
Abstract
Genetic testing for germline mutations in breast cancer predisposition genes can potentially identify individuals at a high risk of developing breast and/or ovarian cancer. There is a paucity of such mutational information for Asians. Panel testing of 25 cancer susceptibility genes and BRCA1/2 deletion/duplication analysis was performed for 220 Asian breast cancer patients or their family members referred for genetics risk assessment. All 220 participants had at least one high-risk feature: having a family history of breast and/or ovarian cancer in first- and/or second-degree relatives; having breast and ovarian cancer in the same individual or bilateral breast cancer; having early-onset breast cancer or ovarian cancer (⩽40 years of age). We identified 67 pathogenic variants in 66 (30.0%) patients. Of these, 19 (28.3%) occurred in BRCA1, 16 (23.9%) in BRCA2, 7 (10.4%) in PALB2, 6 (9.0%) in TP53, 2 (3.0%) in PTEN, 2 (3.0%) in CDH1 and 15 (22.4%) in other predisposition genes. Notably, 47.8% of pathogenic variants were in non-BRCA1/2 genes. Of the 66 patients with pathogenic mutations, 63.6% (42/66) were under the age of 40 years. Family history of breast and/or ovarian cancer is enriched in patients with BRCA1/2 pathogenic variants but less predictive for non-BRCA1/2 related pathogenic variations. We detected a median of three variants of unknown significance (VUS) per gene (range 0–21). Custom gene panel testing is feasible and useful for the detection of pathogenic mutations and should be done in the setting of a formal clinical cancer genetics service given the rate of VUS.
Similar content being viewed by others

INTRODUCTION
In this era of precision medicine, gene-directed risk stratification and management is a common aspiration for modern clinical practice.1 This is reflected in the U.S. Department of Health and Human Services’ genomic objectives of Healthy People 2020 emphasising the importance of obtaining a family and genetic history as a potential and powerful guide for clinical and public health initiatives. The first genomic recommendation is that women with a family history of breast or ovarian cancer should receive genetic counselling. These genomic recommendations are based on the premise that gene-enabled management could improve health outcomes of affected individuals and allow family members to make proactive choices with their health. Indeed, at the recently launched BRCA Challenge at UNESCO, global expert faculty met to discuss ways to expedite this process through data sharing and to address the urgent need for data from diverse populations such as ours (http://www.unesco.org/new/en/media-services/single-view/news/breast_cancer_brca_challenge_officially_launched/).
Breast cancer susceptibility is associated with germline mutations in several genes such as BRCA1, BRCA2, PTEN, TP53, PALB2, CDH1 and STK11, and genes of moderate penetrance like ATM and CHEK2.2 Next-generation sequencing (NGS) technology has enabled panel based genetic testing to the clinic, providing cost savings and the ability to test many genes simultaneously.3 However, the disadvantage of panel testing is the increased probability of encountering a germline VUS. This is particularly problematic in minority populations where there is less data available and/or in regions where the uptake of testing has been traditionally slow, such as in Asia. We present here the largest study undertaken to assess the use of NGS panel testing for breast cancer susceptibility genes in an Asian multi-racial cohort of patients referred for genetic risk assessment in Singapore.
RESULTS
Study population
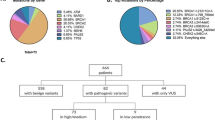
Patients suspected of hereditary breast cancer in this study were referred from Singapore and the region for genetic risk assessment at the National Cancer Centre Singapore. Of the patients with established ethnicity, 181 (82.3%) were Chinese, 17 (7.7%) Malay, and 6 (2.7%) of South Indian descent (Table 1). The remaining 16 (7.3%) were of Burmese, Eurasian, Japanese, Filipino, Vietnamese and other races, respectively. Age at diagnosis of patients with breast and/or ovarian cancer ranged from 19 to 72 years, with an average age of 39 years. Of the 120 patients with available family history information, 104 (86.7%) had at least one first- or second-degree relative with breast cancer, and 16 (13.3%) had a relative with ovarian cancer.
Germline mutations
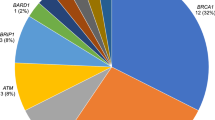
All coding exons and consensus splice sites of 25 known cancer predisposition genes were screened for mutations in the 220 patients. Overall, 67 pathogenic mutations were identified in 66 patients (30.0% (66/220); Table 2). Eight mutations were detected in more than 1 patient, and 10 patients were carriers for more than one mutation (Table 2). Of these, 19 (28.4%) occurred in BRCA1, 16 (23.9%) in BRCA2, 7 (10.4%) in PALB2, 6 (9.0%) in TP53, 2 (3.0%) in PTEN, 2 (3.0%) in CDH1 and 15 (22.4%) in other predisposition genes (Table 2; Figures 1 and 2). Deleterious BRCA1 mutations were detected in 10.5% (23/220) of patients, including 15 truncating (frameshift, nonsense and splice, large deletion/duplication) mutations and 3 known deleterious missense mutations and 1 novel missense mutation. The 16 deleterious BRCA2 mutations (7.7% (17/220)) included 12 truncating mutations, and 4 predicted deleterious missense mutations. Likely deleterious mutations in non-BRCA1/2 predisposition genes were identified in 14.5% of all tested patients (32/220) in the following genes ATM, BARD1, BRIP1, CDH1, CDKN2A, CHEK2, MLH1, MSH6, NF1, PALB2, PMS2, PTEN, RAD51C, RAD51D and TP53. A total of 28 novel potentially pathogenic variants were detected in BRCA1, BRCA2, PALB2, TP53, PTEN, NF1, CDH1, MSH6 and PMS2 (Table 2) by our group in this study and previous studies.4–6

Pathogenic variants detected in 17 genes.

Pie-chart showing the percentage of mutations across the 25 genes.
The mean Manchester score among cases with deleterious mutations was 19.4 (range 1–75) which was higher as compared to cases with no deleterious mutations (mean 9.7; range 1–71). Manchester scores were available for 56 of 66 individuals with deleterious mutations, and 124 of 154 individuals with no mutations.
Family history
We also evaluated whether patients with mutations in the 25 predisposition genes were associated with a greater family history of breast and/or ovarian cancers than non-mutated patient cases (Table 2). Patients with BRCA1 mutations were enriched for a family history of breast (5/23 (21.7%)) and ovarian cancers (2/23 (8.7%)), whereas patient cases with BRCA2 mutations were enriched for a family history of breast (7/17 (41.2%)) but none of the family members had ovarian cancers. (Table 2). This is reflected in the differences in Manchester and Boadicea scores seen between the two groups of patients (Table 3). However, patient cases with mutations in the non-BRCA1/2 genes were not significantly associated with an enriched family history for either breast or ovarian cancer (Table 2). In particular, only 8 (24.2% (8/33)) non-BRCA1/2 gene mutation carriers had a family history of breast or ovarian cancer.
Variants of unknown significance
A total of 94 VUS were identified in 23 genes in 96 of 220 participants. Per participant, the average number of VUS across all genes was 0.67 (s.d., 0.9) (Figure 3a). Of the 220 participants, 103 (46.8%) had at least one VUS among the 25 genes sequenced. Per gene, the median number of VUS detected across all 220 participants was 3, ranging from zero (PTEN and NBN) to 21 (ATM; Figure 3b). Among the 7 high-risk genes, 10 VUS were found in BRCA1, 15 in BRCA2, 10 in PALB2, 2 in CDH1, 2 in STK11, 1 in TP53 and none in PTEN. In the remaining 18 genes, a median of 3.5 VUS per gene (range 0–21) were detected. All VUS were missense mutations and within exonic regions. Of the 94 VUS, 41 (43.6%) were novel, not previously reported in the databases or dbSNP. No statistically significant difference was detected in VUS frequency between ethnicities.

Frequency of variants of uncertain significance (VUS). (a) per participant, across 25 sequenced genes; and (b) per gene, across 220 participants.
DISCUSSION
We present here a comprehensive mutation analysis of Asian patients suspected of having hereditary breast cancer. To our knowledge, this is the largest Asian series to date for the NGS screening of germline mutations using a panel of known breast cancer predisposition genes. We found 67 germline deleterious mutations in 17 of 25 predisposition genes tested. BRCA1 and BRCA2 mutations were found in 17.7% (39/220) of patients, consistent with other studies using panel testing, whereas mutations in 15 other genes were found in 32 (14.5%) patients. The frequency of these mutations, especially in PALB2, which has recently been associated with a high lifetime risk of breast cancer, was similar to the frequency in high- and moderate-risk breast cancer families.7 This is a significant higher yield of potentially actionable results, compared with the 5 to 10% probability threshold endorsed by guidelines for testing for HBOC and Lynch syndrome testing.
In Asia and many parts of the world, while there is a growing appreciation for the testing of patients identified as being at high risk of hereditary cancer, it is still not as yet ‘mainstream’ practice, as such patients are often referred after the development of multiple cancers in a patient. This may account for the relatively high number of TP53 (9.0%) and PTEN (3.0%) germline mutations seen in our cohort. Notably, only 63.6% (42/66) of patients with pathogenic variants were under the age of 40 years at the age of first cancer diagnosis, suggesting that age alone as a cut-off may miss significant numbers of patients (Table 2).
Currently, there is no data as yet on the risk-benefit ratio of increased breast surveillance among patients with pathogenic variants in genes of moderate penetrance (e.g., CHEK2, ATM and BLM). There is remaining uncertainty in penetrance estimates for such variants, and, therefore, the optimal breast screening protocol and age of initiation remain unknown thus limiting the clinical utility of panel testing (for the present) to highly penetrant mutations. To better understand the role of these moderately penetrant genes will require population-based studies of mutation penetrance and clinical trials of risk-reducing interventions to guide clinical decisions. It is a major concern that while the practice of clinical cancer genetics is largely limited in developed countries to trained clinical cancer geneticists, this is not the case for the rest of the world.
The discovery of VUS that do not contribute to risk, may prompt anxiety and overtreatment particularly if the managing clinician is unfamiliar with genetics. Although our experience of finding ~3 VUS per gene is consistent with that from other studies,8 it also highlights the fact that the more we sequence, the more VUS we will unravel. This is particularly so in a population like Singapore, where we have multi-ethnic minority groups for whom there is limited publicly available sequencing data for variant reclassification. In the present study, consistent with our IRB–approved protocol, we did not re-contact any patient about VUS as there are no immediate clinical implications or recommendations to convey. In the clinical setting, where VUS results will be reported back to the patient, it is critical therefore that multigene panel testing is conducted in a dedicated genetics service with a genetics team familiar with cancer risk assessment and who are able to provide adequate pretest and post-test counselling.9
This study was conducted within a formal clinical cancer genetics practice adherent to evidence-based testing guidelines, and using the definition of pathogenic variants as recommended by the American College of Medical Genetics.10 With the clinical availability of multiple-gene sequencing panels and the concurrent decreasing cost of panel testing, it is anticipated that an increased demand for such gene-directed risk stratification will occur. These genetic testing costs are borne by the patient and not by any third-party payer, especially in Asian countries with no insurance coverage or government subsidies for genetic testing for most countries at present. With the reducing costs of genetic testing, many of these health policies are ripe for review if we wish to harness the power of gene-enabled care.
Our study has limitations. The 25 genes that we selected reflect published literature but an optimal multiple-gene panel for routine diagnostic use remains to be defined. Patients were enrolled from within a specialized clinical cancer genetics service and do not reflect general oncology practice nor the general population at large.
To the best of our knowledge, our study is the first to describe multiple-gene testing in an Asian setting within a formal clinical cancer genetics service. Although further research is required to guide practice, our study may help provide a framework for the clinical relevance of multiple-gene sequencing in cancer-risk assessment for other nascent centres in Asia embarking on multigene testing for patients referred for hereditary breast and ovarian cancer syndrome.
Materials and Methods
Patients
We studied 220 cases referred to the Cancer Genetics Service at the National Cancer Centre Singapore. Of these, 210 had a personal history of breast and/or ovarian cancer (192 had breast cancer, 9 had ovarian cancer, and 9 had breast and ovarian cancer). The subjects fulfilled at least one of the following criteria: (1) having a family history of breast and/or ovarian cancer in first- and/or second-degree relatives; (2) having breast and ovarian cancer in the same individual or bilateral breast cancer; (3) having early-onset breast cancer or ovarian cancer (⩽40 years of age). Clinical information including personal and family cancer histories, cancer histology and receptor status, were retrieved from case notes and clinical databases. All patients consented to participate in this study, which was approved by the SingHealth Centralized Institutional Review Board (CIRB 2008/435/B; CIRB 2010/406/B).
Mutation detection using next-generation sequencing (NGS)
An optimised in-house method was used to extract DNA from peripheral blood.5,11 Capture was performed using the SureSelect XT2 target enrichment kit (Agilent, Santa Clara, CA, USA), targeting 25 genes (Supplementary Table 1). The Covaris S2 system (Covaris, Woburn, MA, USA) was used to fragment the genomic DNA samples as recommended by the manufacturer. The exome-enriched libraries were sequenced on the Illumina HiSeq platform (San Diego, CA, USA), with 100-bp paired-end reads.
Deletion/duplication analysis
Detection of large genomic rearrangements in the BRCA1 and BRCA2 genes was done for all 220 samples using the Multiplex Ligation-dependent Probe Amplification test kits (P002-C2 BRCA1 and P045-BRCA2/CHEK2) and confirmation kits (P087-BRCA1 and P077-BRCA2; MRC-Holland, Amsterdam, Netherlands). DNA fragment analysis was performed on the ABI 3130 Genetic Analyzer (ABI-Life Technologies, Thermo Fisher Scientific Corporation, MA, USA) and analysed using the Coffalyser freeware v.131123.1303 (MRC-Holland).
Bioinformatic analysis
The raw reads were aligned to the hg19 reference genome using BWA.12 BAM files were processed to identify variants using the Genome Analysis Tool kit. The variants were annotated using the ANNOVAR tool.12 The mean depth of coverage was ×315 (range: ×97–858). Population allele frequencies were extracted from the Exome Variant Server (http://evs.gs.washington.edu/EVS), 1000 Genomes (http://www.1000genomes.org), and dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP). Frameshift and nonsense mutations were considered to be deleterious. Missense variants were classified as damaging or benign using predictions from SIFT,13 PolyPhen-II HDIV,14 PolyPhen-II HVAR,14 LRT and Mutation Taster.15 If three or more of the five tools predicted the missense mutation to be damaging, then the mutation was classified as damaging. All deleterious or damaging variants were verified visually using the Integrative Genomics Viewer (IGV; Broad Institute), and collectively classified as pathogenic variants.
Variants that were synonymous, or classified as benign, unknown, uncertain or unspecified in the Breast Cancer Information Core, HGMD, ClinVar databases, were excluded. Also excluded were variants with an allele frequency greater than 1% as documented in the Exome Variant Server, 1000 Genomes, dbSNP and ExAC databases. All remaining variants were classified as VUS, and were verified visually using IGV.
Validation of variants detected by NGS
All frameshift, nonsense and damaging missense mutations were validated by Sanger sequencing. PCR amplification using HotstarTaq (Qiagen, Hilden, Germany) using primers flanking mutations was performed as previously described.11 The BigDye Terminator v3.1 cycle sequencing kit (ABI-Life Technologies, Thermo Fisher Scientific Corporation) was used for the incorporation of dye-labelled dNTPs followed by Sanger sequencing using a 3130xl Genetic Analyzer (ABI-Life Technologies, Thermo Fisher Scientific Corporation). The chromatograms were visualised using the Seqman Pro v.12 (Lasergene; DNASTAR, Madison, WI, USA) software.
Statistical analysis
Participant characteristics and sequencing results were tabulated, with descriptive statistics including medians, means and ranges.
References
Easton, D. F., Pharoah, P. D., Antoniou, A. C., Tischkowitz, M., Tavtigian, S. V., Nathanson, K. L. et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N Engl J Med 372, 2243–2257 (2015).
Rahman, N. Realizing the promise of cancer predisposition genes. Nature 505, 302–308 (2014).
LaDuca, H., Stuenkel, A. J., Dolinsky, J. S., Keiles, S., Tandy, S., Pesaran, T. et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med 16, 830–837 (2014).
Lee, A. S. & Ang, P. Breast-cancer risk in families with mutations in PALB2. N Engl J Med 371, 1650–1651 (2014).
Ang, P., Lim, I. H., Lee, T. C., Luo, J. T., Ong, D. C., Tan, P. H. et al. BRCA1 and BRCA2 mutations in an Asian clinic-based population detected using a comprehensive strategy. Cancer Epidemiol Biomarkers Prev 16, 2276–2284 (2007).
Wong, E. S., Shekar, S., Chan, C. H., Hong, L. Z., Poon, S. Y., Silla, T. et al. Predictive Factors for BRCA1 and BRCA2 Genetic Testing in an Asian Clinic-Based Population. PloS one 10, e0134408 (2015).
Antoniou, A. C., Casadei, S., Heikkinen, T., Barrowdale, D., Pylkas, K., Roberts, J. et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med 371, 497–506 (2014).
Kurian, A. W., Hare, E. E., Mills, M. A., Kingham, K. E., McPherson, L., Whittemore, A. S. et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol 32, 2001–2009 (2014).
Hall, M. J., Forman, A. D., Pilarski, R., Wiesner, G. & Giri, V. N. Gene panel testing for inherited cancer risk. J Natl Compr Canc Netw 12, 1339–1346 (2014).
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–423 (2015).
Chan, M., Ji, S. M., Yeo, Z. X., Gan, L., Yap, E., Yap, Y. S. et al. Development of a next-generation sequencing method for BRCA mutation screening: a comparison between a high-throughput and a benchtop platform. J Mol Diagn 14, 602–612 (2012).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081 (2009).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7, Unit7 20 (2013).
Schwarz, J. M., Rodelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7, 575–576 (2010).
Roa, B. B., Boyd, A. A., Volcik, K. & Richards, C. S. Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nature genetics 14, 185–187 (1996).
Bar-Sade, R. B., Kruglikova, A., Modan, B., Gak, E., Hirsh-Yechezkel, G., Theodor, L. et al. The 185delAG BRCA1 mutation originated before the dispersion of Jews in the diaspora and is not limited to Ashkenazim. Human molecular genetics 7, 801–805 (1998).
Simard, J., Tonin, P., Durocher, F., Morgan, K., Rommens, J., Gingras, S. et al. Common origins of BRCA1 mutations in Canadian breast and ovarian cancer families. Nature genetics 8, 392–398 (1994).
Struewing, J. P., Abeliovich, D., Peretz, T., Avishai, N., Kaback, M. M., Collins, F. S. et al. The carrier frequency of the BRCA1 185delAG mutation is approximately 1 percent in Ashkenazi Jewish individuals. Nature genetics 11, 198–200 (1995).
Elit, L., Jack, E., Kwan, E., Baigal, G. & Narod, S. A unique BRCA1 mutation identified in Mongolia. International journal of gynecological cancer: official journal of the International Gynecological Cancer Society 11, 241–243 (2001).
Lee, M. S., Green, R., Marsillac, S. M., Coquelle, N., Williams, R. S., Yeung, T. et al. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer research 70, 4880–4890 (2010).
Robertson, L., Hanson, H., Seal, S., Warren-Perry, M., Hughes, D., Howell, I. et al. BRCA1 testing should be offered to individuals with triple-negative breast cancer diagnosed below 50 years. British journal of cancer 106, 1234–1238 (2012).
Castilla, L. H., Couch, F. J., Erdos, M. R., Hoskins, K. F., Calzone, K., Garber, J. E. et al. Mutations in the BRCA1 gene in families with early-onset breast and ovarian cancer. Nature genetics 8, 387–391 (1994).
Friedman, L. S., Ostermeyer, E. A., Szabo, C. I., Dowd, P., Lynch, E. D., Rowell, S. E. et al. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nature genetics 8, 399–404 (1994).
Szabo, C., Masiello, A., Ryan, J. F. & Brody, L. C. The breast cancer information core: database design, structure, and scope. Human mutation 16, 123–131 (2000).
Ho, G. H., Phang, B. H., Ng, I. S., Law, H. Y., Soo, K. C. & Ng, E. H. Novel germline BRCA1 mutations detected in women in singapore who developed breast carcinoma before the age of 36 years. Cancer 89, 811–816 (2000).
Khoo, A. S., Balraj, P., Volpi, L. & Nair, S. A new BRCA1 germline mutation (E879X) in a Malaysian breast cancer patient of Chinese descent. Human mutation 15, 485 (2000).
Al-Sukhni, W., Rothenmund, H., Borgida, A. E., Zogopoulos, G., O'Shea, A. M., Pollett, A. et al. Germline BRCA1 mutations predispose to pancreatic adenocarcinoma. Human genetics 124, 271–278 (2008).
Tavtigian, S. V., Simard, J., Rommens, J., Couch, F., Shattuck-Eidens, D., Neuhausen, S. et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nature genetics 12, 333–337 (1996).
Foster, K. A., Harrington, P., Kerr, J., Russell, P., DiCioccio, R. A., Scott, I. V. et al. Somatic and germline mutations of the BRCA2 gene in sporadic ovarian cancer. Cancer research 56, 3622–3625 (1996).
Lubinski, J., Phelan, C. M., Ghadirian, P., Lynch, H. T., Garber, J., Weber, B. et al. Cancer variation associated with the position of the mutation in the BRCA2 gene. Familial cancer 3, 1–10 (2004).
Vehmanen, P., Friedman, L. S., Eerola, H., McClure, M., Ward, B., Sarantaus, L. et al. Low proportion of BRCA1 and BRCA2 mutations in Finnish breast cancer families: evidence for additional susceptibility genes. Human molecular genetics 6, 2309–2315 (1997).
Lim, M. C., Kang, S., Seo, S. S., Kong, S. Y., Lee, B. Y., Lee, S. K. et al. BRCA1 and BRCA2 germline mutations in Korean ovarian cancer patients. Journal of cancer research and clinical oncology 135, 1593–1599 (2009).
Bodian, D. L., McCutcheon, J. N., Kothiyal, P., Huddleston, K. C., Iyer, R. K., Vockley, J. G. et al. Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing. PloS one 9, e94554 (2014).
Yeo, Z. X., Wong, J. C., Rozen, S. G. & Lee, A. S. Evaluation and optimisation of indel detection workflows for ion torrent sequencing of the BRCA1 and BRCA2 genes. BMC genomics 15, 516 (2014).
Consortium SNIBB. BRCA1 and BRCA2 mutations in Scotland and Northern Ireland. British journal of cancer 88, 1256–62 (2003).
Eeles, R. A. Germline mutations in the TP53 gene. Cancer surveys 25, 101–124 (1995).
Toguchida, J., Yamaguchi, T., Dayton, S. H., Beauchamp, R. L., Herrera, G. E., Ishizaki, K. et al. Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med 326, 1301–1308 (1992).
Monti, P., Ciribilli, Y., Jordan, J., Menichini, P., Umbach, D. M., Resnick, M. A. et al. Transcriptional functionality of germ line p53 mutants influences cancer phenotype. Clinical cancer research: an official journal of the American Association for Cancer Research 13, 3789–3795 (2007).
Bianchi, F., Galizia, E., Bracci, R., Belvederesi, L., Catalani, R., Loretelli, C. et al. Effectiveness of the CRCAPRO program in identifying patients suspected for HNPCC. Clinical genetics 71, 158–164 (2007).
Plon, S. E., Eccles, D. M., Easton, D., Foulkes, W. D., Genuardi, M., Greenblatt, M. S. et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Human mutation 29, 1282–1291 (2008).
Acknowledgements
We are grateful to all our clinical colleagues who have referred cases for cancer genetics assessment, and to the participants of this study. This work was supported by a grant from the National Medical Research Council (NMRC) of Singapore (NMRC/CBRG/0034/2013) to AL and a NMRC Transition Award to JN.
Author information
Authors and Affiliations
Contributions
AL, YSY and PA conceived the study. AL and JN designed the study. PA and MHT provided genetic counselling and accrued participants for the study. EW, CC and MS contributed to acquisition of data. EW, SS, MM, CC, MS, SR, JN and AL contributed to data analysis and interpretation of data. All authors contributed to manuscript writing and approved the final version of the article. JN and AL are the guarantors of this manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the npj Genomic Medicine website (http://www.nature.com/npjgenmed)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wong, E., Shekar, S., Met-Domestici, M. et al. Inherited breast cancer predisposition in Asians: multigene panel testing outcomes from Singapore. npj Genomic Med 1, 15003 (2016). https://doi.org/10.1038/npjgenmed.2015.3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/npjgenmed.2015.3
- Springer Nature Limited
This article is cited by
-
BRCA1 and BRCA2 germline mutations in Chinese Hakka breast cancer patients
BMC Medical Genomics (2024)
-
The Association of PTEN Gene Mutations with the Breast Cancer Risk: A Systematic Review and Meta-analysis
Biochemical Genetics (2024)
-
Germline variants of uncertain significance, their frequency, and clinico-pathological features in a cohort of Sri Lankan patients with hereditary breast cancer
BMC Research Notes (2023)
-
Germline variants profiling of BRCA1 and BRCA2 in Chinese Hakka breast and ovarian cancer patients
BMC Cancer (2022)
-
Whole-exome sequencing of BRCA-negative breast cancer patients and case–control analyses identify variants associated with breast cancer susceptibility
Human Genomics (2022)