mechanism underlying loss-of-cell polarity-triggered cell invasion, we have conducted a candidate screen using the ptc>scrib-IR invasion phenotype. We have screened >500 UAS-RNAi lines from the Bloomington and National Institute of Genetics (NIG) stock centers targeting potential factors upstream of JNK, or factors that interact with JNK pathway components genetically or biochemically as reported in the literature. The screen is still ongoing, as more RNAi lines are being added to the stock centers. We called specific attention to two genes identified in the screen: Rho1 and wnd. Rho1 belongs to the GTPase family and is a major regulator of actin cytoskeleton dynamics and cell migration.29 Wnd encodes a MAPKKK homolog of vertebrate dual leucine zipper kinase (DLK), a protein implicated in neuronal cell radical migration and apoptosis.30, 31 We found that knocking down either Rho1 or wnd dramatically suppressed ptc>scrib-IR-induced cell invasion and MMP1 activation (Figures 1b–d and l, Supplementary Figures 1A–C) as well as lgl loss-induced cell invasion (Figures 1e–g and l) in wing discs, whereas loss of Rho1 or wnd alone gave no obvious phenotype (Supplementary Figures 1D and E). The knockdown efficiencies of RNAi lines were verified by quantitative reverse transcription polymerase chain reaction (Supplementary Figures 2E and F). To exclude potential off-target effects of RNAi, we examined at least two independent UAS-RNAi lines for each gene, including the previously described lines for Rho1 and wnd,32, 33 and obtained similar results (data not shown). Furthermore, we confirmed the suppression by wnd and Rho1 mutant alleles (Supplementary Figures 1G–K).

Rho1 and Wnd are required for impaired cell polarity or Src42A-induced cell invasion. Fluorescence micrographs of wing discs are shown. Compared with the control (a, a′), loss of scrib (b) or lgl (e) induced cell invasion is suppressed by knocking down Rho1 (c, f) or wnd (d, g). Compared with the control (h–h”), ectopic Src42A-induced cell invasion and MMP1 upregulation (i–i”) are dramatically impeded by knocking down Rho1 (j–j”) or wnd (k–k”). (l) and (m) Quantification data of cell migration in panels a–g and h–k. ***P<0.001. (mean+s.d., n=10). UAS-LacZ was included (b, e and i) as a negative control to demonstrate that the suppression by UAS-RNAi is specific, not a result of Gal4 titration by another UAS line.
The oncoprotein Src is known to regulate various aspects of tumor progression ranging from proliferation and survival to migration and metastasis.17, 25, 34 Recent work has shown that expression of Drosophila Src42A recapitulates loss-of-cell polarity-induced JNK-mediated cell invasion.15, 17, 25 Consistently, we found Src42A-induced cell invasion and MMP1 activation (Figures 1i, i',i” and m) were significantly suppressed by knocking down Rho1 (Figures 1j, j',j” and m) or wnd (Figures 1k, k',K” and m). Together, these data indicate that both Rho1 and Wnd are crucial regulators for cell invasion in Drosophila wing discs.
Wnd induces JNK-mediated EMT
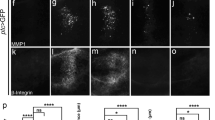
To further explore the role of Wnd in cell invasion, we drove Wnd expression by ptc-Gal4 in the wing disc with tub-Gal80ts, a temperature-sensitive form of the Gal4 inhibitor Gal80 (Gal80ts) that blocks Gal4 activity at low temperature (18 °C) but is inactivated at high temperature (29 °C).35 After Wnd induction, we observed that a large number of cells migrated significantly apart from the ptc domain toward the posterior part, coupled with intensive MMP1 activation (Figure 2e), whereas co-expression of a wnd RNAi completely suppressed the invasion behavior (Supplementary Figures 2A and B). Furthermore, Wnd-expressing cells display reduced cell adherent junction marker E-cadherin (Figure 2f) and increased accumulation of F-actin and β-integrin (Figures 2g and h). Taken together, these results suggest that Wnd-expressing epithelia cells exhibit molecular characteristics of EMT, including MMP1 induction, loss of E-cadherin and actin remodeling.36 In contrast, expression of a kinase dead form of Wnd (WndKD) shows no obvious EMT effects (Figures 2i–l), indicating that Wnd requires its kinase activity to induce cell invasion.

Wnd promotes cell invasion and EMT. Fluorescence micrographs of wing discs are shown. Compared with the controls (a–d), cells expressing Wnd along the A–P boundary show invasive migration and MMP1 secretion (e), reduced E-cadherin (f), increased actin (g) and β-integrin (h) accumulation, whereas cells expressing WndKD fail to produce such phenotypes (i–l).
MMP1 serves as a transcriptional reporter for JNK signaling in tumor cell invasion.20, 21 Consistent with the MMP1 data, expression of Wnd (Figures 3a–d), but not WndKD (data not shown), promotes JNK phosphorylation and puc-LacZ expression, another reporter for JNK signaling. Conversely, Wnd-induced EMT behavior (Figures 3f, i, j and k) is fully suppressed by co-expression of BskDN (Figures 3g, i, l and m), a dominant negative form of Drosophila JNK homolog Bsk, suggesting that JNK signaling is indispensable for Wnd-triggered cell invasion.

JNK signaling is required for Wnd-induced cell invasion and EMT. (a and b) Light micrographs of wing discs are shown. Compared with the control (a), Wnd expression upregulates puc transcription (b). (c–h and j–p) Fluorescence micrographs of wing discs are shown. (c–e) Compared with the control (c), ectopic Wnd-induced JNK phosphorylation (d) is dramatically suppressed by depletion of MKK4 and hep (e). (f–h) Wnd-induced cell invasion and MMP1 activation (f) are significantly suppressed by co-expression of BskDN (g) or knocking down MKK4 and hep (h). (i) Quantification data of cell invasion in panels f–h (mean+s.d., n=10). (j–m) Wnd-induced integrin accumulation (j) and E-cad reduction (k) are suppressed by co-expression of BskDN (l and m). (n) Co-expression of p35 cannot block Wnd-induced cell invasion and MMP1 activation. (o and p) Representative Flp-out clones expressing Wnd and p35 have ‘filopodia’ like structure and display intensive MMP1 activation (o) and F-actin accumulation (p).
To determine how JNK signaling is regulated by Wnd, we analyzed the upstream components of the JNK pathway. Drosophila encodes two JNK kinases, the MKK7 homolog Hemipterous (Hep) and MKK4. Hep is involved in dorsal closure, cell death and tumor cell invasion,37, 38, 39 and MKK4 is a regulator of Eiger-induced cell death.40 Although knocking down either hep or mkk4 alone has no significant suppression on Wnd-induced cell invasion (data not shown), simultaneously knocking down both genes dramatically impedes Wnd-triggered JNK phosphorylation, MMP1 activation and cell invasion (Figures 3e, h and i), suggesting a functional redundancy between hep and mkk4 in mediating Wnd activity. Collectively, these data suggest that Wnd induces JNK-mediated EMT in wing epithelial through both Hep and MKK4.
Cell invasion is frequently accompanied with apoptosis.17 In accordance with this notion, we found that Wnd expression also induced strong cell death, especially in invading cells (Supplementary Figures 3A–D). Previous work showed that caspase activation below cell death threshold also induces JNK-dependent cell invasion,41 indicating that cell invasion is uncoupled from cell death. Consistent with this conclusion, we found that blocking cell death by expressing the caspase inhibitor p35 or deleting one copy of the proapoptotic RGH (reaper, hid and grim) genes did not impede Wnd-induced cell invasion and MMP1 expression (Figure 3n and Supplementary Figures 3E and F), confirming that the invasion behavior is not a secondary effect of cell death. To rule out the possibility that Wnd may have a non-autonomous effect on MMP1 activation, we generated Flp-out clones that co-express Wnd with p35 to block cell death, and observed a clear cell autonomous MMP1 induction (Supplementary Figure 3I). Furthermore, some cells within the clone displayed filopodia like structures and exhibited molecular characteristics of EMT,36 including intensive MMP1 activation and F-actin accumulation in a cell autonomous manner (Figures 3o and p). Some weak MMP1 staining was also observed outside the clones (Figure 3o”), likely representing their secretion to the extra cellular matrix.
Wnd is required for Rho1-induced cell invasion and JNK activation
Rho1 has essential roles in regulating cell invasion and tumorigenesis.17, 42, 43 In accordance with this, we found that ptc-Gal4-driven Rho1 expression also induced JNK activation, MMP1 upregulation, cell death and invasion in an autonomous manner (Figures 4b–b”, Supplementary Figures 4F, I and L),27, 42 whereas depletion of Rho1 completely blocked the invasion phenotype (Supplementary Figures 2C and D). Co-expression of the caspase inhibitor p35 kept Rho1-expressing cells alive (the so called ‘undead cells’44) and generated a broader ptc domain with enhanced MMP1 production, whereas the ‘undead cells’ could still invade into the posterior part (Supplementary Figures 4G–G”), suggesting that cell death is uncoupled from invasion. Consistent with the notion that invasive behavior is associated with disruption of epithelial integrity,36 Rho1-expressing cells exhibited increased integrin and actin accumulation (Figure 4g and Supplementary Figure 4B). We found that Rho1-triggered EMT markers, including cell invasion, integrin and actin remodeling, and MMP1 activation, were significantly suppressed by knocking down wnd (Figures 4c, e and h and Supplementary Figures 4C and E) or inactivation of JNK (Figures 4d, e and i). It is worth noting that Rho1-induced actin accumulation remained unaffected by co-expression of BskDN (Supplementary Figures 4D and E, see Discussion). Consistently, loss of wnd also suppresses Rho1-induced JNK activation and cell death, as indicated by puc-LacZ and acridine orange staining respectively (Supplementary Figures 4H–J and K–M). Taken together, we conclude that Rho1 induces JNK-dependent cell death and invasion through Wnd. Intriguingly, Rudrapatna and colleagues recently reported a positive feedback loop between JNK and Rho1 in cell invasion.25 In accordance with this finding, we found that Wnd-induced cell invasion was also blocked by depletion of Rho1 (Supplementary Figures 3G and H).

Wnd is required for Rho1-induced cell invasion. Fluorescence micrographs of wing discs are shown. (a–d) Compared with the control (a), ectopic Rho1-induced cell invasion and MMP1 activation (b) are dramatically suppressed by knocking down wnd (c) or expressing BskDN (d). (e) Quantification data of cell invasion in panels a–d (mean+s.d., n=10). (f–i) Compared with the control (f), Rho1-induced integrin accumulation (g) is strongly suppressed by reducing wnd (h) or Bsk (i) activity.
Wnd physically interacts with Rho1
As MAPKKK proteins are typically activated by GTPases through direct binding,45 we reasoned that Rho1 may regulate Wnd activity via physical interaction. To test this hypothesis, we expressed enhanced green fluorescent protein (EGFP)-tagged Wnd (EGFP–Wnd) or HA-tagged Rho1 (HA–Rho1) in Drosophila S2 cells. Both Wnd and Rho1 were distributed uniformly in the cytoplasm (Figures 5a and b), and a significant co-localization was observed when the two proteins were co-expressed (Figure 5c). Consistently, co-immunoprecipitation experiments indicated that Wnd co-precipitated with Rho1 (Figure 5d), confirming a physical interaction between Rho1 and Wnd in vivo.

Rho1 physically interacts with Wnd. (a–c) Fluorescence micrographs of S2 cells are shown. Wnd (a) or Rho1 (b) is evenly distributed in the cytoplasm, and show cytoplasmic co-localization when co-expressed (c). (d) Rho1 interacts with Wnd in S2 cells. HA-Rho1 is co-precipitated with EGFP-Wnd in immunoprecipitation assay.
Wnd is dispensable for Rac1-triggered cell invasion
Apart from Rho1, Rac1 and Rac2 are known Rho-GTPase family members that regulate cell migration processes including dorsal closure and wound healing.29, 46, 47 Indeed, ectopic expression of Rac1, but not Rac2, driven by ptc-Gal4 induces JNK-dependent cell invasion behavior (Supplementary Figures 5A, F, K and data not shown). However, Rac1-triggered invasion cannot be suppressed by loss of wnd (Supplementary Figure 5B), indicating that Wnd is dispensable for Rac1-induced cell invasion. Consistent with this, we found that Wnd does not physically interact with Rac1 in immunoprecipitation experiments (Supplementary Figure 5L).
Wnd belongs to the MAPKKK family that also includes Slipper (Slpr) and dTAK1, both of which have been implicated in Rho1-induced cell death.27 Whereas knocking down slpr or dTAK1 alone had no visible effect on Rac1-induced cell invasion (Supplementary Figures 5C, D and K), depletion of both slpr and dTAK1 significantly suppressed Rac1-induced invasion (Supplementary Figures 5E and K). Similarly, knocking down both slpr and dTAK1 dramatically impeded Rho1-induced cell invasion (Supplementary Figures 5G–K), suggesting that dTAK1 and Slpr act redundantly downstream of Rac1 and Rho1 in regulating cell invasion.
Wnd cooperates with p35 or RasV12 to promotes cell proliferation and tissue growth
JNK signaling cooperates with oncogenic Ras to promote growth and invasion.14, 20, 39, 48, 49 Consistent with its ability to activate JNK signaling, Wnd could cooperate with p35 or oncogenic Ras to stimulate cell proliferation. Co-expression of Wnd and either p35 or oncogenic Ras resulted in increased phospho-histone 3 (PH3) staining (Figure 6), increased CycE expression (Figures 7i–l), and increased tissue growth (Figure 6) as compared with expression of either p35 or oncogenic Ras alone. Moreover, co-expression of Wnd with RasV12 in eye disc clones resulted in massive tumor growth in larva heads (Supplementary Figures 6A–C), with about 20% of the larvae showing a phenotype of tumor invasion into the ventral nerve cord, which were never seen in larvae expressing RasV12 or Wnd alone (Supplementary Figures 6A'–C').

Wnd cooperates with p35 and RasV12 to induce proliferation and growth. Fluorescence micrographs of wing discs are shown. Compared with the control (a), or expression of Wnd (b), p35 (c) or RasV12 (e) alone, co-expression of Wnd with p35 (d) or RasV12 (f) results in a significant increase of PH3+ cells and GFP+ region. (g) Ratio of GFP+ region versus the whole disc in a–f were shown. **P<0.01, *P<0.05 (mean+s.d., n⩾5).

Wnd induces JNK-dependent Wg expression. Fluorescence micrographs of wing discs are shown. Figures a–d are higher magnification of the wing pouch region. (a–d) Compared with the control (a), Wnd-induced Wg expression (b) is suppressed by BskDN (c), which does not affect the expression of endogenous Wg (d). (e–h) Ectopic expression of p35 (e) does not affect Wg expression, expression of or RasV12 slightly induces non-autonomously Wg serection (g), whereas co-expression of Wnd with p35 (f) or RasV12 (h) induces intensive Wg expression. (i–l) Compared with control (i), expression of Wnd or RasV12 fails to induce CycE expression (j and k), whereas co-expression of both synergistically upregulates CycE expression (l).
Wg has been reported to act downstream of JNK signaling to stimulate cell proliferation, and Wg expression is upregulated by JNK signaling.44 Consistently, ectopic Wnd is sufficient to induce Wg expression in a JNK-dependent manner (Figures 7a–d). In addition, oncogenic cooperation between Wnd and p35 or RasV12 is coupled with dramatic increase of Wg induction (Figures 7e–h). Induction of Wg expression has an important role during compensatory proliferation,50 and consistent with this view, we found that co-expression of Wg and RasV12 is sufficient to induce an overgrowth phenotype (Supplementary Figures 4N–P). Taken together, these data suggest that Wnd is able to promote cell proliferation and tissue growth by upregulating the expression of CycE and Wg, and collaborates with oncogenic Ras to induce tumor growth and invasion.
Discussion
Maintenance of cell polarity is critical for tissue homeostasis and normal morphogenesis, whereas loss-of-cell polarity is associated with tumor invasion in both fly and mammal.4, 18 The JNK pathway is evolutionarily conserved and has a crucial role in regulating many aspects of tumor progression.39, 51 Mounting evidence suggests that JNK signaling is required for loss-of-cell polarity-triggered tumor invasion,14, 17, 20, 22, 39 yet the molecular links between cell polarity disruption and JNK activation remain largely elusive. By performing a genetic screen using loss of scrib-induced cell invasion phenotype in Drosophila wing epithelia, we have identified the missing link between loss-of-cell polarity and cell invasion, and have shown that Rho1–Wnd signaling connects cell polarity genes and the MKK-JNK cascade (Supplementary Figure 7). We provide in vivo evidence that both Rho1 and Wnd are crucial for cell invasion in Drosophila wing epithelia, and that by forming a complex with Wnd, Rho1 promotes JNK-mediated cell invasion through both Hep and MKK4. This study, though carried out exclusively in Drosophila, also advances our limited understanding toward the upstream regulators of mammalian DLK.52
Interestingly, a recent study suggested a Rho1–ARP (actin remodeling protein)–JNK-positive feedback loop in regulating wing epithelial cell migration.25 In contrast, our data demonstrate that although both cell invasion and MMP1 induction caused by loss-of-cell polarity or Rho1 activation can be dramatically suppressed by blocking Wnd–Bsk signaling, the actin remodeling process could only be impeded by depletion of wnd, but remains unaffected by inactivation of Bsk (Supplementary Figures 4A–E), suggesting that this Wnd function is mediated by downstream factors other than the JNK pathway. Consistent with this notion, Khoo et al.28 have shown that Rho1-Myosin II signaling regulates actin accumulation independent of JNK. As recent studies have linked cytoskeleton regulation with the Hippo pathway,53, 54 and our unpublished data show that Wnd is able to inactivate Hippo signaling, we cannot exclude the possibility that Rho1–Wnd-induced cell invasion also depends on Hippo pathway activity. However, it is worth noting that unlike JNK activation, expression of Hippo pathway transcription co-activator Yorkie (Yki) along the anterior/posterior boundary of wing disc cannot induce a cell invasion phenotype, despite massive MMP1 induction.55 Given that the regulatory mechanism between JNK and Hippo in diverse physiological functions are context dependent,56 it would be interesting to investigate the potential roles of Hippo–Yki signaling in cell invasion and tumor metastasis.
Another promising downstream candidate that modulates the actin remodeling activity of Wnd is p38, which is known to have essential roles in regulating actin polymerization and microtubule dynamics.57, 58 In support of this hypothesis, studies in C aenorhabditis elegans suggest that DLK-1 (homolog of Wnd) requires p38 activity to regulate axon regeneration.59 Further investigation will be of great interest to clarify the role of p38 in Rho1–Wnd signaling-induced actin remodeling and cell invasion.
In this study, we provide the first in vivo evidence that Wnd is both necessary and sufficient for controlling cell invasion, epithelial organization and tumorigenesis. Our genetic data suggest that Wnd is a key regulator of Rho1 and Src42A-induced cell invasion, and that Wnd functions to induce cell invasion and EMT in a JNK-dependent manner. These data raise the possibility that DLK, the mammalian homolog of Wnd, may exhibit a conserved role in regulating cell migration and invasion via a mechanism similar to what we reported in Drosophila. Indeed, although most studies on DLK are focused on neurodegeneration and regeneration,52 some work indicates that DLK may regulate radial migration of neuronal cells during mouse corticogenesis.30 Further investigation is required to clarify the potential role of DLK in tumor cell invasion.
In conclusion, our study in Drosophila has uncovered a novel physiological function for Wnd in modulating cell invasion and EMT, and has identified a molecular link between impaired cell polarity and JNK-mediated cell invasion, which further emphasizes the value of the Drosophila model system to gain insight into human cancer biology. Given that the pathway and components identified in Drosophila are highly conserved, similar molecules and mechanisms could be involved in human cancer progression.
Materials and methods
Drosophila stocks and genetics
All stocks were raised on standard Drosophila media and crosses were performed at 25 °C unless otherwise indicated. For experiments involving tub-Gal80ts, flies were raised at 18 °C to restrict Gal4 activity for 5–6 days, then shifted to 29 °C for 2–3 days to inactivate Gal80ts. The following stocks were used: GMR-Gal4, ptc-Gal4, UAS-p35, UAS-GFP, UAS-Rac1 (#6680), UAS-Rho1 (#7334), UAS-LacZ (#3956), UAS-Rho1-IR (#27727), UAS-wnd-IR (#27525), UAS-lgl-IR (#31089), tub-Gal80ts (Bloomington Stock Center, Bloomington, IN, USA), UAS-Wnd, UAS-WndKD (gifts from Aaron DiAntonio60), pucE69, UAS-dTAK1-IR, UAS-BskDN, UAS-hep-IR, UAS-Puc,61 UAS-wnd-IR,33 UAS-Rho1-IR,32 UAS-Src42A,15 UAS-MKK4-IR,62 UAS-slpr-IR27 and UAS-scrib-IR22 were previously described.
Immunostaining
Third instar larvae wing discs, eye discs and S2 cells were fixed in 4% paraformaldehyde and stained as described previously,63 using mouse anti-MMP1 (1:200), rat anti-DE-cadherin (1:100), mouse anti-β-Gal (1:1000), mouse anti-β-integrin (1:100), mouse anti-Wg (1:300, DSHB; Developmental Studies Hybridoma Bank, Iowa City, IA, USA), rabbit anti-phospho-JNK (1:200, Calbiochem, San Diego, CA, USA), rabbit anti-PH3 (1:100), rabbit anti-active Caspase 3 (1: 400) and Alexa Fluor555 (1:100; Cell Signaling Technology, Danvers, MA, USA), Rat anti-CycE (gift from HE Richardson, 1:200). Secondary antibodies were anti-rabbit-Alexa (1:1000, CST), anti-Rat-Cy3 (1:1000, Jackson Immuno Research, West Grove, PA, USA) and anti-mouse-Cy3 (1:1000, Jackson Immunochemicals, West Grove, PA, USA).
X-gal staining
Eye and wing discs were dissected from third instar larvae in PBST (1x phosphate-buffered saline pH 7.0, 0.1% Triton-X 100) and stained for β-galactosidase activity.
AO staining
Eye and wing discs were dissected from third instar larvae in PBST and incubated in 1 × 10−5 M acridine orange for 5 min at room temperature before imaging.
Plasmids, cell culture and transfection
wnd, Rho1 and Rac1 cDNA were subcloned into pUAST to generate UAS-EGFP-Wnd, UAS-HA-Rho1 and UAS-Flag-Rac1 constructs. S2 cells were cultured in Insectagro DS2 (Corning, Manassas, VA, USA) supplemented with 10% fetal bovine serum (Thermo Scientific, Waltham, MA, USA), 50 U/ml of penicillin and 50 μg/ml of streptomycin. Transfection was performed using Effectene Transfection Reagent (QIAGEN, Valencia, CA, USA) according to manufacturer’s instructions. An actin-Gal4 construct was co-transfected with the pUAST expression vector for all the transfection experiments.
Immunoprecipitation and western blot analysis
S2 cells were transfected with indicated plasmids for 36 h, and immunoprecipitation and Western analyses were performed as previously described.63 In brief, pre-cleared cell lysates were incubated with the indicated antibodies followed by precipitation with protein G sepharose beads (Sigma, St Louis, MO, USA). Immune complexes were washed with lysis buffer, eluted in 2 × SDS sample buffer, and then subjected to Western blot using corresponding antibodies. Antibodies used in this study were as follows: rabbit anti-Wnd antibody (a kind gift from Aaron DiAntonio, St Louis, MO, USA), mouse anti-EGFP, rabbit anti-HA and rabbit anti-Flag (CMCTAG, San Diego, CA, USA), goat anti rabbit/mouse IgG-HRP (CST).
Statistical analysis
Quantification of the data was presented in bar graphs created with Graphpad Prism 5 (San Diego, CA, USA). Data represents mean values+s.d. We used one-way analysis of variance with corrected Bonferroni multiple comparison test to calculate statistical significance in Figures 1 and 6.
References
Muthuswamy SK, Xue B . Cell polarity as a regulator of cancer cell behavior plasticity. Annu Rev Cell Dev Biol 2012; 28: 599–625.
Lee M, Vasioukhin V . Cell polarity and cancer—cell and tissue polarity as a non-canonical tumor suppressor. J Cell Sci 2008; 121: 1141–1150.
Royer C, Lu X . Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ 2011; 18: 1470–1477.
Martin-Belmonte F, Perez-Moreno M . Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 2012; 12: 23–38.
Macara IG, McCaffrey L . Cell polarity in morphogenesis and metastasis. Philos Trans R Soc Lond B Biol Sci 2013; 368: 20130012.
Elsum I, Yates L, Humbert PO, Richardson HE . The Scribble-Dlg-Lgl polarity module in development and cancer: from flies to man. Essays Biochem 2012; 53: 141–168.
Pagliarini RA, Xu T . A genetic screen in Drosophila for metastatic behavior. Science 2003; 302: 1227–1231.
Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB et al. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 2008; 135: 865–878.
Vaira V, Faversani A, Dohi T, Maggioni M, Nosotti M, Tosi D et al. Aberrant overexpression of the cell polarity module scribble in human cancer. Am J Pathol 2011; 178: 2478–2483.
Pearson HB, Perez-Mancera PA, Dow LE, Ryan A, Tennstedt P, Bogani D et al. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J Clin Invest 2011; 121: 4257–4267.
Miles WO, Dyson NJ, Walker JA . Modeling tumor invasion and metastasis in Drosophila. Dis Model Mech 2011; 4: 753–761.
Gonzalez C . Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat Rev Cancer 2013; 13: 172–183.
Rudrapatna VA, Cagan RL, Das TK . Drosophila cancer models. Dev Dyn 2012; 241: 107–118.
Ma X, Yang L, Yang Y, Li M, Li W, Xue L . dUev1a modulates TNF-JNK mediated tumor progression and cell death in Drosophila. Dev Biol 2013; 380: 211–221.
Ma X, Shao Y, Zheng H, Li M, Li W, Xue L . Src42A modulates tumor invasion and cell death via Ben/dUev1a-mediated JNK activation in Drosophila. Cell Death Dis 2013; 4: e864.
Cordero JB, Macagno JP, Stefanatos RK, Strathdee KE, Cagan RL, Vidal M . Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev Cell 2010; 18: 999–1011.
Vidal M, Larson DE, Cagan RL . Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev Cell 2006; 10: 33–44.
Bergstralh DT, Johnston D St . Epithelial cell polarity: what flies can teach us about cancer. Essays Biochem 2012; 53: 129–140.
Grifoni D, Garoia F, Schimanski CC, Schmitz G, Laurenti E, Galle PR et al. The human protein Hugl-1 substitutes for Drosophila lethal giant larvae tumour suppressor function in vivo. Oncogene 2004; 23: 8688–8694.
Uhlirova M, Bohmann D . JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J 2006; 25: 5294–5304.
Srivastava A, Pastor-Pareja JC, Igaki T, Pagliarini R, Xu T . Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc Natl Acad Sci U S A 2007; 104: 2721–2726.
Ma X, Li W, Yu H, Yang Y, Li M, Xue L et al. Bendless modulates JNK-mediated cell death and migration in Drosophila. Cell Death Differ 2014; 21: 407–415.
Ohsawa S, Sato Y, Enomoto M, Nakamura M, Betsumiya A, Igaki T . Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature 2012; 490: 547–551.
Srivastava A, Pastor-Pareja JC, Igaki T, Pagliarini R, Xu T . Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc Natl Acad Sci USA 2007; 104: 2721–2726.
Rudrapatna VA, Bangi E, Cagan RL . A Jnk-Rho-Actin remodeling positive feedback network directs Src-driven invasion. Oncogene 2014; 33: 2801–2806.
Fernandez BG, Jezowska B, Janody F . Drosophila actin-capping protein limits JNK activation by the Src proto-oncogene. Oncogene 2014; 33: 2027–2039.
Neisch AL, Speck O, Stronach B, Fehon RG . Rho1 regulates apoptosis via activation of the JNK signaling pathway at the plasma membrane. J Cell Biol 2010; 189: 311–323.
Khoo P, Allan K, Willoughby L, Brumby AM, Richardson HE . In Drosophila, RhoGEF2 cooperates with activated Ras in tumorigenesis through a pathway involving Rho1-Rok-Myosin-II and JNK signalling. Dis Model Mech 2013; 6: 661–678.
Raftopoulou M, Hall A . Cell migration: Rho GTPases lead the way. Dev Biol 2004; 265: 23–32.
Hirai S, Cui de F, Miyata T, Ogawa M, Kiyonari H, Suda Y et al. The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J Neurosci 2006; 26: 11992–12002.
Itoh A, Horiuchi M, Wakayama K, Xu J, Bannerman P, Pleasure D et al. ZPK/DLK, a mitogen-activated protein kinase kinase kinase, is a critical mediator of programmed cell death of motoneurons. J Neurosci 2011; 31: 7223–7228.
Billuart P, Winter CG, Maresh A, Zhao X, Luo L . Regulating axon branch stability: the role of p190 RhoGAP in repressing a retraction signaling pathway. Cell 2001; 107: 195–207.
Xiong X, Collins CA . A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAP kinase signaling cascade. J Neurosci 2012; 32: 610–615.
Zhang S, Yu D . Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol Sci 2012; 33: 122–128.
McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL . Spatiotemporal rescue of memory dysfunction in Drosophila. Science 2003; 302: 1765–1768.
Thiery JP, Acloque H, Huang RY, Nieto MA . Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139: 871–890.
Glise B, Bourbon H, Noselli S . hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell 1995; 83: 451–461.
Ma X, Huang J, Yang L, Yang Y, Li W, Xue L . NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res 2012; 22: 425–431.
Igaki T, Pagliarini RA, Xu T . Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol 2006; 16: 1139–1146.
Geuking P, Narasimamurthy R, Lemaitre B, Basler K, Leulier F . A non-redundant role for Drosophila Mkk4 and hemipterous/Mkk7 in TAK1-mediated activation of JNK. PLoS One 2009; 4: e7709.
Rudrapatna VA, Bangi E, Cagan RL . Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Rep 2013; 14: 172–177.
Brumby AM, Goulding KR, Schlosser T, Loi S, Galea R, Khoo P et al. Identification of novel Ras-cooperating oncogenes in Drosophila melanogaster: a RhoGEF/Rho-family/JNK pathway is a central driver of tumorigenesis. Genetics 2011; 188: 105–125.
Kulshammer E, Uhlirova M . The actin cross-linker Filamin/Cheerio mediates tumor malignancy downstream of JNK signaling. J Cell Sci 2013; 126: 927–938.
Ryoo HD, Gorenc T, Steller H . Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell 2004; 7: 491–501.
Cuevas BD, Abell AN, Johnson GL . Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene 2007; 26: 3159–3171.
Baek SH, Kwon YC, Lee H, Choe KM . Rho-family small GTPases are required for cell polarization and directional sensing in Drosophila wound healing. Biochem Biophys Res Commun 2010; 394: 488–492.
Parri M, Chiarugi P . Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal 2010; 8: 23.
Brumby AM, Richardson HE . Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J 2003; 22: 5769–5779.
Uhlirova M, Jasper H, Bohmann D . Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc Natl Acad Sci USA 2005; 102: 13123–13128.
Morata G, Shlevkov E, Perez-Garijo A . Mitogenic signaling from apoptotic cells in Drosophila. Dev Growth Differ 2011; 53: 168–176.
Ebelt ND, Cantrell MA, Van Den Berg CL . c-Jun N-terminal kinases mediate a wide range oftargets in the metastatic ascade. Genes Cancer 2013; 4: 378–387.
Tedeschi A, Bradke F . The DLK signalling pathway—a double-edged sword in neural development and regeneration. EMBO Rep 2013; 14: 605–614.
Rauskolb C, Sun S, Sun G, Pan Y, Irvine KD . Cytoskeletal tension inhibits Hippo signaling through an Ajuba-Warts complex. Cell 2014; 158: 143–156.
Gaspar P, Tapon N . Sensing the local environment: actin architecture and Hippo signalling. Curr Opin Cell Biol 2014; 31: 74–83.
Ma X, Chen Y, Xu W, Wu N, Li M, Cao Y et al. Impaired Hippo signaling promotes Rho1-JNK-dependent growth. Proc Natl Acad Sci USA 2015; 112: 1065–1070.
Ma X . Context-dependent interplay between Hippo and JNK pathway in Drosophila. AIMS Genet 2014; 1: 20–33.
Pichon S, Bryckaert M, Berrou E . Control of actin dynamics by p38 MAP kinase - Hsp27 distribution in the lamellipodium of smooth muscle cells. J Cell Sci 2004; 117: 2569–2577.
Lewcock JW, Genoud N, Lettieri K, Pfaff SL . The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 2007; 56: 604–620.
Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M . Axon regeneration requires a conserved MAP kinase pathway. Science 2009; 323: 802–806.
Collins CA, Wairkar YP, Johnson SL, DiAntonio A . Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 2006; 51: 57–69.
Xue L, Igaki T, Kuranaga E, Kanda H, Miura M, Xu T . Tumor suppressor CYLD regulates JNK-induced cell death in Drosophila. Dev Cell 2007; 13: 446–454.
Lesch C, Jo J, Wu Y, Fish GS, Galko MJ . A targeted UAS-RNAi screen in Drosophila larvae identifies wound closure genes regulating distinct cellular processes. Genetics 2010; 186: 943–957.
Zhang L, Ren F, Zhang Q, Chen Y, Wang B, Jiang J . The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev Cell 2008; 14: 377–387.
Acknowledgements
We thank Drs Duojia Pan, Tian Xu, Helena Richardson, Aaron DiAntonio, Bloomington, VDRC and NIG stock centers for fly stocks and reagents, Dr Margaret Ho and members of Xue lab for discussion and comments, Dr Jonathan Phillips for critically reading the manuscript. This research was supported by the National Basic Research Program of China (973 Program) (2011CB943903), National Natural Science Foundation of China (31071294, 31171413, 31371490), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20120072110023, 20120072120030), and Shanghai Committee of Science and Technology (09DZ2260100, 14JC1406000). Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Ma, X., Chen, Y., Zhang, S. et al. Rho1–Wnd signaling regulates loss-of-cell polarity-induced cell invasion in Drosophila. Oncogene 35, 846–855 (2016). https://doi.org/10.1038/onc.2015.137
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.137
- Springer Nature Limited
This article is cited by
-
Slik maintains tissue homeostasis by preventing JNK-mediated apoptosis
Cell Division (2023)
-
CtBP modulates Snail-mediated tumor invasion in Drosophila
Cell Death Discovery (2021)
-
Yorkie-Cactus (IκBα)-JNK axis promotes tumor growth and progression in Drosophila
Oncogene (2021)
-
A novel regulator of ER Ca2+ drives Hippo-mediated tumorigenesis
Oncogene (2020)
-
Identification of the Wallenda JNKKK as an Alk suppressor reveals increased competitiveness of Alk-expressing cells
Scientific Reports (2020)