
Abstract
Somatic mutations in the gene encoding the catalytic subunit of protein phosphatase 6 (Ppp6c) have been identified in malignant melanoma and are thought to function as a driver in B-raf- or N-ras-driven tumorigenesis. To assess the role of Ppp6c in carcinogenesis, we generated skin keratinocyte-specific Ppp6c conditional knockout mice and performed two-stage skin carcinogenesis analysis. Ppp6c deficiency induced papilloma formation with 7,12-dimethylbenz (a) anthracene (DMBA) only, and development of those papillomas was significantly accelerated compared with that seen following DMBA/TPA (12-O-tetradecanoylphorbol 13-acetate) treatment of wild-type mice. NF-κB activation either by tumor necrosis factor (TNF)-α or interleukin (IL)-1β was enhanced in Ppp6c-deficient keratinocytes. Overall, we conclude that Ppp6c deficiency predisposes mice to skin carcinogenesis initiated by DMBA. This is the first report showing that such deficiency promotes tumor formation in mice.
Similar content being viewed by others

Introduction
Although the effect of protein phosphorylation on cancer-related signaling pathways is well documented, little is known about the role of serine/threonine phosphoprotein phosphatases (PPPs) in tumor development. Within the PPP family, and based on similarity of catalytic subunits, protein phosphatase 6 (PP6), PP2A and PP4 are subgrouped as PP2A subfamily enzymes.1, 2 PP6 is trimeric holoenzyme consisting of catalytic, structural and regulatory subunits.3 PP6 is conserved among all eukaryotes, from yeast to humans. Its fission yeast homolog, Ppe1, is important for equal chromosome segregation,4, 5 whereas the budding yeast homolog, Sit4, is required for G1 to S progression.6,7 The Caenorrabditis elegans homolog, PPH-6, regulates spindle positioning.8 Experiments in these organisms suggest that PP6 functions at cell cycle checkpoints.6, 7, 8 Biochemical and knockdown-based experiments using several mammalian cell lines suggest that PP6 dephosphorylates and thus inactivates a MAP kinase known as TAK19 and suppresses IκBɛ degradation in response to tumor necrosis factor (TNF)-α.10 PP6 also may function in repair of double-stranded DNA breaks following ionizing radiation.11 These data suggest that PP6 integrates signaling from multiple pathways.
Recently, two key studies reported somatic mutations in the gene encoding the PP6 catalytic subunit (Ppp6c) in ~10% of malignant melanoma patients harboring B-raf or N-ras mutations.12, 13 Ppp6c mutations were often accompanied by loss of heterozygosity, strongly suggesting that Ppp6c loss functions in tumor formation in the presence of B-raf or N-ras mutations.12 Importantly, based on the Catalogue of Somatic Mutations in Cancer (COSMIC) database, Ppp6c mutations occur in carcinomas other than malignant melanoma. However, whether PP6 has a tumor-suppressive function has not yet been demonstrated in animal models.
Mouse two-stage skin carcinogenesis models, which are widely used to study epithelial carcinogenesis, represent one of the best-established in vivo models.14 Following one-time application of an initiator such as 7,12-dimethylbenz (a) anthracene (DMBA), the same area of skin is painted repeatedly with promoter such as 12-O-tetradecanoylphorbol 13-acetate (TPA), an activator of protein kinase C, giving rise to papillomas. A single DMBA treatment does not lead to papillomas, and survival and growth of skin papillomas require continued TPA painting, as cessation of TPA treatment causes tumor regression.14 Using this system, Fujiki and colleagues15, 16, 17 showed that okadaic acid (OA), an inhibitor of PP2A-type phosphatase, had tumor-promoting activities as potent as TPA, suggesting that PPPs have tumor-suppressive activity. They also showed that tumor promotion by OA or TPA was critically dependent on TNF-α.18, 19 However, it has not yet been shown how and which OA-sensitive PPP, including PP2A, PP4 or PP6 functions in skin carcinogenesis.
To determine whether PP6 deficiency functions in carcinogenesis in vivo, we employed a mouse skin 2-stage carcinogenesis model. To do so we first generated a conditional knockout mouse in which the phosphatase domain of Ppp6c can be deleted in skin keratinocytes in order to determine the consequences of Ppp6c loss. Unexpectedly, we observed tumor formation in the presence of Ppp6c deficiency following application of DMBA only, that is, in the absence of chemical tumor promoters. Furthermore, tumor appearance was significantly accelerated relative to tumor onset following DMBA/TPA treatment.
Results
Skin keratinocyte-specific induction of Ppp6c deletion
To analyze PP6 function in carcinogenesis in vivo, we initially developed mice that globally lack the phosphatase domain of the PP6 catalytic subunit (Ppp6c; Ogoh et al. in preparation). However, homozygotes showed developmental anomalies by embryonic day (E) 7.5 and died embryonically (Ogoh et al. in preparation), making them unsuitable for studies of carcinogenesis. Thus, we developed mice that conditionally lack Ppp6c in skin keratinocytes. To do so, we generated tamoxifen-inducible, keratinocyte-specific homozygous mutant mice (K14-CreERtam; Ppp6cflox/flox) (Figure 1a). In those mice K14-CreERtam expression is driven by the keratin14 promoter, which is activated by 4-hydroxytamoxifen (4HT) administration.20 Cre then excises floxed exon 4, which encodes the Ppp6c phosphatase domain (Figure 1a).

CreERtam-mediated Ppp6c disruption. (a) Schematic representation of floxed and exon 4-deleted Ppp6c alleles. The position of primers (a, b, c and d) is indicated. WT, wild-type. (b) PCR analysis of genomic DNA to detect the exon 4-deleted Ppp6c allele using primers (a and b). Lane 1: genomic DNA from keratinocytes obtained from 4HT-untreated (4HT-) K14-CreERtam; Ppp6cflox/flox mice. Lane 2: genomic DNA from keratinocytes obtained from equivalent 4HT-treated (4HT+) mice. Lanes 3–8: various mixtures of genomic DNAs from tails of Ppp6c+/− and Ppp6cflox/+. Percentages of Ppp6c+/− DNA were 100% (lane 3), 80% (lane 4), 60% (lane 5), 40% (lane 6), 20% (lane 7) and 0% (lane 8). Fragments of 796 and 275 bp correspond to floxed and exon 4-deleted alleles, respectively. On the basis of analysis shown in lane 2, we estimate a recombination rate of ~70%. (c) Immunoblot of keratinocyte lysates using antibodies targeting the Ppp6c N terminus, the Ppp6c C terminus and β-actin. The Ppp6c/β-actin intensity ratio was calculated for each condition and compared.
To assess this system, we purified keratinocytes from day 3 K14-CreERtam; Ppp6cflox/flox mice pretreated with or without 4HT (Figures 1a and b). Keratinocyte DNA was examined by PCR as shown in Figure 1b. Control keratinocytes not treated with 4HT showed a 796-bp-long PCR fragment representing the floxed allele when primers a and b were used (Figure 1b, lane 1). On the other hand, the equivalent genomic DNA from 4HT-treated keratinocytes showed both the 796-bp fragment and the 275-bp fragment corresponding to the exon 4-deleted allele. (Figure 1b, lane 2). To estimate the recombination rate we compared the intensity of both bands obtained in 4HT-treated keratinocytes (lane 2) with that seen in various mixtures of genomic DNA prepared from tails of Ppp6c+/− and Ppp6cflox/+ mice (lanes 3–8). On the basis of that comparison, we estimate the recombination rate to be ~70%.
We next examined levels of Ppp6c protein by immunoblotting of extracts prepared from keratinocytes with an antibody against the Ppp6c N terminus (Figure 1c, upper). Band intensity comparisons of the full-length protein indicated that Ppp6c protein levels in wild-type and 4HT-untreated Ppp6cflox/flox mouse keratinocytes were comparable, indicating that sequences contained in the floxed allele do not alter protein expression. We then examined intensity of the full-length Ppp6c band between 4HT-untreated and -treated keratinocytes and found that protein levels in 4HT-treated keratinocytes were ~25% of those seen in untreated cells. Importantly, we could not detect truncated Ppp6c protein in 4HT-treated cells, suggesting that levels of truncated Ppp6c protein are negligible. Experiments performed using an antibody against the Ppp6c C terminus confirmed a 25% reduction in levels of full-length Ppp6c in the 4HT-treated keratinocytes (Figure 1c, middle).
Ppp6c deficiency enhances carcinogenesis in a two-stage skin cancer model
We next examined the effect of Ppp6c deletion in a mouse two-stage skin carcinogenesis model. To do so, we used 6- to 7-week-old mice and applied 4HT (4HT+) or vehicle (4HT−) for five consecutive days. Two days later we treated the mice once with DMBA and then with TPA twice a week for 20 weeks (Figure 2a).

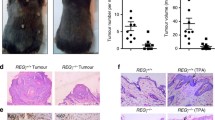
Ppp6c deficiency accelerates tumorigenesis in a two-stage skin carcinogenesis model. (a) Schedule for two-stage carcinogenesis. Before initiation, K14-CreERtam; Ppp6cflox/flox mice were shaved and pretreated with (4HT+) or without (4HT−) 4HT. TPA or OA was then applied as a promoter. w, weeks. (b) 4HT+ (n=11) and 4HT− (n=7) mice were used for DMBA/TPA carcinogenesis. The upper panel shows the percentage (%) of mice that developed any papillomas; the lower shows the average number of papillomas per mouse in the total number of mice tested. (c) 4HT+ (n=8) and 4HT− (n=6) mice were used for DMBA/OA carcinogenesis. Upper panel shows the % of mice that developed any papillomas, and lower shows the average number of papillomas per mouse in the total mice used.
In control (4HT−) mice, papillomas began to appear at ~15 weeks after DMBA treatment, corresponding to the period required for papilloma formation in this model in C57BL/6 mice.21 However, unexpectedly, papillomas appeared at ~5 weeks after DMBA treatment in Ppp6c conditionally deleted mice pretreated with 4HT (4HT+; Figure 2b, upper). It is noteworthy that Ppp6c deficiency induced early tumor formation but did not increase tumor number (Figure 2b, lower), suggesting that Ppp6c loss accelerates papilloma formation in the DMBA/TPA model. We next asked whether such acceleration was TPA-specific. To do so, we used OA, which has a different mode of action compared with TPA, as a tumor promoter. As shown in Figure 2c, in DMBA/OA carcinogenesis, we observed similar early papilloma formation with a slight increase in tumor number in the skin of Ppp6c-deficient (4HT+) relative to control (4HT−) mice (Figure 2c). These data show that Ppp6c deficiency enhances tumor promotion by DMBA irrespective of whether the tumor promoter is TPA or OA.
Ppp6c deficiency predisposes mice to skin carcinogenesis
We next examined the effect of DMBA on Ppp6c-deficient skin tissue in the absence of chemical tumor promoters. Mice (either 4HT+ or 4HT−) were painted with DMBA and monitored for tumor formation over 20 weeks (Figure 3a). Control (4HT−) mice showed no tumors within 20 weeks (Figures 3b and c); however, in Ppp6c-deficient (4HT+) skin, tumors appeared at ~5 weeks (Figure 3b), a time course of papilloma incidence (% of papilloma-bearing mice) almost identical to that seen in DMBA/TPA or DMBA/OA experiments (Figures 2b and c).

Ppp6c deficiency predisposes skin tissue to carcinogenesis. (a) Schedule for carcinogenesis induced by single DMBA treatment. Before initiation, K14-CreERtam; Ppp6cflox/flox mice were shaved and pretreated with (4HT+) or without (4HT−) 4HT. w, weeks. (b) 4HT+(n=15) and 4HT− (n=7) mice were used. Upper panel shows % of mice that developed any papillomas and lower shows the average number of papillomas per mouse in the total mice used. (c) Macroscopic appearance of a representative tumor 20 weeks after a single DMBA treatment. (d) Hematoxylin and eosin staining of the tumor shown in c. Scale bar in d: 500 μm.
Mice were killed at 20 weeks, and tumors were examined histochemically (Figures 2b and c and Figure 3). A total of 24 tumors produced in Ppp6c-deficient mice were diagnosed as papillomas by two independent pathologists and classified as one of five types (hyperkeratotic papilloma (n=10), early follicular papilloma (n=4), exophytic papilloma (n=4), mixed papilloma (n=4) and fibropapilloma (n=2)). These tumor types are reportedly typical of papillomas seen after DMBA/TPA treatment.21 Macroscopic and microscopic images of a hyperkeratotic papilloma are shown in Figures 3c and d. Overall, this analysis strongly suggests that Ppp6c deficiency functions as a tumor promoter.
Ppp6c-deficient keratinocytes express genes associated with oncogenesis
We next analyzed genes expressed in tumors larger than 5 mm in diameter in three tumors each from DMBA- (Figure 3b), DMBA/TPA- (Figure 2b) or DMBA/OA-treated mice (Figure 2c) in which papillomas appeared within 6 weeks of DMBA treatment. In each condition, Ppp6c mRNAs were truncated, suggesting that tumors arose from keratinocytes lacking a fully functional Ppp6c gene (Figure 4a). We then evaluated potential mutation of H-ras at codon 61, a CAA (glutamine) to CTA (leucine) transversion typically seen following use of DMBA as an initiator.14 We detected this mutation in every tumor examined (Figure 4b), indicating that mutation of this codon together with Ppp6c loss-of-function could account for tumor cell phenotypes.

Tumors derived from Ppp6c-deficient keratinocytes show enhanced expression of the keratinocyte-derived cytokine GROα and of cyclin D1. (a) Ppp6c exon 4 transcripts are truncated in tumor tissues. PCR was performed using template cDNA made from control skin, from representative tumors seen following a single DMBA treatment, from DMBA/TPA tumors or from DMBA/OA tumors—from 4HT+ mice (Figures 2b and c, Figure 3b). Primers c and d (recognizing exons 1 and 6 of Ppp6c, respectively) were used (Figure 1a). The 604- and 462-bp fragments likely correspond to wild-type and exon 4-deleted Ppp6c cDNA, respectively. (b) Detection of the H-ras codon 61 mutation. The same cDNA products defined in a were used. For PCR-restriction fragment length polymorphism analysis, H-ras exon 2-specific primers were used, and the amplicon was digested with XbaI to distinguish normal (119 bp) from mutated (63 bp/56 bp) alleles following agarose gel electrophoresis. (c) Transcript expression in corresponding tumors. As controls, we used normal skin from five different littermates. Results were normalized to 18S ribosomal RNA and shown as fold differences in transcript level relative to that seen normal skin, a value arbitrarily set to 1. Values were calculated based on analysis of three tumors and represent the mean±s.e. Data were analyzed by a one-tailed Student’s t-test; P-values less than 0.05 are designated with an asterisk. (d) Detection of γH2AX in papillomas that had developed after a single DMBA treatment. The papilloma sample shown in Figure 3d was subjected to hematoxylin and eosin (HE) staining (upper) or immunohistochemistry (using anti-γH2AX antibody (lower).
Growth-Regulated Oncogene α (GROα) functions in proliferation and progression of malignant keratinocytes and is upregulated by oncogenic ras.22, 23, 24 Thus, we examined its expression in three tumors from either DMBA-, DMBA/TPA- or DMBA/OA- treated mice. All tumors derived from Ppp6c-deficient keratinocytes exhibited GROα expression higher than control (DMBA: P=0.036). Expression of Cyclin D1, a marker of cell proliferation,25 also significantly increased in DMBA-treated cells (DMBA+TPA: P=0.018, DMBA+OA: P=0.029) relative to controls, suggesting that tumors emerging from Ppp6c-deficient keratinocytes harboring H-ras mutations have a growth advantage over Ppp6c-deficient keratinocytes lacking H-ras mutations or wild-type keratinocytes. We then undertook immunohistochemistry (using anti-γH2AX antibody, as γH2AX-positive cells appear following induction of reactive oxygen species by DMBA and are seen in papillomas and squamous cell carcinomas.26 As shown in Figure 4d, in Ppp6c-deficient skin, cells in papilloma tumors induced by DMBA were highly positive for γH2AX, suggesting accumulation of double-stranded breaks.
Ppp6c-deficient skin tissues show an enhanced inflammatory response following DMBA administration
To further assess DMBA effects on Ppp6c-deficient skin, we performed histological examination of the skin after DMBA administration using four mice each for Ppp6c-deficient (4HT+) and control (4HT−) samples. By 48 h after DMBA treatment, all Ppp6c-deficient samples showed a similar response, as illustrated in a representative sample (Figure 5): relative to controls, Ppp6c-deficient samples exhibited (1) thicker epidermis associated with hyperkeratosis, (2) cellular infiltration of the dermis and (3) disappearance of subcutaneous fat tissue, effects indicative of inflammatory and proliferative responses.

Ppp6c deficiency facilitates DMBA-induced inflammation in skin. Six-week-old K14-CreERtam; Ppp6cflox/flox mice were pretreated with (4HT+) or without (4HT−) 4HT and then initiated with DMBA as indicated in Figure 3a. Images show hematoxylin and eosin staining of skin samples before (0 h) or 48 h after (48 h) DMBA treatment. Scale bars: 200 μm. Representative data from four independent experiments are shown.
To examine whether and how Ppp6c deficiency contributes to DMBA-induced proliferation and inflammation, we analyzed expression of proliferation- and inflammation-related genes in skin tissues, including the early response genes c-jun and c-fos (Figure 6). Relative to controls (4HT−), c-jun expression was upregulated in Ppp6c-deficient (4HT+) skin (P=0.007 at 3 h and P=0.01 at 6 h), as was c-fos between 1 and 6 h. In terms of proinflammatory cytokines, the gene encoding TNF-α, which is reportedly indispensable in the two-stage DMBA/TPA model,19 was similarly induced by DMBA in control (4HT−) and Ppp6c-deficient (4HT+) skin. However, IL-1β and IL-6 transcript levels increased starting 24 h after DMBA treatment, an upregulation enhanced in Ppp6c-deficient (4HT+) skin (IL-6:P=0.045 at 48 h). Expression of other inflammation-related genes, including GM-CSF, GROα and MMP-3, significantly increased in Ppp6c-deficient (4HT+) compared with control (4HT−) skin by 48 h after DMBA treatment (granulocyte–monocyte colony-stimulating factor (GM-CSF): P<0.029 at 6 h, P<0.041 at 48 h; GROα: P<0.015 at 48 h; MMP-3: P<0.006 at 24 h). These results collectively show that expression of inflammation-related genes is upregulated by DMBA treatment and that this effect that is more robust in Ppp6c-deficient skin (Figure 6).

Ppp6c deficiency promotes expression of proliferation- and inflammation-related genes following DMBA treatment. Six-week-old K14-CreERtam; Ppp6cflox/flox mice were pretreated with (4HT+) or without (4HT−) 4HT, and initiated with DMBA as indicated in Figure 3a. Skin was then removed at times shown and analyzed for expression of the indicated mRNAs using qPCR. Results were normalized to β-actin levels and are shown relative to levels seen before DMBA treatment (0 h). Transcript levels at 0 h were arbitrarily set to 1. Data are the mean values derived from four independent experiments±s.e. Data were analyzed by a one-tailed Student’s t-test; P-values less than 0.05 are designated with an asterisk.
TNF-α- and TNF-β-dependent NF-κB signaling is enhanced in Ppp6c-deficient keratinocytes
As the proinflammatory cytokine TNF-α is upregulated in the skin by DMBA stimulation (Figure 6), we assessed the effect of DMBA treatment on NF-κB signaling using cultured Ppp6c-deficient 4HT+ and 4HT− control keratinocytes (Figure 7). To do so, we employed immunoblotting to assess the time course of pTAK1 phosphorylation, degradation of IκBα and IκBɛ proteins and phosphorylation of p65/relA following TNF-α (250 ng/ml) stimulation (Figure 7a). Although we detected pTAK1 phosphorylation from 15 to 60 min following TNF-α stimulation in Ppp6c-deficient (4HT+) keratinocytes, neither IκBα nor IκBɛ proteins were downregulated in those cells, whereas control keratinocytes showed slight upregulation of IκBɛ proteins. We observed robust p65/relA phosphorylation between 5 and 60 min following TNF-α stimulation of Ppp6c-deficient (4HT+) keratinocytes, whereas 4HT− controls showed robust phosphorylation between 5 and 15 min (Figures 7a and b).

Ppp6c-deficient keratinocytes show enhanced prolonged NF-κB activation. 4HT− or 4HT+ keratinocytes were treated with 250 ng/ml TNF-α (a, b) or 250 ng/ml IL-1β (c, d) for the indicated times. Representative immunoblot of keratinocyte lysates using anti-phospho TAK1, anti-IκBα, anti-IκBɛ, anti-p65/relA, anti-phospho p65/relA and anti-β-actin antibodies. Phosphorylated p65/relA and β-actin protein levels were quantified based on immunoblots shown in a, c. The p-p65/relA/β-actin ratio at 0 min of control (4HT−) keratinocytes was set to 1. Values represent fold differences relative to that value (b, d). Data represent the mean values derived from three independent experiments±s.e. Data were analyzed by one-tailed Student’s t-test, and P-values less than 0.05 are designated with an asterisk. p-p65/relA protein levels following TNF-α treatment were upregulated in Ppp6c-deficient relative to control skin (P=0.015 at 30 min; P=0.004 at 60 min). p-p65/relA levels following IL-1β treatment were also upregulated in Ppp6c-deficient relative to control skin (P=0.023 at 15 min; P=0.003 at 30 min; P=0.009 at 60 min).
As IL-1β upregulation by DMBA is enhanced in Ppp6c-deficient skin (Figure 6), we examined the effect of IL-1β on NF-κB signaling using cultured Ppp6c-deficient 4HT+ and 4HT− control keratinocytes (Figure 7c). IL-1β (250 ng/ml) induced significant IκBα degradation in 4HT− and 4HT+ keratinocytes over a similar time course (Figure 7b). p65/relA phosphorylation, followed by IκBα degradation, was observed in both 4HT− and 4HT+ keratinocytes; however, p65/relA phosphorylation was more prolonged (from 5 to 60 min) in 4HT+ compared with control keratinocytes. (Figure 7d). These data suggest that in DMBA-treated Ppp6c-deficient keratinocytes, both TNF-α- and IL-1β−dependent NF-κB signaling is prolonged relative to that seen in DMBA-treated wild-type cells.
Discussion
It is generally accepted that a single treatment with DMBA does not produce papillomas without a chemical promoter such as TPA or OA. Here, using a two-stage skin carcinogenesis model, we found that a single DMBA application was sufficient to produce papillomas in Ppp6c-deficient skin (Figure 3). We found only one comparable example in the literature: namely, metallothionein-deficient mice reportedly develop papillomas after a single treatment with DMBA at a dose of 1 mg, a dose 10 times higher than that we used here.27 We also found that papilloma formation is rapid in Ppp6c-deficient skin (Figures 2 and 3): DMBA treatment alone induced papillomas in Ppp6c-deficient mice at around 5–6 weeks, whereas DMBA/TPA or DMBA/OA induced papillomas at 15–16 weeks. These findings suggest that tumor promotion activity associated with Ppp6c deficiency is more robust than that seen after repeated application of TPA or OA. The data also suggest that Ppp6c-deficient skin is predisposed to carcinogenesis initiated by DMBA.
When we examined tumors greater than 5 mm in diameter in DMBA, DMBA/TPA or DMBA/OA mice in our Ppp6c-deficient model we found that every tumor examined contained an oncogenic mutation at H-ras codon 61 (Figure 4a) and truncation of Ppp6c (Figure 4b). These data suggest that both Ppp6c loss-of-function and the H-ras codon 61 mutation give keratinocytes a growth advantage and promote papilloma development in the presence of DMBA. These data support the hypothesis proposed by Hodis et al.12 based on whole-exome sequencing of malignant melanoma tumors, namely that Ppp6c mutations act together with B-raf or N-ras mutations to promote tumorigenesis. Recent COSMIC data indicate that Ppp6c mutations are present in the lung, large intestine and endometrial cancers. In the future, it will be important to generate Ppp6c knockout mice harboring other oncogenic mutations to verify whether Ppp6c loss acts as a driver in oncogene-driven carcinogenesis.12, 13 Currently, we are examining the effect of Ppp6c deficiency on K-ras-driven carcinogenesis.
Chronic inflammation reportedly promotes tumor progression in mice and humans.28, 29 In fact, our histological examination indicated that genes associated with inflammation and proliferation were upregulated in Ppp6c-deficient skin cells following DMBA treatment (Figure 5). Among them were the proinflammatory cytokine genes TNF-α, IL-1β and IL-6 (Figures 6d and e). Ppp6c-deficient skin also showed higher levels of IL-1β and IL-6 transcripts. It is well known that inflammatory stimuli, including DMBA, recruit cells that release TNF-α to areas of inflammation14, 19. Therefore, we analyzed the effect of TNF-α on keratinocytes and found that NF-κB activation was prolonged in Ppp6c-deficient cells (Figure 7b), an effect also seen when IL-1β served as a stimulant (Figure 7d). So far, TAK1 and IκBɛ are reported to be targets of PP6 in the NF-κB pathway;9, 10 however, our data suggest that there could be other targets in mouse primary keratinocytes. Prolonged p65/relA phosphorylation induced by TNF-α or IL-1β in Ppp6c-deficient (4HT+) keratinocytes could be explained by either activation of a kinase(s) other than IKK or blocked dephosphorylation of phospho-p65/relA because of loss of Ppp6 activity (Figures 7a and c).
Other inflammation-related genes, such as GM-CSF, GROα and MMP-3 (Figures 6f–h), were upregulated in Ppp6c-deleted skin upon DMBA treatment (Figure 6). These outcomes may allow tumor-promoting inflammation to persist, induce angiogenesis or activate invasive behavior of tumor cells. Among these factors, we observed particularly high expression of GROα in papilloma samples. GROα regulates inflammation by attracting neutrophils and promotes tumor development by stimulating angiogenesis.30, 31 Increased GROα levels may also explain accelerated papilloma formation.
To determine why papilloma formation in Ppp6c-deficient skin is accelerated, we examined expression of proliferation-related genes. We observed enhanced expression of c-jun and c-fos mRNAs in Ppp6c-deficient skin 1–6 h after DMBA treatment (Figures 6a and b), an early event that may activate AP1 and increase cell proliferation. Although we did not detect phosphorylated p65/rel in papilloma with immunohistochemistry ((data not shown), NF-κB activity may also underlie this proliferation, as it reportedly upregulates cyclin D1.32, 33 In papilloma samples, cyclin D1 expression was highly elevated (Figure 4c), possibly accounting in part for sustained proliferation of Ppp6c-deficient keratinocytes and accelerated papilloma growth.
In summary, we found that Ppp6c deficiency predisposes mouse skin tissue to carcinogenesis initiated by DMBA. This is the first report that a Ppp6c loss-of-function acts as a tumor promoter in mice. This finding supports the proposal by others12, 13 that Ppp6c mutation acts as a driver in B-raf- and N-ras-driven melanoma. The next question is whether PP6 loss has tumor-promotion activity in other human cancers. As PP6 is thought to be a druggable target, mechanisms underlying tumor promotion by PP6 deficiency should be examined in detail.
Materials and methods
Generation of knockout mice
Ppp6c-floxed (Ppp6c flox/flox) mice (accession no. CDB0850K: http://www.cdb.riken.jp/arg/mutant%20mice%20list.html, mixed genetic strain of C57BL/6 and CBA) and Ppp6c heterozygous mice were generated, and phenotypes seen in null mice were assessed by Ogoh et al. (in preparation). K14-CreERtam mice (a strain of CD1) were obtained from Jackson Laboratory, crossed with C57BL/6 mice for four generations in our animal facility, and crossed with Ppp6cflox/flox mice to generate epidermal-specific induction of Ppp6c-deficient mice (K14-CreERtam; Ppp6cflox/flox). Littermate controls were used in all experiments. All animal experiments were conducted with the approval of Miyagi Cancer Center Research Institute Animal Care and Use committee.
Reagents
DMBA and TPA were purchased from Sigma Chemical Co (St Louis, MO, USA). OA was isolated from the black sponge Halichondria okadai as described34 with some modifications. Mouse TNF-α was purchased from Sigma Chemical Co. Mouse IL-1β was purchased from Cell Signaling Technology Inc. (Danvers, MA, USA). 4HT was purchased from Toronto Research Chemicals (North York, ON, Canada).
PCR analysis of Ppp6c gene deletion
For PCR analysis of floxed and exon 4-deleted alleles, we used: primer a, 5′-TATCACGAGGCCCTTTCG-3′, and primer b, 5′-TAGTGAACCTCTTCGAGG-3′ (Figure 1a). PCR products from Ppp6c-floxed and -deleted alleles were 796 and 275 bp, respectively. For analysis of Ppp6c cDNA we used: primer c, 5′-TGGATCTGGACAAGTATGTG-3′, and primer d, 5′-CAAGTGTCCACATCTTCAGG-3′ (Figure 1a). Lengths of PCR products from wild-type and exon 4-deleted Ppp6c cDNAs were 604 and 462 bp, respectively.
Skin carcinogenesis induced by DMBA
When mice were 6–7 weeks of age, the dorsal skin of K14-CreERtam; Ppp6cflox/flox mice was shaved and pretreated once a day for five consecutive days with 100 μl acetone containing 4HT (0.4 mg/mouse; 4HT (+) group) or with 100 μl acetone vehicle as the 4HT (−) group. On the fifth day, the skin was shaved again, and 2 days later, 4HT(+) or 4HT (−) mice received a single application of 100 μg DMBA. For the two-step carcinogenesis model (DMBA/TPA or DMBA/OA), separate groups of 4HT (+) or 4HT (−) mice were prepared for each model. One week after DMBA initiation, TPA (1 μg) or OA (5 μg) dissolved in 100 μl acetone was applied topically twice a week until week 20 to 4HT (+) or 4HT (−) mice. The number of tumors exceeding 1 mm in diameter was determined weekly. Analysis of mRNA expression was evaluated in tumors exceeding 5 mm in diameter.
Histological examination
Formalin-fixed, paraffin-embedded tissues were stained with hematoxylin eosin and independently examined microscopically by two pathologists in the Miyagi Cancer Center. Images were acquired using an Olympus BX53 microscope (Tokyo, Japan). Immunohistochemistry was performed as described26. using anti-γH2AX antibody (Cell Signaling Technology Inc.).
Quantitative real-time PCR
Total RNA was prepared from specimens with miRNeasy Mini (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. cDNA was synthesized using random primers with Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA) and subjected to quantitative real-time PCR using LightCycler 480 (Roche Diagnostics, Basel, Switzerland) with the universal library and probe master kit (Roche Diagnostics). Levels of c-jun, c-fos, IL-1β, IL-6, TNF-α, GROα, GM-CSF and MMP-3 transcripts were reported as a ratio relative to β-actin or 18S ribosomal RNA levels. The following probes were used: no. 7 (c-jun), no. 76 (c-fos), no. 26 (IL-1β), no. 6 (IL-6), no. 49 (TNF-α), no. 83 (GROα), no. 79 (GM-CSF), no. 7 (MMP-3), no. 72 (cyclinD1), no. 64 (β-actin) and no. 48 (18S ribosomal RNA; Roche Universal Probe Library). Primer sequences are follows: c-jun 5′-TATTTTGGGGAGCATTTGGA-3′ and 5′-GAGATTTGCAAAAGTTCGCTCT-3′; c-fos 5′-GGGGCAAAGTAGAGCAGCTA-3′ and 5′-AGCTCCCTCCTCCGATTC-3′; IL-1β 5′-TTGACGGACCCCAAAAGAT-3′ and 5′-TTTGAAGCTGGATGCTCTCAT-3′; IL-6 5′-GATGGATGCTACCAAACTGGA-3′ and 5′-CCAGGTAGCTATGGTACTCCAGAA-3′; TNF-α 5′-TCTTCTCATTCCTGCTTGTGG-3′ and 5′-GGTCTGGGCCATAGAACTGA-3′; GROα 5′-ACACTCCAACACAGCACCAT-3′ and 5′-TGACAGCGCAGCTCATTG-3′; GM-CSF 5′-GCATGTAGAGGCCATCAAAGA-3′ and 5′-CGGGTCTGCACACATGTTA-3′; MMP-3 5′-TTGTTCTTTGATGCAGTCAGC-3′ and 5′-GATTTGCGCCAAAAGTGC-3′; cyclin D1 5′-TTTCTTTCCAGAGTCATCAAGTGT-3′ and 5′-TGACTCCAGAAGGGCTTCAA-3′; β-actin 5′-CTAAGGCAACCGTGAAAAG-3′ and 5′-ACCAGAGGCATACAGGGACA-3′; and 18S ribosomal RNA 5′-GCAATTATTCCCCATGAACG-3′ and 5′-GGGACTTAATCAACGCAAGC-3′.
PCR-restriction fragment length polymorphism
The H-ras mutation at codon 61 (CAA to CTA) was analyzed with PCR-restriction fragment length polymorphism with some modifications.35 PCR (sense primer: 5′-CCGGAAACAGGTGGTCATTG-3′ and antisense primer: 5′-AGGAAGCCCTCCCCTGTGCG-3′) was performed using cDNA from tumors as a template. Amplified H-ras exon 2 was digested with XbaI, which cuts only if codon 61 is mutant (CTA). Digested samples were electrophoresed on 3% agarose gels for analysis.
Primary mouse keratinocytes
Newborn K14-CreERtam; Ppp6cflox/flox mice were painted over their entire body with 100 μl acetone containing 4HT (0.4 mg/mouse) for three consecutive days (days 0–2) to trigger deletion of Ppp6c exon 4 in keratinocytes. Controls were painted with acetone only. Keratinocytes were isolated from 3-day-old mice as described by Lichti et al.36 and maintained in CnT-07 medium (CELLnTEC, Bern, Switzerland). Subsequent experiments were performed between 4 and 6 days of culture.
Immunoblotting
Cell lysate preparation and immunoblot analysis were performed as described.37 A polyclonal antibody against a peptide corresponding to the N-terminal 17 amino acids of Ppp6c was obtained from LifeSpan BioScience Inc. (Seattle, WA, USA). A polyclonal antibody against a peptide corresponding to the Ppp6c C-terminal 16 amino acids was generated in our laboratory. IκBα, IκBɛ, p65/relA and β-actin were assessed using anti-IκBα (Cell Signaling Technology Inc.), anti-IκBɛ (Santa Cruz), anti-p65/relA (Cell Signaling Technology Inc.) and anti-β-actin (Sigma) antibodies. TAK1 and p65/relA phosphorylation levels were assessed using an anti-phospho TAK1 (Th187) antibody (Cell Signaling Technology Inc.) and anti-phospho-p65/relA (Ser 536) antibody (Cell Signaling Technology Inc.). Signals were detected by enhanced chemiluminescence using the LAS-4000 mini Fluoro image analyzer (Fujifilm, Tokyo, Japan). Data are representative of three separate experiments.
References
Shi Y . Serine/threonine phosphatases: mechanism through structure. Cell 2009; 139: 468–484.
Brautigan DL . Protein Ser/Thr phosphatases—the ugly ducklings of cell signalling. FEBS J 2013; 280: 324–345.
Stefansson B, Ohama T, Daugherty AE, Brautigan DL . Protein phosphatase 6 regulatory subunits composed of ankyrin repeat domains. Biochemistry 2008; 47: 1442–1451.
Bastians H, Ponstingl H . The novel human protein serine/threonine phosphatase 6 is a functional homologue of budding yeast Sit4p and fission yeast ppe1. J Cell Sci 1996; 109: 2865–2874.
Goshima G, Iwasaki O, Obuse C, Yanagida M . The role of Ppe1/PP6 phosphatase for equal chromosome segregation in fission yeast kinetochore. EMBO J 2003; 22: 2752–2763.
Luke MM, Della Seta F, Di Como CJ, Sugimoto H, Kobayashi R, Arndt KT . The SAP, a new family of proteins, associate and function positively with the SIT4 phosphatase. Mol Cell Biol 1996; 16: 2744–2755.
Sutton A, Immanuel D, Arndt KT . The SIT4 protein phosphatase functions in late G1 for progression into S phase. Mol Cell Biol 1991; 11: 2133–2148.
Afshar K, Werner ME, Tse YC, Glotzer M, Gönczy P . Regulation of cortical contractility and spindle positioning by the protein phosphatase 6 PPH-6 in one-cell stage C. elegans embryos. Development 2010; 137: 237–247.
Kajino T, Ren H, Iemura S, Natsume T, Stefansson B, Brautigan DL et al. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J Biol Chem 2006; 281: 39891–39896.
Stefansson B, Brautigan DL . Protein phosphatase 6 subunit with conserved Sit4-associated protein domain targets IκBɛ. J Biol Chem 2006; 281: 22624–22634.
Mi J, Dziegielewski J, Bolesta E, Brautigan DL, Larner JM . Activation of DNA-PK by ionizing radiation is mediated by protein phosphatase 6. PLoS ONE 2009; 4: e4395.
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP et al. A landscape of driver mutations in melanoma. Cell 2012; 150: 251–263.
Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012; 44: 1006–1014.
Abel EL, Angel JM, Kiguchi K, DiGiovanni J . Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc 2009; 4: 1350–1362.
Suganuma M, Fujiki H, Suguri H, Yoshizawa S, Hirota M, Nakayasu M et al. Okadaic acid: an additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc Natl Acad Sci USA 1988; 85: 1768–1771.
Fujiki H, Suganuma M . Tumor promoters - microcystin-LR, nodularin and TNF-α and human cancer development. Anticancer Agents Med Chem 2011; 11: 4–18.
Suganuma M, Okabe S, Marino MW, Sakai A, Sueoka E, Fujiki H . Essential role of tumor necrosis factor alpha (TNF-alpha) in tumor promotion as revealed by TNF-alpha-deficient mice. Cancer Res 1999; 59: 4516–4518.
Komori A, Suganuma M, Okabe S, Zou X, Tius MA, Fujiki H . Canventol inhibits tumor promotion in CD-1 mouse skin through inhibition of tumor necrosis factor alpha release and of protein isoprenylation. Cancer Res 1993; 53: 3462–3464.
Schioppa T, Moore R, Thompson RG, Rosser EC, Kulbe H, Nedospasov S et al. B regulatory cells and the tumor-promoting actions of TNF-α during squamous carcinogenesis. Proc Natl Acad Sci USA 2011; 108: 10662–10667.
Omori E, Morioka S, Matsumoto K, Ninomiya-Tsuji J . TAK1 regulates reactive oxygen species and cell death in keratinocytes, which is essential for skin integrity. J Biol Chem 2008; 283: 26161–26168.
Sundberg JP, Sundberg BA, Beamer WG . Comparison of chemical carcinogen skin tumor induction efficacy in inbred, mutant, and hybrid strains of mice: morphologic variations of induced tumors and absence of a papillomavirus cocarcinogen. Mol Carcinog 1997; 20: 19–32.
Kolář M, Szabo P, Dvořánková B, Lacina L, Gabius HJ, Strnad H et al. Upregulation of IL-6, IL-8 and CXCL-1 production in dermal fibroblasts by normal/malignant epithelial cells in vitro: immunohistochemical and transcriptomic analyses. Biol Cell 2012; 104: 738–751.
Davalos AR, Coppe JP, Campisi J, Desprez PY . Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev 2010; 29: 273–283.
Ancrile BB, O'Hayer KM, Counter CM . Oncogenic ras-induced expression of cytokines: a new target of anti-cancer therapeutics. Mol Interv 2008; 8: 22–27.
Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G . Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 1993; 7: 812–821.
Valdiglesias V, Giunta S, Fenech M, Neri M, Bonassi S . γH2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat Res 2013; 753: 24–40.
Zhang B, Satoh M, Nishimura N, Suzuki JS, Sone H, Aoki Y et al. Metallothionein deficiency promotes mouse skin carcinogenesis induced by 7,12-dimethylbenz[a]anthracene. Cancer Res 1998; 58: 4044–4046.
Coussens LM, Werb Z . Inflammation and cancer. Nature 2002; 420: 860–867.
Mantovani A, Allavena P, Sica A, Balkwill F . Cancer-related inflammation. Nature 2008; 454: 436–444.
Vandercappellen J, Van Damme J, Struyf S . The role of CXC chemokines and their receptors in cancer. Cancer Lett 2008; 267: 226–244.
Fimmel S, Devermann L, Herrmann A, Zouboulis C . GRO-alpha: a potential marker for cancer and aging silenced by RNA interference. Ann N Y Acad Sci 2007; 1119: 176–189.
Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M . NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol 1999; 19: 2690–2698.
Karin M . Nuclear factor-kappaB in cancer development and progression. Nature 2006; 441: 431–436.
Tachibana K, Scheuer PJ, Tsukitani Y, Kikuchi H, Engen DV, Clardy J et al. Okadaic acid, a cytotoxic polyether from two marine sponges of the genus Halichondria. J Am Chem Soc 1981; 103: 2469–2471.
Suzuki JS, Nishimura N, Zhang B, Nakatsuru Y, Kobayashi S, Satoh M et al. Metallothionein deficiency enhances skin carcinogenesis induced by 7,12-dimethylbenz[a]anthracene and 12-O-tetradecanoylphorbol-13-acetate in metallothionein-null mice. Carcinogenesis 2003; 24: 1123–1132.
Lichti U, Anders J, Yuspa SH . Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc 2008; 3: 799–810.
Masuda K, Katagiri C, Nomura M, Sato M, Kakumoto K, Akagi T et al. MKP-7, a JNK phosphatase, blocks ERK-dependent gene activation by anchoring phosphorylated ERK in the cytoplasm. Biochem Biophys Res Commun 2010; 393: 201–206.
Acknowledgements
We thank Dr Hirota Fujiki for critical advice and Dr Yoshikazu Nishino for statistical analyses. We thank Nozomi Sasaki, Kuniko Komuro and Miyuki Ueki for technical assistance. We thank Dr Elise Lamar for English editing. This work was supported by JSPS KAKENHI grant numbers 24591928 to Yoichiro Kakugawa, 25861168 to Kayoko Fukamachi and 22590298 to Hiroshi Shima and by a Nara Women’s University Intramural Grant for Project Research to Toshio Watanabe.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Hayashi, K., Momoi, Y., Tanuma, N. et al. Abrogation of protein phosphatase 6 promotes skin carcinogenesis induced by DMBA. Oncogene 34, 4647–4655 (2015). https://doi.org/10.1038/onc.2014.398
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2014.398
- Springer Nature Limited
This article is cited by
-
Targeting protein phosphatases for the treatment of inflammation-related diseases: From signaling to therapy
Signal Transduction and Targeted Therapy (2022)
-
PP6 negatively modulates LUBAC-mediated M1-ubiquitination of RIPK1 and c-FLIPL to promote TNFα-mediated cell death
Cell Death & Disease (2022)
-
PpV, acting via the JNK pathway, represses apoptosis during normal development of Drosophila wing
Apoptosis (2018)