

Abstract
Deciding when and whether to move is critical for survival. Loss of dopamine neurons (DANs) of the substantia nigra pars compacta (SNc) in patients with Parkinson’s disease causes deficits in movement initiation and slowness of movement1. The role of DANs in self-paced movement has mostly been attributed to their tonic activity, whereas phasic changes in DAN activity have been linked to reward prediction2,3. This model has recently been challenged by studies showing transient changes in DAN activity before or during self-paced movement initiation4,5,6,7. Nevertheless, the necessity of this activity for spontaneous movement initiation has not been demonstrated, nor has its relation to initiation versus ongoing movement been described. Here we show that a large proportion of SNc DANs, which did not overlap with reward-responsive DANs, transiently increased their activity before self-paced movement initiation in mice. This activity was not action-specific, and was related to the vigour of future movements. Inhibition of DANs when mice were immobile reduced the probability and vigour of future movements. Conversely, brief activation of DANs when mice were immobile increased the probability and vigour of future movements. Manipulations of dopamine activity after movement initiation did not affect ongoing movements. Similar findings were observed for the initiation and execution of learned action sequences. These findings causally implicate DAN activity before movement initiation in the probability and vigour of future movements.
Similar content being viewed by others

Main
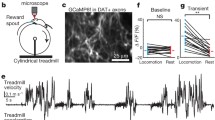
We quantified spontaneous motion of mice placed in an open field (with no external cues, food deprivation or reward) by using inertial sensors that measure high-resolution acceleration and angular velocity (Fig. 1a). We found that the distribution of movements in an open field was not a continuum between arrest and motion, but rather a bimodal distribution (Fig. 1b and Extended Data Fig. 1). Using the minimum acceleration between the two peaks of the bimodal distribution (Fig. 1b), we could separate periods of immobility from periods of overt mobility (Fig. 1b and Extended Data Fig. 1), and precisely identify moments of spontaneous movement initiation.

a, Top left, photoidentification schematics. Top right, movable bundle electrode array with fibre-optic cannula. Bottom, open field set-up. The mouse brain schematic in this panel has been re-drawn with permission from ref. 31. b, Top, distribution of total dynamic acceleration in the open field (mean in blue; 3 ± 2.2 one-hour open-field sessions per mouse, n = 6 mice). Bottom, examples of movement initiation events (red). Dotted line represents acceleration threshold. c, PETH of a positively modulated DAN aligned to movement initiation. d, Left, mean trace for all neurons (n = 25, black), positively modulated neurons (n = 13, green) and negatively modulated neurons (n = 7, red), aligned to movement initiation (93.45 ± 35.99 (mean ± s.e.m.) spontaneous initiations per neuron). Grey shadows denote s.e.m. Right, proportion of positively (+) and negatively (−) modulated and unmodulated (No) neurons. e, Latency of each neuron to be significantly modulated in relation to movement initiation (red, negatively modulated; green, positively modulated). f, Time series of video frames for three movement initiations (1.5 s duration) and their representation in the motion sensor space. Cumulative angular velocity is shown in degrees per second (dg s−1). g, Representation of the initiations of one session using t-distributed stochastic neighbour embedding (t-SNE) dimensionality reduction. Colours represent different clusters determined using affinity propagation clustering. h, Number of initiation clusters in which each positively modulated neuron was significantly activated (n = 13, 30.4 ± 0.19% of initiations were preceded by a significant increase in neuron activity). i, Spread of initiations (mean distance to every other initiation) according to whether positively modulated neurons were active or not (not active, n = 8; active, n = 5). j, Normalized firing rate (firing rate / firing rate of low acceleration trials) for positively modulated neurons of low, medium and high acceleration trials (n = 13). k, Examples of a vigour-related neuron (left) and a neuron unrelated to vigour (right). Red, blue and black represent firing rates during high, medium and low acceleration initiations, respectively. l, Proportion of vigour-related (red) and non-related neurons (grey). P < 0.05, paired t-test comparing firing rate of low and high acceleration trials. Data are mean ± s.e.m. (i, j). *P < 0.05.
We investigated the activity of photoidentified SNc DANs in relation to movement initiation by implanting 16-channel movable electrode bundles coupled to a fibre-optic cannula placed just above the SNc (Fig. 1a). We implanted these bundles in six TH-Cre mice8 (Extended Data Fig. 2a–c) crossed with Ai32 mice9 to obtain expression of channelrhodopsin-2 (ChR2) in TH+ neurons, and used photoidentification10 to identify putative DANs (Extended Data Fig. 3, see Methods for details). Next, we built peri-event time histograms (PETH) of their activity aligned to spontaneous movement initiations. We found that the average activity of all recorded DANs increased transiently before movement initiation (Fig. 1d). Consistently, we found that many photoidentified DANs were significantly modulated by movement initiation and that the majority of these were positively modulated (Fig. 1d). The latency for modulation preceded the initiation of movement for positively modulated neurons, but not for negatively modulated neurons (Fig. 1e). These findings were corroborated using microendoscopic calcium imaging of SNc DANs in freely moving mice (Extended Data Fig. 4; 22 neurons, see Methods and below for details).
We next examined whether the transient activity of individual DANs before action initiation was tuned to the initiation of specific actions, or represented a more general signal before action initiation. To characterize each spontaneous movement initiation, we built trajectories in the motion-sensor space using a combination of total body acceleration (correlated with displacement, see Extended Data Fig. 1), angular velocity (direction of movement) and gravitational acceleration (postural changes)11 (Fig. 1f). Next, we used affinity propagation12 to cluster similar movement initiations13 (Fig. 1g). We found that most DANs were broadly tuned, and were transiently active before rather different initiations (Fig. 1h). Furthermore, we found that initiation trajectories preceded by increased activity of each DAN were as variable as all other initiation trajectories (Fig. 1i). These data strongly indicate that the transient activity of individual DANs before initiation is not very action-specific.
Previous studies in patients with Parkinson’s disease14 and animal models15 have shown that dopamine depletion leads to less vigorous movements. We therefore investigated whether the activity of DANs before movement initiation encoded information about the vigour of the movement that was about to be initiated. We verified that overall, the activity of DANs 300 ms before movement initiation was significantly related to the vigour of future movements (measured by body acceleration; Fig. 1j, k). When doing per-trial analyses comparing all the lower vigour with the higher vigour initiations, we found that 38.5% of the neurons had significantly higher activity before higher vigour movements (Fig. 1l).
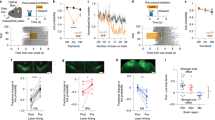
In order to test whether inhibiting DAN activity affects movement initiation, we expressed archaerhodopsin (ArchT)16 specifically in SNc neurons (AAV2/1.CAG.Flex.ArchT-GFP injected into TH-Cre mice, Extended Data Fig. 5). We expressed ArchT in the SNc of 11 TH-Cre mice and YFP in the SNc of 9 TH-Cre mice (control group), and delivered light unpredictably for periods of 15 s (Fig. 2a). Inhibition of SNc DANs increased the probability of mice being immobile (Fig. 2b, c). This was not observed in YFP controls (Fig. 2b, c). To investigate whether DAN inhibition mainly affected movement initiation or reduced ongoing movement, we investigated the effects of DAN inhibition in trials in which the mouse was immobile (for at least 300 ms), or mobile (for at least 300 ms), when the inhibition started (Fig. 2d–h).

a, Schematics showing fibre positioning and trial structure. b, Distribution of acceleration in the open field during laser on and laser off for ArchT (left) and YFP (right) groups. The vertical dashed line denotes the acceleration threshold. c, Left, time spent immobile during laser-off and laser-on periods. Clear bars indicate laser off and filled bars indicate laser on. Right, time spent immobile during laser-on normalized to the baseline. n = 11 ArchT mice, 9 YFP mice. Single asterisk above ArchT indicates significant difference from baseline (1). d, Heat maps of acceleration data of all trials where the mouse was immobile (top) or mobile (bottom) before the start of the trial, in laser-off (left) and laser-on (right) conditions. n = 11 ArchT mice. e, f, Acceleration during laser-off and laser-on trials for immobile trials and mobile trials. n = 7 ArchT mice. The horizontal dotted line denotes the acceleration threshold. There was an interaction between inhibition and mobility state when inhibition started (Supplementary Table 1). g, Left, mean values of the data plotted in e and f. Grey, laser off; green, laser on. Right, same data normalized (laser on / laser off). Single asterisk in Immobile indicates significant difference from 1. h, Mean acceleration per mouse for mobile trials without stops. n = 10 ArchT mice. i, Left, mobile trials aligned to the first stop. Right, normalized mean acceleration (laser on / laser off) before (−4 to −3 s) and after (3–4 s) the first stop for ArchT mice. Single asterisk above ‘After stop’ indicates significant difference from 1. j, Same as in i, but for the YFP group. k, Left, mean cumulative probability of movement initiation for immobile trials. Right, mean probability to initiate movement. n = 7 ArchT mice. l, Mean acceleration for initiations that occurred during immobile trials of ArchT mice. Laser on, n = 16; laser off, n = 30 trials. m, Normalized mean latency to initiate movement. n = 9 YFP mice, 11 ArchT mice. Single asterisk above Move indicates significant difference from 1. Data are mean ± s.e.m. Error bars and shaded areas denote s.e.m. *P < 0.05.
We found a significant impairment in movement initiation when SNc DANs were inhibited during immobility (Fig. 2g). The effect of inhibition was relatively rapid with a significant difference between light and no light after 2.4 s (Fig. 2e). Consistently, we also found that 5 s of inhibition was sufficient to impair movement initiation (Extended Data Fig. 6).
By contrast, there was no significant change in mean acceleration when inhibition happened after movement onset (Fig. 2g). There was no change in the vigour of movement when SNc DANs were inhibited after movement initiation and mice did not stop during the inhibition (Fig. 2h). Furthermore, in mobile trials in which animals stopped during the 15 s (≥300 ms immobile), there was no difference in acceleration between laser-on and laser-off trials before the first stop. However, there was clear impairment in movement initiation after the first stop (Fig. 2i). This was not observed in YFP controls (Fig. 2j).
A more detailed analysis of the effects of inhibition when mice were immobile revealed that there was a significant decrease in the probability of initiating movement during the 15 s of inhibition (Fig. 2k). Furthermore, even when mice were able to initiate movements, the latency to initiate was significantly higher than in laser-off trials, and the initiated movements were less vigorous (Fig. 2l, m). Taken together, these data indicate that DAN activity before movement initiation modulates the probability and vigour of future movements, but activity of these neurons is less critical for the maintenance and vigour of ongoing movements.
Next, we investigated whether brief activation of DANs, when animals were immobile, would be sufficient to promote movement initiation. We expressed ChR2 in SNc DANs using a similar Cre-dependent strategy (DIO-ChR2-YFP in seven mice, and control DIO-eYFP in five mice). Stimulation at 20 Hz for 500 ms (Fig. 3a) delivered when mice were immobile was sufficient to produce overt movement that lasted several seconds (Fig. 3b), in accordance with previous findings7,17,18. The same activation when mice were overtly moving did not significantly affect ongoing acceleration (Fig. 3b, c).

a, Example of three SNc DANs (single units) expressing ChR2, following stimulation at 20 Hz. b, Mean acceleration depending on movement state before the trial. n = 7 ChR2 mice; n = 5 YFP mice. c, Mean acceleration from 0 to 1 s depending on movement state before the trial. Left, immobile; right, mobile. n = 7 ChR2 mice; n = 5 YFP mice. Blue, laser on; grey, laser off. d, Closed loop set-up. e, Heat maps of acceleration data for all trials. Laser-trigger criteria were reached at time 0. White crosses indicate onset of movement. f, Mean acceleration. n = 5 mice per group. Dark blue, ChR2 on; light blue, ChR2 off; black, YFP on; grey, YFP off. g, Mean acceleration from 0 to 1 s. n = 5 mice per group. h, Latency to initiate movement. n = 5 mice per group. i, Percentage of trials with movement initiation between 0 and 1 s. n = 5 mice per group. In h, i, there is a significant effect for group between ChR2 and YFP. j, Mean distribution of acceleration during the first second after movement initiation for laser on (blue) and laser off (black). n = 5 ChR2 mice. k, Mean acceleration during the first second of movement initiation during laser-off and laser-on trials. n = 5 ChR2 mice. Grey bars indicate laser off; blue bars indicate laser on. Data are mean ± s.e.m. Error bars and grey-shaded areas represent s.e.m. *P < 0.05.
To further corroborate this finding, we performed an online closed-loop experiment in which mice received stimulation if they were immobile for at least 900 ms (in 50% of the trials, Fig. 3d). Trials in which light was not delivered (50%) were used as within-animal control (laser-off trials). Average acceleration during the first second after the closed-loop trigger was higher during laser-on than during laser-off trials in the ChR2 group (Fig. 3e–g). Moreover, the latency to initiate movement when SNc DANs were briefly activated was almost three times shorter than in laser-off trials (Fig. 3h). The percentage of trials in which movement was initiated during the first second was also higher in laser-on trials (Fig. 3i). We found no evidence that this closed-loop SNc DAN activation had a reinforcement effect, because immobility states did not become more frequent in ChR2 mice (interval between immobility periods: ChR2, 254.5 ± 116.5 s; YFP, 150.5 ± 65.8 s; t = 1.74, P = 0.12). We found that movements initiated during laser-on trials were more vigorous than movements spontaneously initiated during laser-off trials (Fig. 3j, k). None of the described effects were found in YFP controls.
The results presented above highlight a specific role for the transient activity of DANs for the gating and invigoration of self-paced movement initiation, but not for the modulation of ongoing movements. On the basis of these findings, one prediction would be that if individual spontaneous movements are chunked into a sequence of movements, then the activity of DANs would become preferentially active before sequence initiation, but not during the execution of individual elements within the sequence. To test this prediction, we trained mice on a self-paced operant task in which eight lever presses led to a 20% sucrose solution reward (fixed-ratio eight task (FR8)), without any explicit stimuli signalling the availability of reward4 (Fig. 4a). We implanted a gradient index lens just above the SNc of four TH-Cre mice (Extended Data Fig. 7d), and injected a virus that expressed GCaMP6f19 in a Cre-dependent manner20 (AAV2/5.CAG.Flex.GCaMP6f). We then used a miniaturized epifluorescence microscope21 to image calcium transients in genetically identified SNc DANs while mice were performing the FR8 task (Fig. 4b, c). Similarly to previous findings4, by creating lever press time histograms using normalized fluorescence traces (z score of ΔF), we found that the proportion of modulated neurons was different between press events with the highest proportion of neurons being modulated by the first press (Fig. 4d–f). As predicted, this higher proportion of neurons that were related to the first press was not apparent early in training, and developed with sequence learning4,22 (Extended Data Fig. 8).

a, Example of the behaviour microstructure during the FR8 task late in training. Red, black and blue circles indicate first, middle and last press, respectively. Red and blue bars indicate licks and head entries, respectively. Dashed line denotes reward delivery. b, Field of view (projection of pixel standard deviation) of a TH-Cre mouse expressing GCaMP6f in the SNc. Regions of interest correspond to traces in c. Scale bar, 20 μm. c, Example traces obtained using the CNMF-E algorithm during FR8 task. d, Percentage of neurons modulated by press events. n = 4 mice. e, Venn diagram representing reward- and first press-related neurons. f, PETH of positively modulated neurons for each press event (bottom) and the corresponding heat maps (top). Grey shadow denotes s.e.m. g, Activity of first lever press-responsive neurons aligned to the cross from the magazine to the lever, before the first lever press. n = 22. h, Left, latency to initiate lever press sequence. Right, normalized latency to sequence initiation. ArchT, n = 11 mice; YFP, n = 9 mice. Single asterisk above ArchT indicates significant difference from 1. i, Top, distribution of latencies to sequence initiation. Bottom left, percentage of early initiations. Right, normalized percentage of early initiations (latency <5 s). ArchT, n = 11 mice; YFP, n = 9 mice. There is a significant effect for group between ArchT and YFP. j, Left, press rate. Light was delivered after the first press. Right, mean press rate normalized. ArchT, n = 11 mice; YFP, n = 9 mice. *P < 0.05. Clear bars indicate laser-off and filled indicate laser-on blocks in h–j. Data are mean ± s.e.m.
Recent studies claimed that SNc neurons are more modulated by movement, whereas ventral tegmental area neurons are more modulated by reward7. However, we found that around 35% of neurons in the SNc responded to reward (Fig. 4e). This is almost similar to the percentage of neurons modulated during sequence initiation (approximately 40% first press neurons; Fig. 4d, e). Importantly, there was little overlap between reward- and first-press-modulated neurons, and the overlap was not significantly different than what would be expected by chance (Extended Data Fig. 9). These data suggest that different populations of SNc DANs are related to movement versus reward, but it remains to be determined whether these correspond to populations that project to the dorsal versus ventral striatum, as has previously been suggested23.
Next, we tested whether SNc DAN activity was necessary for sequence initiation. TH-Cre mice expressing ArchT (n = 11) or YFP (n = 9) were trained on the FR8 task for 12–14 days. Mice developed a structured behaviour with predictable sequence initiations and trajectory after reward consumption24 (Fig. 4a). To inhibit SNc neurons before the first press of a sequence, the laser was triggered when mice broke an infrared beam placed between the reward magazine and the lever (Fig. 4g, h), corresponding to the moment of minimal DAN activity (before the increase in activity of first press neurons; Fig. 4g). We compared a block of inhibition (laser-on block) with a previous block without inhibition (laser-off block) during the same session. Inhibition during 5 s before the first lever press resulted in a significant increase in the latency to initiate the action sequence when compared to laser-off trials (Fig. 4h). Moreover, the probability of initiating a sequence decreased during the 5 s of DAN inhibition (Fig. 4i). Consistent with the experiments presented above, when the inhibition happened after sequence initiation (triggered by the first press), the inter-press interval and number of presses during the 5-s inhibition were not altered (Fig. 4j). No effects were observed in YFP controls (Fig. 4h–j). These results were replicated using DAT-IRES-Cre mice using a different inhibitory opsin (Jaws25) when pseudorandomly inhibiting 30% of the trials (Extended Data Fig. 10). Taken together, these results indicate that SNc dopamine activity before the initiation of the action sequence modulates the probability and latency of sequence initiation, but is not critical for the execution of ongoing sequences.
Here we show that SNc DAN activity modulates self-paced movement initiation. Importantly, precisely timed and state-dependent optogenetic manipulations did not change ongoing movements, indicating a specific role for SNc DAN activity for initiation. These results were corroborated using more complex movement sequences.
It has been proposed that dopamine release in the dorsal striatum is important for the regulation of movement vigour2,14,15, but it was thought that this effect was mostly due to the ongoing tonic levels of dopamine release. Our results indicate that the activity of DANs before movement onset modulates future movement vigour. This could explain why patients with Parkinson’s disease select less vigorous movements to initiate14. It is also in accordance with recent studies that have shown that activity of DAN terminals in the dorsal striatum preceded spontaneous movement initiation but did not precede and even followed acceleration bursts during ongoing movement7. Our results suggest that transient changes in dopamine can function as a fast system that acts on top of tonic release to increase the probability (and vigour) of initiating movements, presumably by modulating the excitability of striatal projection neurons24,26, which receive information about the movements that are ‘planned’ at that exact time via glutamatergic inputs from cortex and/or thalamus. This suggests a role for dopamine in gating and invigorating movements that were planned elsewhere27,28, and is consistent with the observation that DAN activity is not very action-specific. More sustained changes in DAN activity could represent states in which the ‘gate’ is more permissive, increasing the probability of action initiation during longer periods of time, therefore promoting movement29. This would translate into more movement variability with exploration of the action space, which could be important in situations of uncertainty or learning.
These results highlight that approaches aimed at providing transient modulations of basal ganglia circuitry tied to movement initiation, for example, via closed-loop deep-brain stimulation30 triggered by activity in cortical areas related to motor planning, could be beneficial to patients with Parkinson’s disease.
Methods
Animals
All experiments were approved by the Portuguese DGAV and Champalimaud Centre for the Unknown Ethical Committee and performed in accordance with European guidelines. TH-Cre male mice from the FI12 mouse line8 between 3 and 5 months, DAT IRES-Cre32 and TH-Cre;Ai329 between 2.5 and 6 months were used.
Sample sizes, randomization and blinding
The number of animals in each experiment was based on previous studies using a power of 0.7 and α = 0.05. n was larger than 5 in all experiments (except imaging experiments) as required for the use of parametric statistics. No formal method of randomization was used; littermates were equally divided among the groups that were compared. There was no blinding of experimental groups. Every experiment contained all experimental groups that were tested concomitantly. The timing of optogenetic manipulations was controlled automatically, not by the experimenter.
Recombinant adeno-associated viral vectors
The following Cre-dependent adeno-associated viral vectors were used in the experiments: AAV2/5.CAG.Flex.GCaMP6f.WPRE.SV40 (titre 1.19 × 1013, University of Pennsylvania); AAV2/1.CAG.Flex.ArchT-GFP (titre 1.4 × 1012, University of Pennsylvania); AAV2/1.ChR2-eYFP (titre 1.4 × 1013; University of North Carolina), AVV2/1.EF1a.DIO.eYFP (titre 1.4 × 1013, University of North Carolina); AAV8/hSyn.Flex.Jaws-GFP (titre 4.2 × 1012, University of North Carolina); rAAV8/hSyn.DIO.eGFP, (titre 4.9 × 1012, University of North Carolina).
Virus injections, electrode, lens and fibre placement
Surgeries were performed using a stereotaxic system (Kopf). Mice were kept in deep anaesthesia using a mixture of isoflurane and oxygen (1–3% isoflurane at 1 l min−1).
For imaging experiments, a 1 μl of virus solution was injected in the right substantia nigra compacta at the following coordinates: −3.16 mm anteroposterior, 1.40 mm lateral from Bregma and 4.20 mm deep from the brain surface. The injection was done through a glass pipette using a Nanojet II (Drummond Scientific) with a rate of injection of 4.6 nl every 5 s. After the injection was finished, the pipette was left in place for 10–15 min. The virus solution was kept at −80 °C and thawed at room temperature just before the injection. A 500-μm diameter, 8.2-mm long gradient index (GRIN) lens (GLP-0584, Inscopix) was implanted at the same coordinates as the injection. Before the lens was lowered, a blunt 28G needle was lowered to 3 mm deep from the brain surface to facilitate the lowering of the GRIN lens. The GRIN lens was then lowered (4.2 mm deep). The lens was fixed in place using cyanoacrilate and black dental cement (Ortho-Jet). One 1/16-inch stainless-steel screw (Antrin miniatures) was attached to the skull to provide a scaffold to build a dental-cement-based cap that protected and fixed the lens to the skull.
Three weeks after surgery, the mouse was anaesthetized and fixed with head bars. A baseplate (BPC-2, Inscopix) attached to a mini epifluorescence microscope (nVista HD, Inscopix) was positioned above the GRIN lens. To correctly position the baseplate, brain tissue was imaged through the lens to find the appropriate focal plane using 20% LED power, a frame rate of 5 Hz and a digital gain of 4. Once the focal plane was set, the baseplate was cemented to the rest of the cap using the same dental cement. Imaging started 2–3 days after this final step.
The same stereotaxic system and anaesthesia protocol was used for electrode and fibre placement. In the case of TH-Cre;Ai32 mice, no virus injection was used.
The same coordinates were used for optrode placement, except for a depth of 3.8–3.9 mm from the brain surface. The ground wire was attached to a 1/16-inch stainless-steel screw (Antrin miniatures), touching the surface of the brain.
For optogenetic experiments (ChR2 and ArchT groups), the same procedure and coordinates were used, except that a 1.5-μl virus solution was injected bilaterally at 2.3 nl every 5 s and optical fibres with a diameter of 230 μm and a NA of 0.39 (Thorlabs FMT 200 EMT) were placed bilaterally at a depth of 3.9 mm from the brain surface. Optical fibres were built based on a published protocol33. Two TH-Cre mice underwent the same virus injection protocol as the ArchT group and were used to obtain the data presented in Extended Data Fig. 5.
For the Jaws experiment, the same procedure was followed except that 1 μl of virus was used and optical fibres with a diameter of 400 μm and a NA of 0.5 (Thorlabs FP400URT) were implanted at the same depth.
Optogenetic set-ups
For ChR2-expressing mice and corresponding controls, light from a free-launched 200-mW, 473-nm, diode-pumped, solid-state laser (Laserglow Technologies), controlled using an AOM (AA Optolectronic), was delivered after being captured by a collimator and split using a one-input to two-outputs rotary joint (Doric Lenses). In addition, 200-nm, 0.22 NA optical fibre patch cords were used to guide the light to the fibres implanted in the mice.
For the ArchT and the corresponding controls, the same set-up was used, but with a different light source (free-launched 500-mW, 556-nm, diode-pumped, solid-state laser from CNI Lasers).
For the Jaws group and corresponding controls, we used a red LED (around 100 mW maximum output, approximately 625 nm, Prizmatix). The light was captured by a large diameter optical fibre (1 mm), which connected to a one-input to one-output rotary joint. A branched 500-μm optical fibre was then used to connect to the fibres that had been implanted in the mice.
Light intensity was measured before and during experiments using a fibre similar to the ones implanted and a power meter (PD1000-S130C, Thorlabs). The power was adjusted at the tip of the fibre to be around 15 mW for photoidentification experiments (Fig. 1 and Extended Data Fig. 3), approximately 3 mW for ChR2 (Fig. 3), around 35 mW for ArchT experiments (Figs 2, 4 and Extended Data Fig. 5), and approximately 9 mW in Jaws experiments (Extended Data Fig. 10).
Open field
We used a 39 × 39 cm open field with black walls (17.5 cm height) and white acrylic floor to assess the spontaneous movement of mice. The open field was inside a sound-attenuating chamber. Illumination was provided by white (2700 K) LEDs (Dioder, Ikea) that were placed on the floor and symmetrically around the open field in a way that illumination of the open field was uniform and indirect (135 lx).
FR8 operant task
Behaviour training and testing took place in operant chambers as described previously4. In brief, each chamber (23 cm L × 20 cm W × 19.5 cm H) was housed within a sound-attenuating box (Med-Associates) and equipped with one retractable lever on the left side of the food magazine and a house light (3 W, 24 V) mounted on the left lateral wall. Sucrose solution (10%) was delivered into a metal cup in the magazine through a syringe pump (20 μl per reward). Magazine entries were recorded using an infrared beam and licks using a contact lickometer. Mice were placed on food restriction throughout training, and fed daily after the training sessions with approximately 2 g of regular food to allow them to maintain a body weight of around 85% of their baseline weight.
Training started with a 30-min magazine training session, in which the reinforcer was delivered on a random time schedule, on average every 60 s (30 reinforcers). The following day lever-pressing training started with continuous reinforcement (CRF), in which animals obtained a reinforcer after each lever press. The session began with the illumination of the house light and insertion of the lever, and ended with the retraction of the lever and by turning off the house light. On the first day of CRF, the sessions lasted 45 min or until mice received five reinforcers, the second day of CRF lasted 45 min or until mice received 15 reinforcers, and the last day of CRF lasted 45 min or until mice received 30 reinforcers. This last CRF session was repeated if mice failed to obtain 30 rewards within the time limit. After CRF, animals started to be trained (day 1) on a fixed ratio schedule in which eight presses earn a reinforcer (FR8), without any stimulus signalling when eight presses were completed or when the reinforcer was delivered; this training continued for 12–14 days.
All timestamps of lever presses, magazine entries and licks for each animal were recorded with a 10-ms resolution. The same training chamber was used during imaging and optogenetic experiments.
Acceleration and video recordings
In the experiments where photoidentified TH+ neurons were recorded, acceleration was recorded using a digital 9-axis inertial sensor with a sampling rate of 200 Hz (MPU-9150, Invensense) assembled on a custom-made PCB and connected to a computer via a custom-made USB interface PCB (Champalimaud Foundation Hardware Platform).
For the other experiments, an analogue 3-axis inertial sensor was used with a sampling rate of 1,000 Hz (LIS331AL, ST) assembled on a custom PCB (Champalimaud Foundation Hardware Platform), and the signals were fed to the analogue inputs of a Cerebus recording system (Blackrock Microsystems).
Acceleration data obtained from these sensors aggregates acceleration from two sources: acceleration generated from gravity and from the body. To separate these two components we processed data obtained from both types of inertial sensor using a custom MATLAB code. We used a standard approach34 that relies on filtering the 3-axis data using a Butterworth filter. In our analysis we used a median filter (7 bins wide) to remove noise peaks. Then we used a 1-Hz high-pass fifth-order Butterworth filter to separate the static (gravitational) component of the signal. Unless stated otherwise, we used the sum of the three vectors of acceleration as a global measurement of body acceleration for our analysis. The dynamic (body acceleration) component accurately tracked animal movement, and correlated well with pixel change in video measurements (Extended Data Fig. 1b–d).
Video recordings were obtained using a charge-coupled device camera (DFK 31BF03, Imaging Source) and a custom-developed software in Labview (National Instruments) at a rate of 15 frames per second. This software allowed us to introduce signals to the video frames to sync acceleration, neural recordings and light delivery periods.
Classification of movement state
We used the average distribution of acceleration in the open field for each experimental group to define the movement state of the mice. This was possible, because the average distribution of the logarithm of total body acceleration was clearly bimodal, with a very low acceleration distribution corresponding to immobility periods (with the possible exception of small and slow postural adjustments) and a high acceleration distribution corresponding to periods of mobility (see Fig. 1b and Extended Data Fig. 1 for a comparison between video and motion-sensor acceleration data). The acceleration threshold was defined as the lowest acceleration value between the two distributions. Unless stated otherwise in the methods, initiation events were defined as transitions between periods of at least 300 ms below the threshold followed by at least 300 ms above the threshold.
Extracellular recordings of photoidentifed SNc DANs
We used a single-drive movable microbundle (sixteen 23-μm tungsten electrodes) with an optic-fibre guide cannula (Innovative Neurophysiology). An optical fibre with 230 μm diameter and a NA of 0.39 (Thorlabs FMT 200 EMT) was inserted in the cannula just to the top of the electrode microbundle cannula (see Fig. 1a for schematics). The neural activity and the timestamps from the light stimulation were recorded using a Cerebus recording system (Blackrock Microsystems).
Experiments were started one week after electrode placement. Every day, we sorted putative units using an online sorting algorithm (Central Software, Blackrock Microsystems) while the mouse was in its home cage. If putative single units were isolated, we delivered a screening protocol consisting of a train of 100 blue light pulses with a 10-ms width delivered at 1 Hz. Using neurophysiology data analysis software (NeuroExplorer V4), we built PETH that were aligned to the train pulses. If any of the isolated units appeared to be modulated by the light train, the mouse was introduced to the open field and neurons were recorded for 1 h. The stimulation protocol was run again at the end of the open field session for confirmation. At the end of the experiment, the microbundle was advanced 50 μm to record next day. We used six mice in these experiments. During this experiment, microbundles were moved on average 433 ± 93.1 μm.
Units were resorted using an offline sorting algorithm (Offline Sorter V3, Plexon Inc.) to isolate single units on the basis of waveform characteristics, inter-spike intervals and clustering. Single units together with the timestamps of the light stimulation provided by a pulse generator (Master 8, AMPI) were exported to MATLAB for analysis.
Criteria used to photoidentify DANs
Neural activity referenced to light pulse onset was averaged in 1-ms bins, and averaged across trials to construct a PETH, which was the basis for analysing amplitude and latency of light-related firing activity. Distributions of the PETH from −900 to −10 ms before light onset were considered baseline activity. We then determined which bins, slid in 1-ms steps during an epoch spanning from light onset to 50 ms after, met the criteria for significant firing rate increases. A significant increase in firing rate was defined as at least four consecutive bins had a firing rate larger than a threshold of five standard deviations above baseline activity. The latency in modulation of photoidentification was defined as the time between light onset and the first significant bin. On the basis of the distribution of latencies of significantly modulated neurons and in accordance with previous studies4,35, we used a very short latency (≤7 ms) for neural response to light, combined with at least a 30% increase in firing rate during the light pulse, and a high correlation coefficient between spike waveforms during light on and off (>0.9) as criteria for positive photoidentification (Extended Data Fig. 3).
Criteria to identify neurons modulated by movement initiation
We built a PETH for each photoidentified single unit spanning from 1,500 ms before and after movement-initiation events. Neural activity was averaged in 100-ms bins, shifted by 1 ms (100 bins, centred on current bin). Distributions of the PETH from −1,000 to −500 ms before light onset were considered baseline activity. We then determined which bins, slid in 1-ms steps during an epoch spanning from −500 ms to 500 ms after movement initiation, met the criteria for significant firing rate changes. A significant change in firing rate was defined when at least 50 consecutive bins had a firing rate higher or lower than a threshold of 2.56 standard deviations above or below baseline activity (99% confidence interval). The latency to modulation was defined as the time between movement onset and the first of the 50 consecutive significant bins.
Area under the receiver operating characteristic curve (auROC) analysis
For this analysis, we used a method similar to the one described previously35. We convolved the spike trains with a function similar to a post-synaptic potential36. To produce the ROC curves, we compared the firing rate of each 50-ms bin to the firing rates during baseline (–1,500 to −1,000 ms before movement initiation) across trials. The auROC for each bin was calculated using trapezoidal numerical integration.
Identification of neuron types using affinity propagation clustering
We used the affinity propagation algorithm12 to look for subtypes of significantly modulated neurons (Extended Data Fig. 3f). It is an efficient clustering algorithm that takes as inputs the similarities between pairs of observations in the dataset (in this case, the auROC traces for each modulated neuron), and finds exemplars and the clusters around them by exchanging real-valued messages between data points. We used the MATLAB (Mathworks) function made available by the authors of ref. 12 at http://www.psi.toronto.edu/index.php?q=affinity%20propagation. We used the correlation between neuron auROC traces as the measure of similarity used by the algorithm. We also used maxits = 1,000, convits = 100; lam = 0.9 and a preference equal to the median similarity of the dataset.
Trial-by-trial analysis of significant increases in neuron activity before movement initiation
We defined a baseline distribution of the number of spikes per 200-ms bin for a period of 1 s (from 2,200 ms before movement initiation to 1,200 ms before movement initiation) for all trials. On the basis of this distribution, we determined a criterion for each neuron such that 90% of the 200-ms bins in the baseline had a lower number of spikes than the criteria. Trials were considered to present a significant increase in neuron activity when the number of spikes during the 200-ms bin immediately preceding movement initiation was above this criterion.
Definition of movement initiation clusters and spread
We used a methodology previously developed by our group to use motion-sensor data to classify behaviour11.
We defined 1-s movement initiation trajectories specified by three motion-sensor variables: total body acceleration, the angular velocity of the axis most parallel to the dorsal–ventral axis of the mice and the gravitational acceleration of the same axis. We divided these trajectories in two 500-ms bins and for each bin we defined a vector composed by a concatenation of a normalized histogram for each motion-sensor variable. In the end, each trajectory was defined by the concatenation of two vectors, the first representing the distributions of the motion-sensor variables for the first 500 ms of initiation, and the second for the last 500 ms. Consequently, the final initiation vector was composed by six different motion-sensor variable distributions. We then determined a matrix representing the distances from each initiation vector to every other vector. To calculate the distances, the motion-sensor variable distribution of one vector was compared to the same motion-sensor variable distribution of another vector (range of 0–1; with 0 indicating that the vectors are exactly the same and 1 indicating that the vectors are maximally different). The differences between each motion-sensor variable distribution were then squared and summed together to find the distance between the two initiation vectors that were evaluated (range 0–6). The final distance matrix was built by comparing each initiation vector in this way and squaring the final result, obtaining a matrix that varied from 0 (exactly the same) to 36 (maximally different). The spread values presented in Fig. 1i were determined by averaging the distances between movement initiations.
After determining an initiation distance matrix for each session, we used affinity propagation12 to find clusters of initiations (see the description above regarding this methodology). We provided the affinity clustering algorithm with the additive inverse of the distance matrix as a similarity matrix. We also used maxits = 1,000, convits = 100; lam = 0.9. To make sure that we were selecting a consistent structure for the data, we used a value for preference between the minimum and the maximum similarity of each similarity matrix that provided the highest but at the same time most stable number of clusters (that is, a value was chosen in the middle of an interval of consecutive values that provided the same number of clusters).
t-Distributed stochastic neighbour embedding for visualization of initiations
We used t-distributed stochastic neighbour embedding (t-SNE)13 to visualize and assess the existence of structure within the movement initiations of each session (see the example in Fig. 1h). We implemented this using a MATLAB code provided by the authors of ref. 13 (https://lvdmaaten.github.io/tsne/). We used a 2D t-SNE using a perplexity of 15 to produce the image in Fig. 1h.
DAN activity and movement vigour analysis
We used the mean acceleration during the first 500 ms of each spontaneous initiation as a measurement of movement vigour, and separated trials into low acceleration trials (lower tertile), medium and high acceleration trials (upper tertile). We then calculated the activity of positively modulated neurons during 300 ms before movement initiation for each acceleration tertile. A neuron was considered to be vigour related if the activity during the lower tertile trials was significantly lower than the activity during the upper tertile trials.
GCaMP6f imaging using a mini-epifluorescence microscope
Mice were briefly anaesthetized using a mixture of isoflurane and oxygen (1% isoflurane at 1 l min−1) and the mini-epifluorescence microscope was attached to the baseplate. This was followed by a period of 20–30 min of recovery in the home cage before experiments started. Fluorescence images were acquired at 10 Hz and the LED power was set 10–20% (0.1–0.2 mW) with a gain of 4. Image acquisition parameters were set to the same values between sessions to be able to compare the activity recorded. Three GCaMP6f-expressing TH-Cre mice were imaged while freely exploring an open field. The same mice and one more were also imaged during the FR8 task. Data shown in Fig. 4 were obtained in two consecutive late training sessions (between days 7 and 13).
Calcium image processing and analysis
GCaMP6f image processing and fluorescence trace extraction
All fluorescence movies were initially processed using the Mosaic Software (v.1.1.2, Inscopix). First, all frames were spatially binned by a factor of 4. To correct the movie for translational movements and rotations, the frames were registered to a reference image consisting of an average of the raw fluorescence movie. This was achieved by implementing the TurboReg registration engine37 within the mosaic software. The movie was cropped after registration to remove the post-registration black borders.
GCaMP6f fluorescence trace extraction
Although calcium imaging using miniscopes enables researchers to image neurons in freely moving mice, it is a challenge to adequately extract neuronal signals without background contamination. Because of this, we implemented the ‘constrained non-negative matrix factorization for endoscopic data’ (CNMF-E) framework11,38. This recently described framework is an adaptation of the CNMF algorithm39. It can reliably deal with the large fluctuating background from multiple sources in the data, enabling accurate source extraction of cellular signals. It includes four steps: (1) initialize spatial and temporal components of single neurons without the direct estimation of the background; (2) estimate the background given the estimated spatiotemporal activity of the neurons; (3) update the spatial and temporal components of all neurons while fixing the estimated background fluctuations; (4) iteratively repeat step 2 and 3.
Criteria to identify DANs modulated by movement initiation using GCaMP6f imaging
We built a PETH for each neuronal trace spanning from 3 s before to 3 s after movement-initiation events. For this analysis, we considered movement initiations as transitions between a period of at least 500 ms below to a period of at least 500 ms above the acceleration threshold. Distributions of the PETH from −3 to −1 s before movement onset were considered baseline activity. We then determined which bins, during an epoch spanning from −0.5 before to 0.5 s after movement initiation, met the criteria for significant ΔF changes. A significant change in ΔF was defined if at least 2 consecutive bins had ΔF higher or lower than a threshold of 99% above or below baseline ΔF.
Criteria to identify lever-press-related and reward-related DANs using GCaMP6f imaging
We constructed a PETH for each neuron trace spanning from −3 to 3 s from lever press onset for the first, second, third, third to final, second to final and final press, and also for the first lick of reward. Distributions of the PETH from −5 to −3 s before first lever press were considered baseline activity for all press-related activity and distributions from −5 to −3 s of the first lick of reward PETH was considered as baseline for reward-related activity. We then searched each PETH during an epoch spanning from −0.5 s to 0.5 s for bins that were significantly different from the baseline. A significant change in fluorescence was defined as at least three consecutive bins with fluorescence higher than a threshold of 99% above baseline ΔF.
Extracellular recordings during SNc DAN inhibition
Optic fibres and electrodes were positioned 300 μm above the SNc. Light intensity was around 35 mW at the tip of the fibre, corresponding to an estimated40 irradiance of 70–206 mW mm−2 at SNc depth (200 to 400 μm from the fibre tip). Neural activity was recorded daily and the electrodes were moved 50 μm at the end of each recording session. We were thus able to record neural activity from different depths (−3.90 mm to −4.60 mm from the brain surface), with neurons being recorded above, within and below the SNc. We recorded from 140 units and observed that at depths where the SNc is located, more than 60% of recorded units were inhibited (Extended Data Fig. 5), whereas above and below the SNc, very few neurons were modulated. Furthermore, we only observed one single unit that was modulated by light at the depth closest to the fibre where light intensity is higher (−3.9 to −3.95mm, 0.7% of all units recorded, 7.7% of all units recorded at this depth), indicating that light delivery per se was not sufficient to change neural activity at this power. The same positioning of the fibres and light intensity was used during the open field and operant task inhibition experiments.
We used the same set-up and methodology described for the extracellular recordings of photoidentifed SNc DANs, but instead of using blue light, we used green light (see ‘Optogenetics set-ups’). The mean auROC traces presented in Extended Data Fig. 5a, b were calculated as described for the photoidentification experiments. The anatomical scheme depicted in Extended Data Fig. 5b was based on the histological determined position of the electrode cannula and the amount of electrode travel at each recording session.
Open field
Mice were introduced to the open field and green light was delivered continuously for periods of 15 s (mean of 22 ± 3 trials with a mean inter-trial interval (ITI) of 64 ± 28 s per mouse in the ArchT group and a mean of 23 ± 4 trials with a mean ITI of 65 ± 33 s per mouse in the YFP group). An open-field session was done on the day before to habituate mice to the open field and to light delivery (with similar trial structure except for light duration, which was 5 s). Data regarding this first session are shown in Extended Data Fig. 6.
Operant task
The same groups of ArchT and YFP mice used in the open-field inhibition experiment were trained to perform the FR8 task. Optogenetic experiments were started at the end of the FR8 training. We used two different light-delivery schedules: continuous light delivery for 5 s before the first lever press in a sequence; and continuous light delivery for 5 s after the first lever press in a sequence. For the first condition, we made the triggering of the light contingent on the breaking of an infrared beam (IRB) positioned right next to the magazine, on the side of the lever. This way, mice coming from consuming the reward would break the IRB before they started the next sequence. Sessions were divided into two blocks: a first block of 15 trials without light delivery; and a second block of 10 trials with light delivery. The last 10 trials with no light delivery were used to compare with the 10 trials with light delivery. In a few trials, the mouse failed to break the IRB before starting the action sequence. These trials were discarded and not included in the analysis. Sessions with no light delivery were interspersed between sessions with light delivery and a first session with light delivery was only used to habituate the mice to the delivery of light and it was not analysed. The same methodology was used in Jaws experiments with the exception that instead of light on and light off blocks there was a 30% probability of switching on the laser for each trial and data were collected in three consecutive sessions for each experimental condition (inhibition before initiation and inhibition after first press).
SNc DAN activation
Open field
Mice were introduced to the open field and a train of blue laser light (10 ms pulses at 20 Hz, during 0.5 s) was delivered with a variable interval: after 90 s there was a 33% probability that the light was delivered and this was repeated every 10 s until light was delivered.
Closed loop experiment in the open field
For closed loop experiments, the same light pulse train was used, but it was delivered depending on the acceleration state of the mouse in the following way: acceleration of mice was monitored online by feeding the analogue accelerometer data through a Cerebus recording system (Blackrock Microsystems) into MATLAB (Mathworks) using Blackrock’s MATLAB interface (CBMEX). Using a custom MATLAB code, we processed accelerometer data as described above. We monitored acceleration of mice using bins of 300 ms and when mice reached 900 ms below the threshold used to identify immobility, light was delivered with a 50% probability. There was a minimum of 30 s between trials. In this experiment, we considered the maximum acceleration during the first second after each movement initiation as a measurement of initiation vigour.
Anatomical verification
Animals were euthanized after completion of the behavioural tests. First, animals were anaesthetized with isoflurane, followed by intraperitoneal injection of ketamine–xylazine (around 5 mg kg−1 xylazine; 100 mg kg−1 ketamine). Animals were then perfused with 1× phosphate-buffered saline (PBS) and 4% paraformaldehyde, and brains were extracted for histological processing. Brains were kept in 4% paraformaldehyde overnight and then transferred to 1× PBS solution. Brains were sectioned coronally in 50-μm slices (using a Leica vibratome (VT1000S) and kept in PBS solution before mounting or immunostaining experiments). Images were taken using a wide-field fluorescence microscope (Zeiss AxioImager) and the tip of the longest track found was used to determine the anatomical location of the implants (lenses, fibres and electrodes), which was represented in the corresponding Allen Brain Atlas41 slice (Extended Data Fig. 7).
To estimate the specificity of the TH-Cre line, we crossed TH-Cre mice with ROSA26-eGFP mice. TH-Cre;ROSA26-eGFP mice express GFP in neurons expressing Cre recombinase. We used slices from the brain of one TH-Cre;ROSA26-eGFP mouse and used a tyrosine hydroxylase antibody (ImmunoStar) to label the ventral tegmental area (VTA) and SNc DANs (Extended Data Fig. 2). We imaged the VTA and SNc in three different slices (approximately −2.9 mm, −3.3 mm, −3.8 mm from Bregma) using a confocal microscope equipped with a Diode 405-nm, Argon multi-line 458−488−514-nm and DPSS 561-nm lasers (LSM710, Zeiss). We acquired z stacks (354 μm × 354 μm × 5 μm; 5-μm interslice interval) in a tile that covered the VTA, SNc and areas 200 μm above and below the SNc. We imported these images into the Stereo Investigator software (MBF Bioscience) and used a stereological approach to count labelled cells (TH+ and Cre+) and evaluate co-localization, using a 100 μm × 100 μm counting frame (Extended Data Fig. 2a–c).
We also used a stereological approach to estimate the rate and specificity of infection of the AAV2/1.CAG.Flex.ArchT-GFP and the density of TH+ neurons in the SNc of ArchT and YFP TH-Cre mice (ArchT, n = 2; YFP, n = 2). We imaged every six slices in which the VTA and/or SNc were found, using the confocal microscope described above (three slices per mouse). Using a 40× magnification, we acquired z stacks (354 μm × 354 μm × 5 μm; 5-μm interslice interval) in a tile that covered the whole SNc. These tile z stacks were imported into the stereo investigator software (MBF Bioscience) and quantification of the TH+, eYFP+ or GFP+ cells was performed using a 100 μm × 100 μm counting frame (Extended Data Fig. 2d–h).
Statistics
Statistical hypothesis testing was done at a 0.05 significance level (except for classification of neurons, which was done at a 0.01 significance level as explained above). Parametric testing was used whenever possible to test differences between two or more means. Normality was tested using the Shapiro–Wilk test, wheras F-tests (for unpaired t-tests) and Levene’s tests (for ANOVA) were used to assess equality of variance. If data were not normally distributed, we first tried to transform the data using the natural logarithm. If the distribution of the transformed data was still not normally distributed or there was a significant difference in variance, an alternative non-parametric test was used. ANOVA and linear mixed models were used to check for main effects and interactions in experiments with repeated measures and more than one factor. The assumptions for linear mixed models were checked by careful inspection of the model residuals to check for normality and equality of variances. When main effects or interactions were significant, we did planned comparisons according to experimental design (for example, comparing laser on and off). Fisher’s least significant difference tests were used for comparisons after ANOVA tests and least square means tests were used for comparison when linear mixed models were significant. Details on the statistical analysis used for hypothesis testing in the main figures can be found in the Supplementary Table 1. Statistical tests were done using Prism (GraphPad), MATLAB (MathWorks) statistical toolbox and R (R core team 2015, v.3.1.3, lme442).
Code availability
MATLAB (MathWorks) codes used for data analysis are available from the corresponding author.
Data availability
Source Data for Figs 1, 2, 3, 4 have been provided with the online version of the paper and all other data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376 (2008)
Niv, Y., Daw, N. D. & Dayan, P. How fast to work: response vigor, motivation and tonic dopamine. Adv. Neural Inf. Process. Syst. 18, 1019–1026 (2005)
Schultz, W. Multiple dopamine functions at different time courses. Annu. Rev. Neurosci. 30, 259–288 (2007)
Jin, X. & Costa, R. M. Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature 466, 457–462 (2010)
Syed, E. C. J. et al. Action initiation shapes mesolimbic dopamine encoding of future rewards. Nat. Neurosci. 19, 34–36 (2016)
Dodson, P. D. et al. Representation of spontaneous movement by dopaminergic neurons is cell-type selective and disrupted in Parkinsonism. Proc. Natl Acad. Sci. USA 113, E2180–E2188 (2016)
Howe, M. W. & Dombeck, D. A. Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature 535, 505–510 (2016)
Gong, S. et al. Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J. Neurosci. 27, 9817–9823 (2007)
Madisen, L. et al. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci. 15, 793–802 (2012)
Lima, S. Q., Hromádka, T., Znamenskiy, P. & Zador, A. M. PINP: a new method of tagging neuronal populations for identification during in vivo electrophysiological recording. PLoS ONE 4, e6099 (2009)
Klaus, A. et al. The spatiotemporal organization of the striatum encodes action space. Neuron 95, 1171–1180 (2017)
Frey, B. J. & Dueck, D. Clustering by passing messages between data points. Science 315, 972–976 (2007)
Van Der Maaten, L. & Hinton, G. H. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008)
Mazzoni, P., Hristova, A. & Krakauer, J. W. Why don’t we move faster? Parkinson’s disease, movement vigor, and implicit motivation. J. Neurosci. 27, 7105–7116 (2007)
Panigrahi, B. et al. Dopamine is required for the neural representation and control of movement vigor. Cell 162, 1418–1430 (2015)
Han, X. et al. A high-light sensitivity optical neural silencer: development and application to optogenetic control of non-human primate cortex. Front. Syst. Neurosci. 5, 18 (2011)
Barter, J. W. et al. Beyond reward prediction errors: the role of dopamine in movement kinematics. Front. Integr. Neurosci. 9, 39 (2015)
Hamid, A. A. et al. Mesolimbic dopamine signals the value of work. Nat. Neurosci. 19, 117–126 (2016)
Chen, T.-W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013)
Atasoy, D., Aponte, Y., Su, H. H. & Sternson, S. M. A FLEX switch targets channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J. Neurosci. 28, 7025–7030 (2008)
Ghosh, K. K. et al. Miniaturized integration of a fluorescence microscope. Nat. Methods 8, 871–878 (2011)
Wassum, K. M., Ostlund, S. B. & Maidment, N. T. Phasic mesolimbic dopamine signaling precedes and predicts performance of a self-initiated action sequence task. Biol. Psychiatry 71, 846–854 (2012)
Parker, N. F. et al. Reward and choice encoding in terminals of midbrain dopamine neurons depends on striatal target. Nat. Neurosci. 19, 845–854 (2016)
Tecuapetla, F., Jin, X., Lima, S. Q. & Costa, R. M. Complementary contributions of striatal projection pathways to action initiation and execution. Cell 166, 703–715 (2016)
Chuong, A. S. et al. Noninvasive optical inhibition with a red-shifted microbial rhodopsin. Nat. Neurosci. 17, 1123–1129 (2014)
Kravitz, A. V. et al. Regulation of Parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626 (2010)
Wong, A. L., Lindquist, M. A., Haith, A. M. & Krakauer, J. W. Explicit knowledge enhances motor vigor and performance: motivation versus practice in sequence tasks. J. Neurophysiol. 114, 219–232 (2015)
Thura, D. & Cisek, P. The basal ganglia do not select reach targets but control the urgency of commitment. Neuron 95, 1160–1170 (2017)
Spielewoy, C. et al. Behavioural disturbances associated with hyperdopaminergia in dopamine-transporter knockout mice. Behav. Pharmacol. 11, 279–290 (2000)
Rosin, B. et al. Closed-loop deep brain stimulation is superior in ameliorating Parkinsonism. Neuron 72, 370–384 (2011)
Franklin, K. B. J. & Paxinos, G. The Mouse Brain in Stereotaxic Coordinates (Academic, 2008)
Bäckman, C. M. et al. Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis 44, 383–390 (2006)
Sparta, D. R. et al. Construction of implantable optical fibers for long-term optogenetic manipulation of neural circuits. Nat. Protoc. 7, 12–23 (2011)
Mathie, M. J., Lovell, N. H., Coster, A. C. F. & Celler, B. G. Determining activity using a triaxial accelerometer. In Proc. 2nd Joint EMBS-BMES Conf. 3, 2481–2482 (2002)
Cohen, J. Y., Haesler, S., Vong, L., Lowell, B. B. & Uchida, N. Neuron-type-specific signals for reward and punishment in the ventral tegmental area. Nature 482, 85–88 (2012)
Thompson, K. G., Hanes, D. P., Bichot, N. P. & Schall, J. D. Perceptual and motor processing stages identified in the activity of macaque frontal eye field neurons during visual search. J. Neurophysiol. 76, 4040–4055 (1996)
Thévenaz, P., Ruttimann, U. E. & Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41 (1998)
Zhou, P ., Resendez, S. L ., Stuber, G. D ., Kass, R. E. & Paninski, L. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Preprint at https://arxiv.org/abs/1605.07266 (2016)
Pnevmatikakis, E. A. et al. Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron 89, 285–299 (2016)
Yizhar, O., Fenno, L. E., Davidson, T. J., Mogri, M. & Deisseroth, K. Optogenetics in neural systems. Neuron 71, 9–34 (2011)
Lein, E. S. et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007)
Bates, D., Mächler, M., Bolker, B. M. & Walker, S. C. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015)
Acknowledgements
We thank A. Vaz for mouse colony management, I. Vaz for the help during photoidentification experiments, L. Perry for help with stereological cell counts, A. Klaus, P. Zhou, L. Paninski for help with the application of the CNMF-E analysis, and the Champalimaud Hardware Platform (F. Carvalho, A. Silva, D. Bento) for help with the development of the motion sensors. This work was supported by fellowships from Gulbenkian Foundation to J.A.d.S. and Grants from Fundação para a Ciência e Tecnologia, Fronteras de la Ciencia-CONACyT-2022 and the IN226517 DGAPA-PAPIIT-UNAM to F.T. and from ERA-NET, European Research Council (COG 617142), and HHMI (IEC 55007415) to R.M.C.
Author information
Authors and Affiliations
Contributions
J.A.d.S. and R.M.C. designed the experiments and analyses and wrote the paper, J.A.d.S. performed all experiments and analyses, F.T. helped with optogenetic and recording experiments, V.P. helped with accelerometer experiments and accelerometer data analyses.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks D. J. Surmeier and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Comparison between video-motion analysis and motion-sensor data in the open field.
a, Example of raw acceleration (static and dynamic acceleration) and angular velocity collected using a six-axis inertial sensor. b, Bivariate histogram of log pixel change and log acceleration in the open field (n = 13 sessions, from three mice). Notice the two clusters that emerge from this bivariate histogram (low acceleration and low pixel change cluster on the left, and high acceleration and high pixel change cluster on the right). The acceleration histogram provides a clear distinction between these two clusters. There is a high correlation between video-tracking measurements and acceleration (r = 0.74, n = 62,984 frames, P < 0.05). This also shows that animals rely on constant acceleration from their limbs to move, and that locomotion at a constant speed (acceleration 0) is highly unlikely. c, Heat maps of mean pixel change of video clips of 300 ms and 10 s (top and bottom, respectively), during which mice showed acceleration that was lower (left) or higher (right) than the threshold used to define immobility. d, Top, comparison between a video-derived motion measurement (pixel change) and total dynamic acceleration aligned to movement initiation determined using the acceleration threshold (n = 454 initiations obtained from three mice during a total of 13 open-field sessions). Bottom, representation of the movement of mice (based on the centre-of-mass) during each trial within the time intervals as indicated on the x axis. The trajectories were aligned to the centre-of-mass of the last frame of each 1-s interval. Different colours denote individual trials.
Extended Data Figure 2 TH-Cre line and ArchT infection characterization.
a, TH-Cre mice were crossed with ROSA26R-YFP mice (expression of YFP in Cre+ cells). This is an example of a midbrain slice of a TH-Cre × ROSA26R-YFP mouse with TH+ neurons labelled in red. The white line delimits the SNc, and the yellow and green lines delimit areas that cover a depth of 200 μm above and below the nigra, respectively, that were also targeted by stereological cell counts. Scale bar, 100 μm. b, Example of a SNc sampling field. Arrowheads denote examples of Cre+ cells that were TH+. Scale bars, 20 μm. c, Quantification of the specificity of the Cre line for tagging TH+ cells (n = 3 slices; 117 counting frames were analysed). d, Representative merged image of VTA and SNc after two weeks of infection. ArchT+ cells are labelled in green, TH+ cells are labelled in red and merged colours in yellow. ArchT+ cells are mainly confined to SNc. Scale bar, 500 μm. e, Detail of a SNc region labelled for TH (red) and ArchT (green) expression. Arrows are examples of TH+ and ArchT+ cells; closed arrowheads denote examples of TH+ and ArchT− cells. Scale bars, 20 μm. f, Efficiency of ArchT virus infection (left). Specificity of ArchT virus infection (right). This was calculated by quantifying the whole SNc stereologically, not only the area closest to the infection (n = 6 slices from two ArchT mice, 122 counting frames). g, Examples of the slices and fields used to do the stereological count shown in f and h. Scale bars, 100 μm. h, Stereological quantification of the number of SNc TH+ cells in YFP- and ArchT-expressing mice after two weeks of infection (ArchT, n = 6 slices from two ArchT mice, 122 counting frames; YFP, n = 6 slices from two YFP mice, 124 counting frames). i, Photomicrograph of a midbrain slice of a ArchT-expressing mouse at the end of the experiments (open field and FR8). Red indicates TH+ cells and green indicates ArchT+ cells. Scale bar, 100 μm. Data are mean ± s.e.m. (c, f, h).
Extended Data Figure 3 Photoidentification and clustering of SNc dopamine neurons.
a, Photomicrograph of a midbrain slice of a TH-Cre;Ai32 mouse denoting the right SNc and VTA. ChR2 in green and TH+ cells in red. Initial electrode position (dashed square) and distance travelled (dashed triangle). Scale bar, 100 μm. b, Example of continuous recording of a photoidentified neuron. Blue triangles denote 10-ms light pulses of blue light that were delivered at 1 Hz. c, PETH of the neuron in b aligned to blue-light delivery (100 pulses). d, Histogram of latencies to modulation by light delivery. A threshold of 7 ms and an increase in at least 30% firing rate was used to define neurons as photoidentified (blue bars). e, Mean spike traces for all photoidentified neurons used in Fig. 2. The black trace represents the mean of spikes obtained without light delivery and the blue trace represents the mean trace of spikes obtained during light delivery. f, Left, the area under the ROC curve (auROC) was calculated for each time bin of each significantly modulated neuron. Right, we used an affinity propagation algorithm to cluster the traces that resulted from the auROC analysis (see Methods for details). Four clusters were found, of which the PETH of the representative neuron is shown. Neurons were: transiently active before the initiation of movement (blue), transiently active before the initiation of movement followed by inhibition after the initiation (grey), sustained increase in activity with movement initiation (green) or negatively modulated (red).
Extended Data Figure 4 Calcium imaging of TH+ neurons from the SNc during movement initiation.
a, Example of the average pixel per pixel ΔF of one mouse aligned to movement initiation (n = 46 trials). Black arrowheads denote three neurons that are significantly activated before movement initiation. b, Proportion of neurons that were positively or negatively modulated and not modulated by movement initiation (n = 22 neurons obtained from three mice). c, Mean auROC trace of positively modulated neurons based on calcium imaging (solid line, n = 7). d, Mean auROC trace of negatively modulated neurons according to calcium imaging (solid line, n = 4). Grey shadow denotes s.e.m. (c, d).
Extended Data Figure 5 In vivo external recordings reveal specific inhibition of neuronal activity in the SNc.
a, Mean unit activity aligned to light onset (values less than 0.5 auROC indicate a decrease compared to baseline and more than 0.5 auROC indicate an increase compared to baseline) at different recording depths. The green rectangle signals the duration of light delivery. Left, mean of all units recorded. Right, mean of all units except negatively modulated units. b, Top, anatomical representation31 of the mean unit activity depending on recording depth and the location of the cannula of the recording electrode (red, decrease from baseline; blue, increase from baseline). The percentage of inhibited cells was not homogeneous throughout all depths (χ24,140 = 18.01, P < 0.05, test based on five levels of depth from –3.9 to –4.6 mm with 150-μm steps). In fact, when we investigated the mean activity of all units recorded at each depth, we found that the mean activity during light delivery changed depending on the depth, and it was only significantly different at the depths at which the SNc is located, for which the percentage of inhibited units was 61.3%. This is anatomically represented in b. Depth (number of neurons): −3.9 mm (3); −4 mm (22); −4.1 mm (14); −4.2 mm (6); −4.3 mm (8); −4.4 mm (10); −4.5 mm (4); −4.6 mm (5). Kruskal–Wallis test: H = 18.22; P = 0.011. Dunn’s multiple comparison test, all means compared to mean at −4.6 mm: 3.9 mm, P > 0.99; −4 mm, P = 0.078; −4.1 mm, *P = 0.017; −4.2 mm, **P = 0.008; −4.3 mm, P = 0.67; −4.4 mm, P = 0.82; −4.5 mm, P > 0.99). Asterisks indicate depths with mean auROC significantly different from −4.6 mm depth. c, Example of a single unit inhibited by green light. The mouse brain has been reproduced with permission from ref. 31.
Extended Data Figure 6 Five-second inhibition of SNc TH+ neurons during mobile and immobile trials.
a, Acceleration during laser-off and brief laser-on trials (5 s inhibition) when ArchT mice were mobile before the start of the trial (n = 217 laser-on trials and n = 212 laser-off trials obtained from 11 ArchT mice). The horizontal dotted line denotes the threshold used to classify acceleration state. b, Acceleration during laser-off and brief laser-on trials (5 s inhibition) when ArchT mice were immobile before trial start (n = 17 laser-on trials from 5 ArchT mice and n = 12 laser-off trials obtained from 7 ArchT mice). Horizontal dotted line denotes the threshold used to classify acceleration state. c, Acceleration during brief laser-on (5 s inhibition) normalized to mean laser-off acceleration for immobile and mobile states (n = 17 laser-on immobile trials obtained from 5 ArchT mice, n = 12 laser-off immobile trials obtained from 7 ArchT mice; n = 217 laser-on mobile trials, n = 212 laser-off mobile trials obtained from 11 mice). Acceleration state significantly affected normalized acceleration (linear mixed model with ‘mouse’ as a random effect, F = 19.57, P < 0.0001).
Extended Data Figure 7 Anatomical position of fibres, electrodes and lens.
a, Fibre placement (green, ArchT mice; yellow, YFP mice). b, Fibre placement (blue, ChR2 mice; yellow, YFP mice). c, Optrode placement. Horizontal blue lines denotes the cannula position and vertical lines the distance travelled by the electrodes. d, GRIN lens placement (green bar indicates the position of the bottom of the lens). The representations of the mouse brain and anatomical structures was obtained from the Allen Mouse Brain Atlas (2004) using API. Top, http://api.brain-map.org/api/v2/svg_download/100960073?groups=28; middle, http://api.brain-map.org/api/v2/svg_download/100960057?groups=28; bottom, http://api.brain-map.org/api/v2/svg_download/100960525?groups=28.
Extended Data Figure 8 Lever-press significant neurons across training days.
a, Field-of view of one of the GCamP6-expressing mice across different training days spanning days from the beginning to the end of training. Scale bars, 100 μm. b, Percentage of neurons significantly modulated by the sequence lever presses throughout training. Data shown were obtained during the training of three out of the four mice used to obtain the calcium imaging data shown in Fig. 4. Data during training was not available for one mouse.
Extended Data Figure 9 First press- and reward-related neurons populations do not overlap.
a, Example of a neuron modulated by first press but not reward (left) and a neuron related to reward but not first press (right), aligned to first press (blue) and reward consumption (red). b, Monte Carlo simulations (10,000 samples) were used to generate a distribution of the number of overlapping neurons for first press and reward, assuming random assignment. Red lines denote the 95% confidence interval. Dashed line represents the number of overlapping neurons found in our experiment.
Extended Data Figure 10 FR8-inhibition experiment using Jaws.
We replicated the result obtained in the FR8 task (Fig. 4h–j), using Jaws (see Methods for details). a, Latency to initiate lever press sequence for laser-off trails and trials with inhibition starting just before sequence initiation for both Jaws (n = 6) and GFP (n = 6) groups. Two-way mixed ANOVA; planned comparisons between laser-on and laser-off trials using Fisher’s least significant difference tests; main effect group F1,10 = 3.16, P = 0.11; main effect laser F1,10 = 9.074, P = 0.0131; interaction effect F1,10 = 0.92, P = 0.36; planned comparisons: Jaws laser off − laser on P = 0.019, GFP laser off − laser on P = 0.18. b, Press rate in trials with no light delivery and trials with light delivery starting after the first press for both Jaws (n = 5) and GFP (n = 6) groups. Two-way mixed ANOVA; planned comparisons between laser-on and laser-off trials using Fisher’s least significant difference tests; main effect group F1,9 = 1.607, P = 0.24; main effect laser F1,9 = 0.53, P = 0.49; interaction effect F1,9 = 1.01, P = 0.34; planned comparisons: Jaws laser off − laser on P = 0.86, GFP laser off − laser on P = 0.23.
Supplementary information
Supplementary Information
This file contains Supplementary Table 1. (PDF 144 kb)
Rights and permissions
About this article

Cite this article
da Silva, J., Tecuapetla, F., Paixão, V. et al. Dopamine neuron activity before action initiation gates and invigorates future movements. Nature 554, 244–248 (2018). https://doi.org/10.1038/nature25457
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature25457
- Springer Nature Limited
This article is cited by
-
Explaining dopamine through prediction errors and beyond
Nature Neuroscience (2024)
-
Constraints on the subsecond modulation of striatal dynamics by physiological dopamine signaling
Nature Neuroscience (2024)
-
Neural inhibition as implemented by an actor-critic model involves the human dorsal striatum and ventral tegmental area
Scientific Reports (2024)
-
A feature-specific prediction error model explains dopaminergic heterogeneity
Nature Neuroscience (2024)
-
Orexin neurons mediate temptation-resistant voluntary exercise
Nature Neuroscience (2024)