

Key Points
-
Approaches to discover genetic causes of congenital anomalies of the kidney and urinary tract (CAKUT) include candidate gene and whole exome sequencing, and genome-wide linkage and copy number variant (CNV) analyses
-
The majority of sporadic cases of CAKUT cannot yet be explained by monogenic causes
-
Certain CNVs are associated with an elevated risk of CAKUT, indicating that CNV analysis should become part of the diagnostic procedure
-
Chromosomal imbalances and single nucleotide variants in non-coding regions contribute to congenital malformations, suggesting that genomic regulatory elements might also function in the pathogenesis of CAKUT
-
Epigenetic and gestational environmental risk factors can influence kidney development and/or fibrosis and might also increase susceptibility to CAKUT
-
Collaborative efforts are needed to collect large cohorts of patients with CAKUT and to integrate the data from epidemiologic, clinical, genomic, epigenomic, transcriptomic, and proteomic studies
Abstract
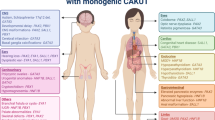
Congenital anomalies of the kidney and urinary tract (CAKUT) refer to a spectrum of structural renal malformations and are the leading cause of end-stage renal disease in children. The genetic diagnosis of CAKUT has proven to be challenging due to genetic and phenotypic heterogeneity and incomplete genetic penetrance. Monogenic causes of CAKUT have been identified using different approaches, including single gene screening, and gene panel and whole exome sequencing. The majority of the identified mutations, however, lack substantial evidence to support a pathogenic role in CAKUT. Copy number variants or single nucleotide variants that are associated with CAKUT have also been identified. Numerous studies support the influence of epigenetic and environmental factors on kidney development and the natural history of CAKUT, suggesting that the pathogenesis of this syndrome is multifactorial. In this Review we describe the current knowledge regarding the genetic susceptibility underlying CAKUT and the approaches used to investigate the genetic basis of CAKUT. We outline the associated environmental risk factors and epigenetic influences on CAKUT and discuss the challenges and strategies used to fully address the involvement and interplay of these factors in the pathogenesis of the disease.


Similar content being viewed by others

Change history
24 November 2015
In the HTML and PDF versions of this article originally published online, the black arrow was missing from the 'Posterior urethral valves' panel in Figure 1. This error has now been corrected in print and online.
References
Brown, T., Mandell, J. & Lebowitz, R. L. Neonatal hydronephrosis in the era of sonography. Am. J. Roentgenol. 148, 959–963 (1987).
Queisser-Luft, A., Stolz, G., Wiesel, A., Schlaefer, K. & Spranger, J. Malformations in newborn: results based on 30,940 infants and fetuses from the Mainz congenital birth defect monitoring system (1990–1998). Arch. Gynecol. Obstet. 266, 163–167 (2002).
Sanna-Cherchi, S. et al. Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr. Nephrol. 22, 1675–1684 (2007).
Pope, J. C. 4th, Brock, J. W. 3rd, Adams, M. C., Stephens, F. D. & Ichikawa, I. How they begin and how they end: classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J. Am. Soc. Nephrol. 10, 2018–2028 (1999).
Vivante, A., Kohl, S., Hwang, D. Y., Dworschak, G. C. & Hildebrandt, F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr. Nephrol. 29, 695–704 (2014).
Schedl, A. Renal abnormalities and their developmental origin. Nat. Rev. Genet. 8, 791–802 (2007).
Blake, J. & Rosenblum, N. D. Renal branching morphogenesis: morphogenetic and signaling mechanisms. Semin. Cell Dev. Biol. 36, 2–12 (2014).
Chesnaye, N. et al. Demographics of paediatric renal replacement therapy in Europe: a report of the ESPN/ERA–EDTA registry. Pediatr. Nephrol. 29, 2403–2410 (2014).
Wuhl, E. et al. Timing and outcome of renal replacement therapy in patients with congenital malformations of the kidney and urinary tract. Clin. J. Am. Soc. Nephrol. 8, 67–74 (2013).
Kerecuk, L., Schreuder, M. F. & Woolf, A. S. Renal tract malformations: perspectives for nephrologists. Nat. Clin. Pract. Nephrol. 4, 312–325 (2008).
Stoll, C., Dott, B., Alembik, Y. & Roth, M. P. Associated nonurinary congenital anomalies among infants with congenital anomalies of kidney and urinary tract (CAKUT). Eur. J. Med. Genet. 57, 322–328 (2014).
Winyard, P. & Chitty, L. S. Dysplastic kidneys. Semin. Fetal Neonatal Med. 13, 142–151 (2008).
Bulum, B. et al. High frequency of kidney and urinary tract anomalies in asymptomatic first-degree relatives of patients with CAKUT. Pediatr. Nephrol. 28, 2143–2147 (2013).
Monn, E. & Nordshus, T. Hereditary renal adysplasia. Acta. Paediatr. Scand. 73, 278–280 (1984).
McPherson, E. et al. Dominantly inherited renal adysplasia. Am. J. Med. Genet. 26, 863–872 (1987).
Kaplan, B. S., Milner, L. S., Jequier, S., Kaplan, P. & de Chadarevian, J. P. Autosomal dominant inheritance of small kidneys. Am. J. Med. Genet. 32, 120–126 (1989).
Doray, B., Gasser, B., Reinartz, I. & Stoll, C. Hereditary renal adysplasia in a three generations family. Genet. Couns. 10, 251–257 (1999).
Sanyanusin, P. et al. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat. Genet. 9, 358–364 (1995).
Horikawa, Y. et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat. Genet. 17, 384–385 (1997).
Weber, S. et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J. Am. Soc. Nephrol. 17, 2864–2870 (2006).
Thomas, R. et al. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr. Nephrol. 26, 897–903 (2011).
Madariaga, L. et al. Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin. J. Am. Soc. Nephrol. 8, 1179–1187 (2013).
Hwang, D. Y. et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 85, 1429–1433 (2014).
Saisawat, P. et al. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int. 81, 196–200 (2012).
Exome Aggregation Consortium (ExAC), http://exac.broadinstitute.org [June 2015 accessed].
Weber, S. et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J. Am. Soc. Nephrol. 19, 891–903 (2008).
Skinner, M. A., Safford, S. D., Reeves, J. G., Jackson, M. E. & Freemerman, A. J. Renal aplasia in humans is associated with RET mutations. Am. J. Hum. Genet. 82, 344–351 (2008).
Gimelli, S. et al. Mutations in SOX17 are associated with congenital anomalies of the kidney and the urinary tract. Hum. Mutat. 31, 1352–1359 (2010).
Vivante, A. et al. Renal hypodysplasia associates with a WNT4 variant that causes aberrant canonical WNT signaling. J. Am. Soc. Nephrol. 24, 550–558 (2013).
Coon, K. D. et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer's disease. J. Clin. Psychiatry 68, 613–618 (2007).
Julier, C. et al. Insulin-IGF2 region on chromosome 11p encodes a gene implicated in HLA-DR4-dependent diabetes susceptibility. Nature 354, 155–159 (1991).
Hall, J. M. et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 250, 1684–1689 (1990).
Williams, G., Fletcher, J. T., Alexander, S. I. & Craig, J. C. Vesicoureteral reflux. J. Am. Soc. Nephrol. 19, 847–862 (2008).
Weng, P. L. et al. A recessive gene for primary vesicoureteral reflux maps to chromosome 12p11-q13. J. Am. Soc. Nephrol. 20, 1633–1640 (2009).
Ashraf, S. et al. Mapping of a new locus for congenital anomalies of the kidney and urinary tract on chromosome 8q24. Nephrol. Dial. Transplant. 25, 1496–1501 (2010).
Sanna-Cherchi, S. et al. Localization of a gene for nonsyndromic renal hypodysplasia to chromosome 1p32–33 Am. J. Hum. Genet. 80, 539–49 (2007).
Chatterjee, R. et al. Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling complex in a cohort of living US patients with urinary tract malformations. Hum. Genet. 131, 1725–1738 (2012).
Kohl, S. et al. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J. Am. Soc. Nephrol. 25, 1917–1922 (2014).
Humbert, C. et al. Integrin alpha 8 recessive mutations are responsible for bilateral renal agenesis in humans. Am. J. Hum. Genet. 94, 288–294 (2014).
Saisawat, P. et al. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int. 85, 1310–1317 (2014).
PhenomeCentral Version 1.0 Milestone-7. A hub for secure data sharing within the rare disorder community [online], (2015).
Swaminathan, G. J. et al. DECIPHER: web-based, community resource for clinical interpretation of rare variants in developmental disorders. Hum. Mol. Genet. 21, R37–44 (2012).
Rehm, H. L. et al. ClinGen - The Clinical Genome Resource. N. Engl. J. Med. (2015).
Fokkema, I. F. et al. LOVD v.2.0: the next generation in gene variant databases. Hum. Mutat. 32, 557–563 (2011).
Matchmaker Exchange. Genomic discovery through the exchange of phenotypic & genotypic profiles [online], (2015).
Sanna-Cherchi, S. et al. Mutations in DSTYK and dominant urinary tract malformations. N. Engl. J. Med. 369, 621–629 (2013).
Vissers, L. E. et al. A de novo paradigm for mental retardation. Nat. Genet. 42, 1109–1112 (2010).
Girard, S. L. et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 43, 860–863 (2011).
O'Roak, B. J. et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589 (2011).
Bower, M. et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 33, 457–466 (2012).
Serra-Juhe, C. et al. Contribution of rare copy number variants to isolated human malformations. PLoS ONE 7, e45530 (2012).
Anad, F. et al. Alagille syndrome and deletion of 20p. J. Med. Genet. 27, 729–737 (1990).
Davies, A. F. et al. Delineation of two distinct 6p deletion syndromes. Hum. Genet. 104, 64–72 (1999).
Kirk, J. M. et al. Unilateral renal aplasia in X-linked Kallmann's syndrome. Clin. Genet. 46, 260–262 (1994).
Mefford, H. C. et al. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am. J. Hum. Genet. 81, 1057–1069 (2007).
Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376 (2011).
de Ligt, J. et al. Detection of clinically relevant copy number variants with whole-exome sequencing. Hum. Mutat. 34, 1439–1448 (2013).
Hoefele, J. et al. A novel interstitial deletion of 10q24.2q24.32 in a patient with renal coloboma syndrome. Eur. J. Med. Genet. 55, 211–215 (2012).
Sanna-Cherchi, S. et al. Copy-number disorders are a common cause of congenital kidney malformations. Am. J. Hum. Genet. 91, 987–997 (2012).
Caruana, G. et al. Copy-number variation associated with congenital anomalies of the kidney and urinary tract. Pediatr. Nephrol. 30, 487–495 (2015).
Uchiyama, Y. et al. Kif26b, a kinesin family gene, regulates adhesion of the embryonic kidney mesenchyme. Proc. Natl Acad. Sci. USA 107, 9240–9245 (2010).
Selleri, L. et al. Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development 128, 3543–3557 (2001).
Vulto-van Silfhout, A. T. et al. An update on ECARUCA, the European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations. Eur. J. Med. Genet. 56, 471–474 (2013).
Riggs, E. R., Jackson, L., Miller, D. T. & Van Vooren, S. Phenotypic information in genomic variant databases enhances clinical care and research: the International Standards for Cytogenomic Arrays Consortium experience. Hum. Mutat. 33, 787–796 (2012).
MacDonald, J. R., Ziman, R., Yuen, R. K., Feuk, L. & Scherer, S. W. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 42, D986–992 (2014).
Ibn-Salem, J. et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 15, 423 (2014).
Kloosterman, W. P. & Hochstenbach, R. Deciphering the pathogenic consequences of chromosomal aberrations in human genetic disease. Mol. Cytogenet. 7, 100 (2014).
Dauber, A. et al. SCRIB and PUF60 are primary drivers of the multisystemic phenotypes of the 8q24.3 copy-number variant. Am. J. Hum. Genet. 93, 798–811 (2013).
Francioli, L. C. et al. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 46, 818–825 (2014).
Gilissen, C. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347 (2014).
Weedon, M. N. et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat. Genet. 46, 61–64 (2014).
Smemo, S. et al. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 21, 3255–3263 (2012).
Li, Q. Y. et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 15, 21–29 (1997).
Kawahara, Y. Human diseases caused by germline and somatic abnormalities in microRNA and microRNA-related genes. Congenit. Anom. (Kyoto) 54, 12–21 (2014).
Drake, K. M. et al. Loss of heterozygosity at 2q37 in sporadic Wilms' tumor: putative role for miR-562. Clin. Cancer Res. 15, 5985–5992 (2009).
Mencia, A. et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat. Genet. 41, 609–613 (2009).
Patel, V. et al. MicroRNAs regulate renal tubule maturation through modulation of Pkd1. J. Am. Soc. Nephrol. 23, 1941–1948 (2012).
Bartram, M. P. et al. Conditional loss of kidney microRNAs results in congenital anomalies of the kidney and urinary tract (CAKUT). J. Mol. Med. 91, 739–748 (2013).
Ho, J. et al. Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J. Am. Soc. Nephrol. 19, 2069–2075 (2008).
Sun, H. et al. MicroRNA-17 post-transcriptionally regulates polycystic kidney disease-2 gene and promotes cell proliferation. Mol. Biol. Rep. 37, 2951–2958 (2010).
Patel, V. et al. miR-17∼92 miRNA cluster promotes kidney cyst growth in polycystic kidney disease. Proc. Natl Acad. Sci. USA 110, 10765–10770 (2013).
Thiagarajan, R. D. et al. Refining transcriptional programs in kidney development by integration of deep RNA-sequencing and array-based spatial profiling. BMC Genomics 12, 441 (2011).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Cordell, H. J. et al. Whole-genome linkage and association scan in primary, nonsyndromic vesicoureteric reflux. J. Am. Soc. Nephrol. 21, 113–123 (2010).
van Eerde, A. M. et al. Genes in the ureteric budding pathway: association study on vesico-ureteral reflux patients. PLoS ONE 7, e31327 (2012).
Djuric, T. et al. MMP-1 and -3 haplotype is associated with congenital anomalies of the kidney and urinary tract. Pediatr. Nephrol. 29, 879–884 (2014).
Reis, G. S. et al. Study of the association between the BMP4 gene and congenital anomalies of the kidney and urinary tract. J. Pediatr. (Rio J.) 90, 58–64 (2014).
Miranda, D. M. et al. Association of angiotensin type 2 receptor gene polymorphisms with ureteropelvic junction obstruction in Brazilian patients. Nephrology (Carlton) 19, 714–720 (2014).
Darlow, J. M. et al. A new genome scan for primary nonsyndromic vesicoureteric reflux emphasizes high genetic heterogeneity and shows linkage and association with various genes already implicated in urinary tract development. Mol. Genet. Genomic Med. 2, 7–29 (2014).
van der Zanden, L. F. et al. Common variants in DGKK are strongly associated with risk of hypospadias. Nat. Genet. 43, 48–50 (2011).
Genovese, G. et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845 (2010).
Kopp, J. B. et al. Clinical features and histology of apolipoprotein L1-associated nephropathy in the FSGS clinical trial. J. Am. Soc. Nephrol. (2015).
Kottgen, A. et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet. 41, 712–717 (2009).
Kottgen, A. et al. New loci associated with kidney function and chronic kidney disease. Nat. Genet. 42, 376–384 (2010).
Woroniecki, R., Gaikwad, A. B. & Susztak, K. Fetal environment, epigenetics, and pediatric renal disease. Pediatr. Nephrol. 26, 705–711 (2011).
Parikh, C. R., McCall, D., Engelman, C. & Schrier, R. W. Congenital renal agenesis: case-control analysis of birth characteristics. Am. J. Kidney Dis. 39, 689–694 (2002).
Hsu, C. W., Yamamoto, K. T., Henry, R. K., De Roos, A. J. & Flynn, J. T. Prenatal risk factors for childhood CKD. J. Am. Soc. Nephrol. 25, 2105–2111 (2014).
Dart, A. B., Ruth, C. A., Sellers, E. A., Au, W. & Dean, H. J. Maternal diabetes mellitus and congenital anomalies of the kidney and urinary tract (CAKUT) in the child. Am. J. Kidney Dis. 65, 684–691 (2015).
Amri, K., Freund, N., Vilar, J., Merlet-Benichou, C. & Lelievre-Pegorier, M. Adverse effects of hyperglycemia on kidney development in rats: in vivo and in vitro studies. Diabetes 48, 2240–2245 (1999).
Hoppe, C. C., Evans, R. G., Bertram, J. F. & Moritz, K. M. Effects of dietary protein restriction on nephron number in the mouse. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1768–R1774 (2007).
Wilkinson, L. J. et al. Renal developmental defects resulting from in utero hypoxia are associated with suppression of ureteric beta-catenin signaling. Kidney Int. 87, 975–983 (2015).
Feil, R. & Fraga, M. F. Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 13, 97–109 (2011).
Oberlander, T. F. et al. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 3, 97–106 (2008).
Teh, A. L. et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 24, 1064–1074 (2014).
Sun, G. et al. Epigenetic histone methylation modulates fibrotic gene expression. J. Am. Soc. Nephrol. 21, 2069–2080 (2010).
Ko, Y. A. et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 14, R108 (2013).
Patel, S. R., Kim, D., Levitan, I. & Dressler, G. R. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev. Cell 13, 580–592 (2007).
Lefevre, G. M., Patel, S. R., Kim, D., Tessarollo, L. & Dressler, G. R. Altering a histone H3K4 methylation pathway in glomerular podocytes promotes a chronic disease phenotype. PLoS Genet. 6, e1001142 (2010).
Castillo-Fernandez, J. E., Spector, T. D. & Bell, J. T. Epigenetics of discordant monozygotic twins: implications for disease. Genome Med. 6, 60 (2014).
Jin, M. et al. Genomic and epigenomic analyses of monozygotic twins discordant for congenital renal agenesis. Am. J. Kidney Dis. 64, 119–122 (2014).
Gribouval, O. et al. Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum. Mutat. 33, 316–326 (2012).
Gribouval, O. et al. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat. Genet. 37, 964–968 (2005).
Weber, S. et al. Muscarinic acetylcholine receptor M3 mutation causes urinary bladder disease and a prune-belly-like syndrome. Am. J. Hum. Genet. 89, 668–674 (2011).
Alazami, A. M. et al. FREM1 mutations cause bifid nose, renal agenesis, and anorectal malformations syndrome. Am. J. Hum. Genet. 85, 414–418 (2009).
Vogel, M. J. et al. Mutations in GRIP1 cause Fraser syndrome. J. Med. Genet. 49, 303–306 (2012).
Daly, S. B. et al. Mutations in HPSE2 cause urofacial syndrome. Am. J. Hum. Genet. 86, 963–969 (2010).
Li, Y. et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am. J. Hum. Genet. 86, 696–706 (2010).
Tufan, F. et al. Clinical and molecular characterization of two adults with autosomal recessive Robinow syndrome. Am. J. Med. Genet. A 136, 185–189 (2005).
Kraus, M. R. et al. Two mutations in human BICC1 resulting in Wnt pathway hyperactivity associated with cystic renal dysplasia. Hum. Mutat. 33, 86–90 (2012).
Tabatabaeifar, M. et al. Functional analysis of BMP4 mutations identified in pediatric CAKUT patients. Pediatr. Nephrol. 24, 2361–2368 (2009).
Unger, S. et al. Mutations in the cyclin family member FAM58A cause an X-linked dominant disorder characterized by syndactyly, telecanthus and anogenital and renal malformations. Nat. Genet. 40, 287–289 (2008).
Trarbach, E. B. et al. Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. J. Clin. Endocrinol. Metab. 95, 3491–3496 (2010).
Dode, C. et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 33, 463–465 (2003).
Ali, A. et al. Functional characterization of GATA3 mutations causing the hypoparathyroidism-deafness-renal (HDR) dysplasia syndrome: insight into mechanisms of DNA binding by the GATA3 transcription factor. Hum. Mol. Genet. 16, 265–275 (2007).
Johnston, J. J. et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am. J. Hum. Genet. 76, 609–622 (2005).
Heidet, L. et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin. J. Am. Soc. Nephrol. 5, 1079–1090 (2010).
Barbacci, E. et al. HNF1beta/TCF2 mutations impair transactivation potential through altered co-regulator recruitment. Hum. Mol. Genet. 13, 3139–3149 (2004).
Guegan, K., Stals, K., Day, M., Turnpenny, P. & Ellard, S. JAG1 mutations are found in approximately one third of patients presenting with only one or two clinical features of Alagille syndrome. Clin. Genet. 82, 33–40 (2012).
Albuisson, J. et al. Kallmann syndrome: 14 novel mutations in KAL1 and FGFR1 (KAL2). Hum. Mutat. 25, 98–99 (2005).
Kamath, B. M. et al. NOTCH2 mutations in Alagille syndrome. J. Med. Genet. 49, 138–144 (2012).
Favor, J. et al. The mouse Pax2(1Neu) mutation is identical to a human PAX2 mutation in a family with renal-coloboma syndrome and results in developmental defects of the brain, ear, eye, and kidney. Proc. Natl Acad. Sci. USA 93, 13870–13875 (1996).
Meeus, L. et al. Characterization of a novel loss of function mutation of PAX8 in a familial case of congenital hypothyroidism with in-place, normal-sized thyroid. J. Clin. Endocrinol. Metab. 89, 4285–4291 (2004).
Bertoli-Avella, A. M. et al. ROBO2 gene variants are associated with familial vesicoureteral reflux. J. Am. Soc. Nephrol. 19, 825–831 (2008).
Yoshida, Y. et al. Increased levels of pigment epithelium-derived factor in aqueous humor of patients with uveitis. Br. J. Ophthalmol. 91, 149–150 (2007).
Hanchate, N. K. et al. SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. 8, e1002896 (2012).
Ruf, R. G. et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc. Natl Acad. Sci. USA 101, 8090–8095 (2004).
Hoskins, B. E. et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am. J. Hum. Genet. 80, 800–804 (2007).
Li, H., Sheridan, R. & Williams, T. Analysis of TFAP2A mutations in Branchio-Oculo-Facial Syndrome indicates functional complexity within the AP-2α DNA-binding domain. Hum. Mol. Genet. 22, 3195–3206 (2013).
Jenkins, D. et al. De novo Uroplakin IIIa heterozygous mutations cause human renal adysplasia leading to severe kidney failure. J. Am. Soc. Nephrol. 16, 2141–2149 (2005).
Acknowledgements
The authors would like to thank Realexis Koutsavaki Christofides for modelling the congenital abnormalities of the kidney and urinary tract phenotypes in a 3D format and kindly providing the images shown in Figure 1. The authors' research is supported by grants from the European Community's Seventh Framework Programme FP7/2009 under grant agreement 305608 (EURenOmics).
Author information
Authors and Affiliations
Contributions
N.N. researched the data for and wrote the article. All authors provided a substantial contribution to discussion of the content and to review and/or editing of the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Nicolaou, N., Renkema, K., Bongers, E. et al. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol 11, 720–731 (2015). https://doi.org/10.1038/nrneph.2015.140
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneph.2015.140
- Springer Nature Limited
This article is cited by
-
GEN1 as a risk factor for human congenital anomalies of the kidney and urinary tract
Human Genomics (2024)
-
Plasma MCP-1 and TGF-β1 Levels are Associated with Kidney Injury in Children with Congenital Anomalies of the Kidney and Urinary Tract
Applied Biochemistry and Biotechnology (2024)
-
Integrated analysis of copy number variation-associated lncRNAs identifies candidates contributing to the etiologies of congenital kidney anomalies
Communications Biology (2023)
-
Acetyl-CoA is a key molecule for nephron progenitor cell pool maintenance
Nature Communications (2023)
-
Sex difference and risk factors in burden of urogenital congenital anomalies from 1990 to 2019
Scientific Reports (2023)