

Abstract
Cells can sense the density and distribution of extracellular matrix (ECM) molecules by means of individual integrin proteins and larger, integrin-containing adhesion complexes within the cell membrane. This spatial sensing drives cellular activity in a variety of normal and pathological contexts1,2. Previous studies of cells on rigid glass surfaces have shown that spatial sensing of ECM ligands takes place at the nanometre scale, with integrin clustering and subsequent formation of focal adhesions impaired when single integrin–ligand bonds are separated by more than a few tens of nanometres3,4,5,6. It has thus been suggested that a crosslinking ‘adaptor’ protein of this size might connect integrins to the actin cytoskeleton, acting as a molecular ruler that senses ligand spacing directly3,7,8,9. Here, we develop gels whose rigidity and nanometre-scale distribution of ECM ligands can be controlled and altered. We find that increasing the spacing between ligands promotes the growth of focal adhesions on low-rigidity substrates, but leads to adhesion collapse on more-rigid substrates. Furthermore, disordering the ligand distribution drastically increases adhesion growth, but reduces the rigidity threshold for adhesion collapse. The growth and collapse of focal adhesions are mirrored by, respectively, the nuclear or cytosolic localization of the transcriptional regulator protein YAP. We explain these findings not through direct sensing of ligand spacing, but by using an expanded computational molecular-clutch model10,11, in which individual integrin–ECM bonds—the molecular clutches—respond to force loading by recruiting extra integrins, up to a maximum value. This generates more clutches, redistributing the overall force among them, and reducing the force loading per clutch. At high rigidity and high ligand spacing, maximum recruitment is reached, preventing further force redistribution and leading to adhesion collapse. Measurements of cellular traction forces and actin flow speeds support our model. Our results provide a general framework for how cells sense spatial and physical information at the nanoscale, precisely tuning the range of conditions at which they form adhesions and activate transcriptional regulation.
Similar content being viewed by others

Main
To explore the spatial sensing of ECM molecules by cells, we investigated a wide array of conditions, considering not only the nanometre-scale distribution of ligands but also substrate rigidity, which is itself a major regulator of focal adhesions12. To this end, we developed a two-step protocol to combine polyacrylamide hydrogels with block co-polymer micelle nanolithography13. We thereby fabricated hydrogels that have on their surface nanopatterned, quasi-hexagonal arrays of gold nanoparticles (nanodots), functionalized with a small peptide (cyclic arginine–glycine–aspartate, cRGD), which is an integrin ligand (Fig. 1a). Owing to steric hindrance3,8, this system allows just one integrin protein to bind to each functionalized nanodot; it also allows us to control both nanodot spacing and substrate rigidity. Because hydrogels swell14, the spacing of the nanodots was greater on gels than on glass surfaces. This swelling was of approximately 20%, and was not affected by gel rigidity (Extended Data Fig. 1). We seeded human breast myoepithelial cells onto the substrates; the cells attached specifically to the nanodots by using α5β1 integrin proteins to bind the cRGD ligands. Blocking these integrins with an antibody, functionalizing the nanodots with a peptide with low affinity for integrin binding (arginine–glycine–glutamate, RGE)15, or functionalizing gels with cRGD in the absence of nanodots all inhibited cell attachment (Extended Data Fig. 2).

a, Top, the nanopatterned polyacrylamide substrates, showing gold nanodots (yellow) on top of polyacrylamide gels with embedded fluorescent beads (purple). A cell adhered through focal adhesions (orange) is shown, with its nucleus in blue. Bottom, spatial configuration of integrin–ECM bonds on the quasi-hexagonal pattern of nanodots coated with cRGD ligands (yellow integrins are in blue). Right, scanning electron micrograph of a 100-nm pattern on a polyacrylamide gel (one of two independent experiments is shown). b, Staining for phosphorylated paxillin protein (a component of focal adhesions) in cells seeded on glass substrates coated with either a homogeneous layer of cRGD or quasi-hexagonal distributed cRGD ligands (30-nm, 50-nm and 100-nm spacing). The right-hand image of each pair corresponds to the red rectangle in the left-hand image. c, Corresponding quantification of focal adhesion length (mean length of at least three focal adhesions per cell from n = 12 cells per condition, from two independent experiments). *** P < 0.001; one-way ANOVA. d, Staining of phosphorylated paxillin in cells seeded on polyacrylamide substrates with rigidities (Young’s moduli) from 0.5 kPa to 150 kPa, for nanodots spaced 50 nm, 100 nm or 200 nm apart. e, Corresponding quantification of focal-adhesion length (mean of at least three focal adhesions per cell from n = 10/10/11, 10/12/11, 10/11/11, 10/10/11, 12/11/11, 10/10/11, 11/11/11, 12/12/− cells for 50/100/200-nm-spaced substrates and increasing rigidity; two independent experiments). The effect of both rigidity and spacing was significant (P < 0.05; two-way ANOVA). f, Staining for the transcriptional regulator YAP in cells seeded on polyacrylamide substrates with rigidities ranging from 0.5 kPa to 150 kPa, with 50-nm-, 100-nm- or 200-nm-spaced nanodots. g, Corresponding quantification of nuclear/cytosolic YAP ratio (n = 15 cells per condition; two independent experiments); the effect of both rigidity and spacing was significant. (P < 0.05, two-way analysis of variance (ANOVA)). Error bars represent mean ± s.e.m. Scale bars represent 200 nm in a and 20 μm in b, d, f.
We then analysed how cells formed adhesion complexes on the substrates as a function of ligand spacing and substrate rigidity. As an initial control, we checked cell behaviour on a stiff substrate (glass). As expected, cells plated on non-patterned glass substrates (which were merely coated with a uniform gold layer and functionalized with cRGD) formed long focal adhesions that were rich in phosphorylated paxillin protein (Fig. 1b, c). On glass nanopatterned substrates with 30-nm spacing between nanodots, focal adhesions still formed, but were shorter. Cells on substrates with 50-nm or 100-nm spacing between nanodots exhibited only small, dotted adhesions. This confirms the reported maximum distance between bound integrins for focal-adhesion formation3,5, and indicates a length of between 30 nm and 50 nm for a potential molecular ruler in our system.
Also as expected and previously described10, cells seeded on very soft polyacrylamide gels formed small adhesive structures resembling nascent adhesions16 when ligands were spaced 50 nm or 100 nm apart, and formed focal adhesions only above a rigidity threshold (Young’s modulus) of 5 kPa (Fig. 1d, e). However, above this threshold we found several striking behaviours. First, focal adhesions formed on gels when ligands were spaced both 50 nm and 100 nm apart, though they did not on glass (Fig. 1b, c). Second, the dependency between focal-adhesion formation and ligand spacing was the reverse of that found on stiff substrates: cells formed longer focal adhesions as ligand spacing increased from 50 nm to 100 nm. Finally, adhesions seemed to collapse (drastically reduce their length) above a second rigidity threshold, which was 30 kPa for 100-nm-spaced substrates, and 150 kPa for 50-nm-spaced substrates. Because focal-adhesion collapse occurred at a lower rigidity for the higher spacing, this led to a regime (150 kPa) in which focal-adhesion length increased with decreasing spacing, reproducing the behaviour found on glass.
Thus, our results show that there is an optimal rigidity for adhesion formation, which decreases as ligand spacing increases. We then confirmed the generality, validity, and implications of these results in different ways. First, we increased ligand spacing on gels to 200 nm. Confirming the trend, this reduced the optimal rigidity even further, to 1.5 kPa (Fig. 1d, e). Second, we checked that different methods of quantifying adhesions led to the same trends. These methods involved transfecting green fluorescent protein (GFP)-labelled paxillin into live cells or staining phosphorylated paxillin in fixed cells; and measuring overall paxillin recruitment or measuring focal-adhesion length (Extended Data Fig. 3). Third, we carried out experiments in other cell types (mouse embryonic fibroblasts, human umbilical vein endothelial cells, and MCF 10A breast epithelial cells) and in myoepithelial cells seeded on nanodots coated with a different ligand, the collagen-mimicking GFOGER peptide16. The specific thresholds varied, but adhesion formation and collapse, and their dependency on ligand spacing and substrate rigidity, were maintained in all cases (Extended Data Fig. 4). Finally, we analysed the nuclear localization of the mechanosensitive transcriptional regulator YAP18, which correlates with focal-adhesion formation10. Indeed, YAP’s nuclear localization closely mirrored focal-adhesion length in all cases, showing a rigidity optimum that depended on ligand spacing (Fig. 1f, g).
Our results—which show adhesion formation on substrates with 50-nm, 100-nm and even 200-nm nanodot spacing, depending on the conditions—are inconsistent with a molecular-ruler mechanism, even if gel deformation were to reduce nanodot spacing. Indeed, although cells could potentially pull on neighbouring ligands to reduce their spacing down to the length of a molecular ruler, this could not explain why, on 1.5 kPa substrates, focal adhesions form only when nanodots are spaced by 200 nm and not shorter distances. To further discard the molecular-ruler hypothesis, we used super-resolution stochastic optical reconstruction microscopy (STORM) of 100-nm-spaced substrates to image paxillin clusters, which correctly reproduced the expected 100-nm spacing (Extended Data Fig. 5). There were no differences in spacing on 30-kPa or 150-kPa substrates, confirming that the increased adhesion formation on the softer 30-kPa substrate was not due to reduced nanodot spacing caused by gel deformability. Nor could the results be explained by differential regulation of cell spreading, which did not correlate well with adhesion formation, particularly on 50-nm- and 100-nm spaced substrates (Extended Data Fig. 3).
A plausible alternative is regulation by force, as suggested theoretically19. Mechanical forces are important in focal-adhesion maturation20,21, and we have shown10,11 that focal-adhesion growth in response to rigidity can be explained by force loading in integrins via a molecular-clutch mechanism. Furthermore, force transmission mediated by a molecular clutch is predicted to depend on ligand density22,23. We thus asked whether our results could be explained by a force-regulated, molecular-clutch mechanism.
Our previous molecular-clutch model involves a number of myosin motor proteins pulling on an actin filament, generating a rearward actin flow towards the cell centre. The substrate is modelled by a set of ECM-binding sites (corresponding to the functionalized gold nanodots here) connected to a spring (representing substrate elasticity). Molecular clutches, which comprise integrins and adaptor proteins, dynamically link the actin filament to the ECM substrate. When clutches are engaged, they become progressively loaded as myosin motors contract the actin filament. Mechanosensitive growth of focal adhesions is modelled by defining a force threshold in each clutch—a force which we previously identified as that leading to unfolding of the actin–integrin adaptor protein talin10. If any individual clutch surpasses this threshold before disengaging, it triggers a mechanosensing event that grows adhesions by increasing integrin recruitment. As integrins are recruited, the fraction of integrin-bound ligands increases, allowing adhesions to better withstand force.
To consider the effects of ligand distribution, we expanded this model in two ways (Fig. 2a; see Methods and Extended Data Table 1 for model description and parameters). First, we modelled ligand spacing by using springs to connect ligands not only to the substrate (with a spring constant ksub), but also to each other (with a spring constant klink). In this way, and as expected for an elastic substrate, forces applied to one ligand also deform its neighbours. Increasing klink increases this effect on neighbours, modelling the increased mechanical coupling between ligands that would result from reduced spacing. Second, we imposed a maximum integrin recruitment, because integrin clustering cannot grow indefinitely, but will be restricted by integrin packing and by the physical size of focal adhesions, stress fibres, and cells themselves.

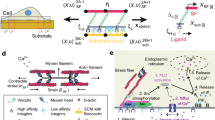
a, The molecular-clutch model. Within cells, myosin motors (black) pull on actin filaments (grey) with velocity v, exerting a force on a set of parallel clutches (formed by adaptor proteins, red, and integrins, blue) which dynamically bind and unbind cRGD ligands (yellow) with on and off rates kon and koff. Mechanosensitivity is introduced by setting a force threshold, Fthreshold, in each clutch that triggers further integrin recruitment when surpassed (brown star). The elastic substrate is represented by springs connecting ligands to the substrate (ksub, black) and to each other (klink, orange). b, The effect of ligand spacing on clutch forces: as the spacing decreases, so does the force loading on each individual clutch. c, Model predictions (solid lines) and experimental average values (data points, from Fig. 1e) of focal-adhesion length as rigidity increases for differently spaced ligands. Model parameters changed were nl (180 for 50 nm, 130 for 100 nm and 5 for 200 nm) and the ratio klink/ksub (10 for 50 nm, 5 for 100 nm and 3 for 200 nm). d, Examples of cell tractions (colour coding) exerted on substrates of differing rigidity and differing ligand spacing. e, Corresponding model predictions (solid lines) and experimental average values of cell tractions (n = 13/13, 11/16, 11/19, 16/13, 13/16, 13/14 cells for 50/100-nm substrates and increasing rigidity; mean of two independent experiments). f, Examples of lifeact-GFP-transfected cells plated on substrates of increasing rigidity. Insets are kymographs showing the movement of actin features along the lines marked in red. The slopes of the traces created by the features (marked with lines) were used to calculate actin speed. g, Corresponding model predictions (solid lines) and experimental average values for actin speed (at least three traces were obtained per cell from n = 7/9, 9/9, 11/9, 8/7, 8/7, 9/9 cells on 50/100-nm-spaced substrates of increasing rigidity; two independent experiments). Scale bars represent 20 μm in the main images and 20 s per 2 μm (x = y axes) in the kymographs. Error bars represent mean ± s.e.m.
We then used this model to evaluate the role of ligand spacing. As the number of clutches is reduced and their spacing increases, the force exerted by myosin is distributed among fewer clutches, increasing the force loading on each individual clutch (Fig. 2b). This has no effect on very soft substrates, where force loading remains too low to reach the force threshold in any case, and adhesions do not grow. However, as rigidity increases, clutches with higher spacing are more likely to reach the force threshold, increasing integrin recruitment. Because these focal adhesions grow more, they also reach their maximum recruitment at a lower rigidity. At this point, the increased force loading caused by increased rigidity can no longer be compensated by further integrin recruitment, and the adhesion collapses. Thus, this framework can explain the experimental differences. Accordingly, running the computational model with a base set of parameters (Extended Data Table 1) and modifying only the number of ECM ligands (nl) and their coupling (klink) correctly reproduces the effect of ligand spacing on focal adhesions (Fig. 2c). For 200-nm substrates, we note that the experimental effects were even larger than the range that the model could predict.
To test this model further, we examined its predictions regarding substrate–cell force transmission and actin flows. Our molecular-clutch model11 predicts an initial increase in force transmission with rigidity, then a plateau or even a slight decrease, and finally an increase until adhesions and forces collapse. As ligand spacing decreases, the plateau is shifted to lower rigidities, and lower forces. We verify these predictions here by measuring them experimentally using traction force microscopy at all rigidities except 150 kPa (for which cell-induced gel displacements were too small to resolve). Although agreement with experiments was not exact in all cases, running the model with the same parameters used above correctly reproduced the trends and relative differences of measured experimental tractions (Fig. 2d, e). Regarding actin flows, the clutch model predicts that they should be anti-correlated with forces24, because increased force transmission impairs and slows myosin function. Indeed, measured actin flows did show opposite trends to forces in response to both rigidity and ligand spacing, and were correctly reproduced by the model using the same parameters (Fig. 2f, g). Interestingly, actin flow measurements were available for 150 kPa substrates, allowing us to visualize the regime inducing adhesion collapse (for 100-nm spacing). As predicted by the model, this collapse resulted in increased actin flows (Fig. 2f, g).
We then explored whether force loading regulated by a molecular clutch could explain cell responses to not only overall ligand density, but also ligand distribution. We used substrates with the same density of nanodots and the same mean interparticle distance, but with a disordered rather than ordered quasi-hexagonal distribution (Extended Data Fig. 6)8. Because of this spatial disorder, the model predicts that force will be distributed less evenly among clutches (Fig. 3a). This would lead some clutches to experience high loads, increasing the likelihood of surpassing the mechanosensing force threshold, favouring focal-adhesion growth, and shifting focal adhesion collapse to lower rigidities. We verified this prediction experimentally using substrates with 50-nm spacing (Fig. 3b, c) and 100-nm spacing (Fig. 3f, g): in both cases, focal-adhesion growth at intermediate rigidities was increased drastically, and focal-adhesion collapse moved to lower rigidities. Of note, and unlike with ordered patterns, disordering the pattern allowed us to visualize focal adhesion collapse on 50-nm polyacrylamide substrates. The effect of pattern disorder was successfully modelled by modifying only the parameter that represents ligand spacing (klink), to which we assigned not a constant value for all ligands, but a distribution of random values centred on a mean (Fig. 3c, g). In terms of force transmission, the model predicts that, owing to the increased adhesion formation, disorder eliminates the plateau observed at intermediate rigidities, leading to a monotonic force increase with rigidity up to focal-adhesion collapse. This was verified experimentally, and was modelled successfully with the same parameters (Fig. 3d, e, h, i). Notably, disordering the pattern on 100-nm substrates shifted the onset of focal adhesion collapse to a rigidity low enough (30 kPa) to measure force transmission. This allowed us to verify the prediction that focal-adhesion collapse at high rigidities is also associated with a decrease in force transmission (Fig. 3i).

a, Effect of ligand disorder on clutch force. b, Staining of phosphorylated paxillin in cells seeded on substrates of varying rigidity, and with ordered or disordered ligand spacing. The left-hand images of each pair are magnifications of the rectangular regions in the right-hand images. c, For 50-nm substrates are shown corresponding model predictions (lines) and experimental average values for adhesion length (means of at least three adhesions per cell from n = 10/10, 10/10, 10/10, 11/10, 11/12, 11/10, 11/11 cells in disordered/ordered conditions and on substrates of increasing rigidity; two independent experiments). Differences between ordered and disordered conditions were significant (P < 0.05; two-way ANOVA). d, e, For the same 50-nm substrates as in c are shown: d, examples of cell tractions, and e, corresponding model predictions (lines) and experimental average traction values (n = 14/13, 12/11, 12/11, 19/16, 16/13, 21/13 cells for disordered/ordered conditions; two independent experiments). Differences between ordered and disordered conditions were significant (P < 0.05; two-way ANOVA). f, g, For 100-nm substrates are shown: f, phosphroylated-paxillin staining on ordered and disordered substrates, and g, corresponding model predictions (lines) and experimental average traction values (at least three adhesions per cell for n = 10/10, 10/12, 10/11, 11/10, 12/11, 11/10, 11/11 cells on disordered/ordered ligands; two independent experiments). h, i, For the same 100-nm substrates as in g are shown: h, examples of cell tractions, and i, corresponding model predictions (solid lines) and experimental average traction values (n = 12/13, 15/16, 18/19, 14/13, 11/16, 15/14 cells on disordered/ordered ligands; two independent experiments). Scale bars represent 20 μm. Error bars represent mean ± s.e.m.
Finally, we verified a fundamental hypothesis of the model: that focal-adhesion collapse at high rigidities is due to excessive loading of integrin–ECM bonds, which can no longer be compensated with adhesion growth. This hypothesis leads to the counterintuitive prediction that in this ‘collapsed’ regime, decreasing force loading (for instance by impairing myosin function) should lead to adhesion growth. To confirm this, we seeded cells on rigid 150 kPa gels with 100-nm-spaced nanodots, thereby generating collapsed focal adhesions. One hour after seeding, we treated cells with the myosin inhibitor blebbistatin. As predicted, using a low concentration of blebbistatin (5 μM) resulted in focal-adhesion growth (Fig. 4a–c) compared with the control condition—effectively putting cells in the intermediate force-loading regime where focal-adhesion growth is favoured (Fig. 4a). Also as expected, using a higher concentration of blebbistatin (15 μM) reversed the effect, as force loading was disrupted enough to bring cells to the low-rigidity regime where focal adhesions are also impaired (Fig. 4a–c). Conversely, increasing myosin contractility using calyculin A25 in cells with the largest adhesions (100-nm spacing, 30 kPa) brought cells to the collapsed regime, decreasing adhesion length (Fig. 4d–f).

a, Using the myosin inhibitor blebbistatin at concentrations of 5 μM or 15 μM progressively decreases contractility in cells with collapsed adhesions (150 kPa substrate, 100-nm ligand spacing); this should first bring adhesion length to its maximum, and then decrease it again. b, Staining for phosphorylated paxillin in control (−) and blebbistatin-treated cells on 150 kPa substrates with 100-nm ligand spacing. c, Corresponding quantification of adhesion length (mean of at least three adhesions per cell from n = 14 cells per condition; two independent experiments). d, Using calyculin A to increase contractility in cells with maximum adhesions (30 kPa substrate; 100-nm spacing) should decrease adhesion length. e, Staining for phosphorylated paxillin in control and calyculin-A-treated cells on 30 kPa substrates with 100-nm spacing. f, Corresponding quantification of adhesion length (mean of at least three adhesions per cell from n = 15 cells per condition; two independent experiments; two-tailed Student’s t-test). Scale bars represent 20 μm. **, P < 0.01; ***, P < 0.001. Error bars, mean ± s.e.m.
The field of cell–matrix adhesion is mature, and several studies have addressed how adhesions are regulated by molecular interactions26,27,28 and physical signals10,20,29,30; there is wide consensus that cells respond to increases in both rigidity and ligand density by promoting adhesion growth3,4,10,11,31. Strikingly, we find here the opposite behaviour in response to both factors. This behaviour is explained not by a distance-sensing mechanism per se, but by regulation of molecular force loading, within a predictive model that integrates the effects of rigidity, ligand distribution, and contractility. The resulting cellular response includes the surprising feature of adhesion collapse under high load, which can explain previous findings on stiff substrates showing how ligand molecular length regulates adhesion stability32, and how increasing ligand spacing renders adhesions unstable7,19 and unable to sustain large forces on integrins over time33. Our results provide a general framework of how cells sense spatial and physical information at the nanoscale, precisely tuning the range of conditions at which they form adhesions and activate transcriptional regulation via YAP. This mechanism might be harnessed by cells in the myriad of physiological and pathological processes that are regulated by mechanical factors and ECM characteristics.
Methods
Preparation of nanopatterned substrates on glass surfaces
Nanopatterned substrates were prepared as described3,4,7. Briefly, polystyrene(x)-b-poly(2-vinylpyridine)(y) diblock copolymers (PolymerSource Inc.) and polystyrene standard (Alfa Aesar) were dissolved in toluene and stirred for 24 hours at room temperature. Different compositions were used to generate ordered and disordered substrates with different spacing (Extended Data Table 2). HAuCL4•3H20 (Sigma-Aldrich) was added to the micellar solutions with a specific loading parameter defined as  . For disordered structures the micellar gold solution was mixed with a polystyrene solution in a 1/1 ratio. 10 μl of the solution were spin-coated (WS-400A-6NPP/Lite, Laurell Technologies Cooperation) onto round coverglasses of 12 mm diameter, previously cleaned with piranha solution. Samples were treated with oxygen plasma (TePla 100-E, 0.4 mbar, 150 W, 10 min) to remove the polymer matrix.
. For disordered structures the micellar gold solution was mixed with a polystyrene solution in a 1/1 ratio. 10 μl of the solution were spin-coated (WS-400A-6NPP/Lite, Laurell Technologies Cooperation) onto round coverglasses of 12 mm diameter, previously cleaned with piranha solution. Samples were treated with oxygen plasma (TePla 100-E, 0.4 mbar, 150 W, 10 min) to remove the polymer matrix.
Transfer of nanopatterns to polyacrylamide gels
Nanostructured glass surfaces were activated with ultraviolet light for 30 min, incubated in 10 mM N,N′-bis-(acryloyl)cystamine (Sigma-Aldrich) in ethanol in the dark for 1 hour, and washed thoroughly with pure ethanol. Next, nanostructured surfaces were dried with nitrogen. Polyacrylamide gels were prepared as described11. Briefly, glass-bottom dishes were incubated with a solution of acetic acid, 3-(trimethoxysilyl)propyl methacrylate (Sigma) and ethanol (1/1/14), and washed three times with 96% ethanol. A solution containing 0.5% ammonium persulphate, 0.2% tetramethylethylenediamine (Sigma), and 2% fluorescent 200-nm red carboxylated nanobeads (Invitrogen) was mixed with different concentrations of acrylamide and bis-acrylamide to make gels of different rigidities (see Extended Data Table 3). 10 μl of this solution were then placed in the centre of glass-bottom dishes and covered with 12-mm nanostructured surfaces. After 20 min of gel polymerization, hydrogels were soaked in phosphate-buffered saline (PBS) and incubated in the oven for 72 hours at 37 °C, allowing them to swell. Hydrogels were then stabilized at room temperature and the patterned glass surfaces were removed carefully from the hydrogel. Hydrogels with nanopatterned nanodots were gently washed with PBS and then incubated with 25 μM cRGD-thiol (cyclo [Arg–Gly–Asp]–d-Phe–Lys(2-aminohexanoic acid–mercaptopropionic acid), PCS-31062-PI, Peptides International) at room temperature for 4 hours. Afterwards, cRGD-conjugated nanopatterned hydrogels were washed five times (for at least 10 min each time) to remove unbound peptides before cell seeding. For GFOGER experiments, after removing the glass surfaces, gels were incubated with 1 mM of the hetero-bifunctional linker 11-mercaptoundecanoyl N-hydroxysuccinimide ester (MU-NHS) (Prochimia) for 3 hours, then with 1 μM of GFOGER peptide overnight. GFOGER-conjugated nanopatterned hydrogels were washed five times (for at least 10 min each time) to remove unbound peptides before cell seeding.
Substrate characterization by scanning electron microscopy (SEM)
Nanostuctured surfaces were sputtered with carbon (low-vacuum coater EM ACE200, Leica) and imaged by SEM (Leo1530, Zeiss) with an in-lens detector and 5 kV acceleration voltage at working distances between 9 mm and 11 mm. Polyacrylamide hydrogels with embedded gold nanodots were vitrified, mounted in a liquid-nitrogen-cooled stage, and transferred to a freeze-fracture system (EM BAF060, Leica). Samples were heated to −90 °C, kept in vacuum for 45 min to sublimate the water at the interfaces, and coated with carbon. Samples were further transferred to the cryo-SEM (Ultra 55 FE-SEM, Zeiss) by an evacuated liquid-nitrogen-cooled shuttle (BAL-TECH VLC 100). Images were recorded at low-temperature conditions (T = −130± 5 °C) and low acceleration voltages of 1–1.5 kV because of the low conductivity of the samples, with a working distance of 3 mm. Resulting electron micrographs were analysed in ImageJ (National Institutes of Health) by measuring the distances between a gold nanoparticle and its k-nearest neighbours (k = 6 for ordered nanostructures; 4 < k < 8 for disordered nanostructures) for at least 300 particles of two or more individual nanostructures per condition.
Cell culture and reagents
Human breast myoepithelial immortalized cell lines have been described previously11,34. We cultured them in Hams-F12 (Sigma, N4888) media supplemented with 10% foetal bovine serum (FBS), 1% penicillin streptomycin, hydrocortisone (1 μg ml−1), epidermal growth factor (EGF; 10 ng ml−1) and insulin (5 μg ml−1). Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (CC-2517) and cultured in endothelial growth medium-2 (EGM-2; Lonza CC-4176). Mouse embryonic fibroblasts (MEFs) have been described35, and were cultured in Dulbecco’s modified eagle medium (DMEM; LifeTechnologies, 41965-039) media supplemented with 10% FBS and 1% penicillin streptomycin. Mammary epithelial cells (MCF 10A) were purchased from ATCC and cultured in DMEM-F12 (LifeTechnologies, 21331-020) with 5% horse serum, 1% penicillin streptomycin, EGF (20 ng ml−1), hydrocortisone (0.5 μg ml−1), cholera toxin (100 ng ml−1), and insulin (10 μg ml−1). Myoepithelial cells were authenticated in their laboratory of origin through expression of the proteins integrin β4, P-cadherin, cytokeratin 17, and desmoglein 3. Other cell lines (used only to verify the generality of our findings) were not authenticated. For all experiments, cells were gently washed with PBS twice, trypsinized, and resuspended in media without FBS. After centrifugation, cells were seeded on hydrogels in media without FBS. To block α5β1 integrins, cells were incubated with an anti-α5β1 antibody (30 μg ml−1, clone JBS5–MAB1969, Millipore) for 30 min before seeding. Cell attachment was evaluated 1 hour after seeding. For blebbistatin experiments, cells were treated with the indicated concentrations of blebbistatin (CalBiochem) for 30 min. For calyculin A experiments, cells were treated with the indicated concentration (Merck Millipore) for 30 min. All cells tested negative for mycoplasma contamination.
Traction force measurements
Traction force measurements were carried out as described10,11. Briefly, cells seeded on gels were placed on an inverted microscope (Nikon Eclipse Ti). Single cells were tracked for 3 hours while we acquired phase-contrast images of the cells and fluorescence images of the embedded nanobeads using a ×40 objective. Cells were then trypsinized, and an image of bead position in the relaxed state of the gel was acquired. By comparing bead positions with and without cells, a map of gel deformations caused by cells was first obtained using custom particle-imaging-velocimetry software. Then, after assuming that gel displacements were caused by forces exerted by cells in the cell–gel contact area, we calculated the corresponding map of cell forces using a previously described Fourier transform algorithm36. The average forces per unit area exerted by each cell were then calculated. Force measurements for each cell were taken once per hour during the measurement, and the average value for all time measurements was used.
Rearward-flow measurements
To measure the rearward flow of actin filaments, we transfected cells with LifeAct-GFP using a jetPRIME transfection kit (Polyplus transfection) one day before measurements. Cells were then plated on gels of varying rigidity, and imaged every second for 2 min with ×60 oil-immersion objective (numerical aperture (NA) 1.40) with spinning-disc confocal microscopy (Andor). For each cell, kymographs were obtained at the cell periphery, and actin speed was measured from the slope of actin features observed in the kymographs. In cells plated on 0.5 kPa gels, actin features were so diffuse that no reliable slopes could be measured in kymographs.
Immunostaining and adhesion quantification
For fluorescence staining of cell–substrate adhesions, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and labelled first with primary antibody against either phosphorylated paxillin (Cell Signaling 2541S, 1/50 dilution) or YAP (clone 63.7 produced in mice; Santa Cruz catalogue no. sc-101199, 1/200 dilution) for 1 hour at room temperature, and then with anti-rabbit or anti-mouse Alexa-conjugated secondary antibody (Invitrogen) for 1 hour at room temperature. Fluorescence images were then acquired with a ×60 oil-immersion objective (NA 1.40) using a Nikon Eclipse Ti microscope. To quantify adhesion lengths, we identified focal adhesions manually and measured their length. To provide an alternative quantification unbiased by the identification of specific structures, we also quantified the average intensity of phosphorylated-paxillin staining in regions at the cell edge containing both adhesions and surrounding areas, as described11. Then, we subtracted the background intensity value calculated from neighbouring cell areas lacking adhesions. The measured trends as a function of both rigidity and ligand density were the same in both quantifications. The degree of nuclear localization of YAP was assessed by calculating the ratio of YAP fluorescence in the nuclear region to YAP fluorescence in the immediately adjacent cytoplasmic region. Nuclear and cytoplasmic regions were previously determined by co-staining the nucleus with Hoechst 33342.
Measurements of gel rigidity
The rigidity (Young’s modulus) of polyacrylamide gels was measured by atomic force microscopy as described37. Briefly, measurements were made with a custom-built atomic force microscope attached to an inverted optical microscope (Nikon TE200). Silicon nitride pyramidal tips with an effective half-angle θ of 20° and a nominal spring constant of k = 0.01–0.03 N m−1 were used (MLCT, Bruker). The actual spring constant was calibrated by thermal tuning using the simple harmonic oscillator model38. The Young’s modulus was measured by recording ten force-displacement curves with a peak-to-peak amplitude of 6 μm and a frequency of 1 Hz. Three points near the gel centre were selected in each gel, separated 5 μm from each other. For each rigidity, six or more gels produced in two batches were measured. To compute the Young’s modulus (E), we fitted the Hertz model equation for pyramidal tips to the force-displacement curves. The equation was fitted for an effective indentation of 1,000 nm for all rigidities except 150 kPa, where 500 nm was used.
STORM imaging and distance assessment
To perform direct STORM (dSTORM) imaging, we mounted immunostained cells on gels of different rigidity on a 24 mm × 24 mm glass coverslip using Vectashield mounting medium (H-1000). For image acquisitions, the samples were flipped and placed on the microscope’s sample holder. This allowed the excitation light to pass through the optically matched layer of Vectashield and to be focused on the focal adhesions located at the interface between the cells and the gel substrate. F8811 spheres (Life Technology) on the gel surface allowed for the correction of mechanical drift during acquisition, while the mounting medium allowed for the photoswitching of the fluorophores necessary to perform dSTORM. dSTORM images were acquired using a Nikon N-STORM 4.0 system configured for total internal reflection fluorescence (TIRF) imaging. Alexa647-labelled secondary antibodies were imaged by means of a 647-nm laser (160 mW) while F8811 spheres were imaged using a 488-nm laser (80 mW). No activation ultraviolet light was used. Fluorescence was collected using a Nikon ×100, 1.49 NA oil-immersion objective and passed through a quad-band pass dichroic filter (97335 Nikon). Images were acquired onto a 128 × 128 pixel region (pixel size 0.16 μm) of a Hamamatsu ORCA–Flash 4.0 camera with an integration time of 5 ms. A total of 50,000 frames was acquired for the 647 channel. Every one hundred imaging frames, one image of the 488 channel was acquired to perform drift correction. STORM images were analysed with the STORM module of the NIS element Nikon software, which generates a list of localizations by Gaussian fitting of blinking dyes in the acquired movie of conventional microscopic images. To avoid overcounting, the software counts blinkings detected in consecutive frames as single. For pattern analysis of focal adhesions, the lists of localizations corresponding to focal-adhesion regions (size about 2.5 μm) were imported and converted in binary images with a pixel size of 3 nm, using a custom Matlab script. Then, binary images were dilated, and clusters identified. A threshold radius was set to discard single blinks not belonging to the cluster. A first analysis of the patterns was then performed with a previously developed Matlab script39,40 to calculate the pair-correlation functions of cluster patterns up to distances of 900 nm from the identified clusters. Additionally, the centroid of each cluster was identified and the distance of the nearest neighbour was calculated with custom Matlab script.
Statistical analysis
Data reported throughout the manuscript are mean ± s.e.m. Statistical analyses were done with two-tailed Student’s t-test when two cases were compared, and with analysis of variance (ANOVA) tests when more cases were analysed. If data did not meet normality criteria, equivalent non-parametric tests were used. No statistical methods were used to predetermine sample size.
Implementation of the molecular-clutch model
Base model. The present implementation of the molecular-clutch model is based on a model that has been described in detail11, which was in turn based on previous implementations22,23,24. Briefly, the model considers a given number of myosin molecules, nm, pulling on an actin fibre, which in the absence of load contracts at a rearward speed vu. The substrate is represented by a set of ligands nl (here, cRGD-functionalized gold nanodots) connected to springs representing substrate elasticity. The actin fibre binds to ligands dynamically through molecular clutches, which represent a complex containing an adaptor protein and integrin. Those clutches have characteristic on and off rates kont and koff. The off rate (in units of s−1) depends on force as a catch bond, which we modelled according to reported experimental values for the strength of fibronectin–α5β1 bonds41. kont (in units of μm2 s−1) is the true on-rate for each ligand, which must be multiplied by the available density of integrins (dint) to provide an effective on rate kon with units of s−1. The model is implemented as a Monte Carlo stochastic simulation that starts with all clutches disengaged and actin flowing freely. As the simulation progresses and clutches engage, they pull on the substrate, loading force on the clutches and affecting koff. In addition, force exerted by the substrate on actin slows the myosin motors linearly, which are assumed to stall and stop completely at a force of nm × Fm, where Fm is the stall force of an individual motor. To model mechanosensitive adhesion growth (reinforcement), if individual clutches exceed a given force threshold, Fthreshold, before disengaging, integrins are recruited. This is implemented by increasing dint by a factor dadd. In previous work, we identified this mechanosensitive event as the unfolding of talin10. The simulation is run for 100 s with time steps of 2 ms, and run 20 times per condition to obtain average results.
Model expansion. To the model described previously and summarized above, we added two main features to model the effect of substrate spacing and distribution. First, we simply introduced a maximum value for dint (dint,max) to model the fact that integrin recruitment has a physical limit. To calculate a parameter that is predictive of adhesion size, in simulations we multiplied dint by the fraction of bound clutches. To compare this with experimental adhesion lengths, we scaled model predictions for cells on 100-nm substrates as a function of rigidity to fall between the maximum and minimum experimental values. All other conditions were scaled by using the same 100-nm reference to retain the relative differences predicted by the model. The second and most important new feature was an improvement of the elastic characterization of the system. In our previous models, the substrate was modelled simply as a set of ligands connected to each other with a rigid rod, which was in turn connected to an elastic spring. Whereas this effectively modelled substrate rigidity, deformation in all ligands was always the same; it did not allow us to model the elastic coupling between ligands—that is, the fact that a force applied to a ligand will deform neighbouring ligands to a lesser extent as the distance increases. To introduce this, we modelled the substrate not as a single spring, but as a network of springs, where each ligand had a spring connecting it to the substrate (ksub) and one connecting it to neighbouring ligands (klink) (Fig. 2a). For each ligand, its force, Fi, and displacement from rest position, xi, were then calculated as:

At each time step, bound ligands were displaced by the amount of actin movement during the step (providing a known xi), and unbound ligands were considered to be under zero load (providing a known Fi). This led to a system with nl linear equations and nl unknowns, corresponding to the forces of bound ligands and the positions of unbound ligands. After resolving the system, the total force exerted by all ligands was calculated. The degree of mechanical coupling between ligands (modelled by klink) will depend on how forces are transmitted between nanobeads both through the cell cytoplasm and through the polyacrylamide gels. Those nanoscale parameters are essentially inaccessible experimentally and thus klink values were merely adjusted to fit the data. Importantly, however, this approach correctly reproduced the fact that local forces will induce decreasing deformations as distance increases. To take into account the fact that the overall rigidity of the system depends on both ksub and klink, we used these values to calculate an effective network constant, knet, corresponding to the spring constant obtained when pulling on one ligand connected to the entire network. We calculated knet as an iterative process as:



The factor 2 in the last expression corresponds to considering the ligands both to the right and to the left of the one being pulled. Although this calculation corresponds to the ligand at the centre of the system, we note that, with the parameters used, knet quickly converged and was largely independent of either nl or ligand position within the system. This knet was then used to calculate an equivalent substrate Young’s modulus by assuming a characteristic adhesion radius, r0, as described11,42. The same characteristic radius was used to convert the one-dimensional model output of force into tractions (force per unit area).
Model parameters and prediction. All model parameters and their origins are described in Extended Data Table 1. The same base set of parameters was used to model all conditions, and the different conditions were modelled by modifying only the relevant parameters in the relevant direction. Specifically, the effect of increased ligand spacing was modelled by decreasing the number of clutches, nl, and the coupling between ligands, klink. More precisely, both ksub and klink scaled with rigidity, and to model ligand spacing we altered their ratio (klink/ksub). The effect of disorder was modelled by introducing a different value of klink to each clutch, rather than a constant value. Following the long-tailed distribution of distances observed in Extended Data Fig. 4, we chose the values of klink to be randomly distributed according to a Poisson distribution, with a peak corresponding to the value used in the ordered simulations. Of note, using a Gaussian rather than a Poisson distribution led to the same relative trends. Regarding model predictions, those concerning adhesion formation and the effect of ligand distribution are discussed in the main text. However, an interesting point to add is that of the two parameters modified in the simulations (nl and klink/ksub), nl is the one responsible for shifting the optimal rigidity for adhesion formation, confirming previous analyses22,23. By contrast, klink/ksub serves to modulate the height of the peak. Predictions regarding rigidity and force/actin flow have been discussed extensively10,11,24. Briefly, in the absence of adhesion reinforcement and recruitment, the molecular-clutch model predicts a biphasic force–rigidity relationship, in which forces first increase and then decrease with rigidity. The introduction of force-dependent reinforcement triggers adhesion growth and force increase above a rigidity threshold, reverting the downward force trend predicted at high rigidities. Depending on the specific threshold, reinforcement can eliminate the high-rigidity downward trend completely if the threshold is low (leading to a monotonically increasing force–rigidity curve), or only partially. In this case, the force–rigidity curve first increases, then plateaus or even decreases slightly, and then increases again because of reinforcement. We see both cases here: in ordered configurations (Fig. 2), we see the plateau; in disordered configurations (Fig. 3), the threshold is shifted to lower rigidities and we see a monotonic curve.
Data availability and code availability
The data that support the findings of this study, and the Matlab code used to generate the computational model, are available from the corresponding author on reasonable request.
References
Lu, P., Weaver, V. M. & Werb, Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395–406 (2012)
Daley, W. P., Peters, S. B. & Larsen, M. Extracellular matrix dynamics in development and regenerative medicine. J. Cell Sci. 121, 255–264 (2008)
Arnold, M. et al. Activation of integrin function by nanopatterned adhesive interfaces. ChemPhysChem 5, 383–388 (2004)
Cavalcanti-Adam, E. A. et al. Lateral spacing of integrin ligands influences cell spreading and focal adhesion assembly. Eur. J. Cell Biol. 85, 219–224 (2006)
Altrock, E., Muth, C. A., Klein, G., Spatz, J. P. & Lee-Thedieck, C. The significance of integrin ligand nanopatterning on lipid raft clustering in hematopoietic stem cells. Biomaterials 33, 3107–3118 (2012)
Amschler, K., Erpenbeck, L., Kruss, S. & Schon, M. P. Nanoscale integrin ligand patterns determine melanoma cell behavior. ACS Nano 8, 9113–9125 (2014)
Cavalcanti-Adam, E. A. et al. Cell spreading and focal adhesion dynamics are regulated by spacing of integrin ligands. Biophys. J. 92, 2964–2974 (2007)
Huang, J. et al. Impact of order and disorder in RGD nanopatterns on cell adhesion. Nano Lett. 9, 1111–1116 (2009)
Schvartzman, M. et al. Nanolithographic control of the spatial organization of cellular adhesion receptors at the single-molecule level. Nano Lett. 11, 1306–1312 (2011)
Elosegui-Artola, A. et al. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 18, 540–548 (2016)
Elosegui-Artola, A. et al. Rigidity sensing and adaptation through regulation of integrin types. Nat. Mater. 13, 631–637 (2014)
Plotnikov, S. V., Pasapera, A. M., Sabass, B. & Waterman, C. M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 151, 1513–1527 (2012)
Lohmüller, T. et al. Nanopatterning by block copolymer micelle nanolithography and bioinspired applications. Biointerphases 6, MR1–MR12 (2011)
Ahmed, E. M. Hydrogel: preparation, characterization, and applications: a review. J. Adv. Res. 6, 105–121 (2015)
Hersel, U., Dahmen, C. & Kessler, H. RGD modified polymers: biomaterials for stimulated cell adhesion and beyond. Biomaterials 24, 4385–4415 (2003)
Lawson, C. et al. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 196, 223–232 (2012); erratum 196, 387 (2012)
Emsley, J., Knight, C. G., Farndale, R. W., Barnes, M. J. & Liddington, R. C. Structural basis of collagen recognition by integrin alpha2beta1. Cell 101, 47–56 (2000)
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011)
de Beer, A. G. et al. Force-induced destabilization of focal adhesions at defined integrin spacings on nanostructured surfaces. Phys. Rev. E 81, 051914 (2010)
Riveline, D. et al. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J. Cell Biol. 153, 1175–1186 (2001)
Wolfenson, H., Bershadsky, A., Henis, Y. I. & Geiger, B. Actomyosin-generated tension controls the molecular kinetics of focal adhesions. J. Cell Sci. 124, 1425–1432 (2011)
Bangasser, B. L. & Odde, D. J. Master equation-based analysis of a motor-clutch model for cell traction force. Cell. Mol. Bioeng. 6, 449–459 (2013)
Bangasser, B. L., Rosenfeld, S. S. & Odde, D. J. Determinants of maximal force transmission in a motor-clutch model of cell traction in a compliant microenvironment. Biophys. J. 105, 581–592 (2013)
Chan, C. E. & Odde, D. J. Traction dynamics of filopodia on compliant substrates. Science 322, 1687–1691 (2008)
Peterson, L. J. et al. Simultaneous stretching and contraction of stress fibers in vivo. Mol. Biol. Cell 15, 3497–3508 (2004)
Humphries, J. D. et al. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J. Cell Biol. 179, 1043–1057 (2007)
Kanchanawong, P. et al. Nanoscale architecture of integrin-based cell adhesions. Nature 468, 580–584 (2010)
Choi, C. K. et al. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 10, 1039–1050 (2008)
Prager-Khoutorsky, M. et al. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat. Cell Biol. 13, 1457–1465 (2011)
Oakes, P. W., Beckham, Y., Stricker, J. & Gardel, M. L. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 196, 363–374 (2012)
Engler, A. et al. Substrate compliance versus ligand density in cell on gel responses. Biophys. J. 86, 617–628 (2004)
Pallarola, D. et al. Interface immobilization chemistry of cRGD-based peptides regulates integrin mediated cell adhesion. Adv. Funct. Mater. 24, 943–956 (2014)
Liu, Y. et al. Nanoparticle tension probes patterned at the nanoscale: impact of integrin clustering on force transmission. Nano Lett. 14, 5539–5546 (2014)
Allen, M. D. et al. Altered microenvironment promotes progression of preinvasive breast cancer: myoepithelial expression of αvβ6 integrin in DCIS identifies high-risk patients and predicts recurrence. Clin. Cancer Res. 20, 344–357 (2014)
Roca-Cusachs, P. et al. Integrin-dependent force transmission to the extracellular matrix by alpha-actinin triggers adhesion maturation. Proc. Natl Acad. Sci. USA 110, E1361–E1370 (2013)
Butler, J. P., Tolic-Norrelykke, I. M., Fabry, B. & Fredberg, J. J. Traction fields, moments, and strain energy that cells exert on their surroundings. Am. J. Physiol. Cell Physiol. 282, C595–C605 (2002)
Alcaraz, J. et al. Microrheology of human lung epithelial cells measured by atomic force microscopy. Biophys. J. 84, 2071–2079 (2003)
Hutter, J. L. & Bechhoefer, J. Calibration of atomic-force microscope tips. Rev. Scient. Instr. 64, 1868–1873 (1993)
Sengupta, P., Jovanovic-Talisman, T. & Lippincott-Schwartz, J. Quantifying spatial organization in point-localization superresolution images using pair correlation analysis. Nat. Protocols 8, 345–354 (2013)
Veatch, S. L. et al. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS One 7, e31457 (2012)
Kong, F., García, A. J., Mould, A. P., Humphries, M. J. & Zhu, C. Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 185, 1275–1284 (2009)
Ghibaudo, M. et al. Traction forces and rigidity sensing regulate cell functions. Soft Matter 4, 1836–1843 (2008)
Molloy, J. E., Burns, J. E., Kendrick-Jones, J., Tregear, R. T. & White, D. C. Movement and force produced by a single myosin head. Nature 378, 209–212 (1995)
Litvinov, R. I. et al. Resolving two-dimensional kinetics of the integrin alphaIIbbeta3-fibrinogen interactions using binding-unbinding correlation spectroscopy. J. Biol. Chem. 287, 35275–35285 (2012)
Roca-Cusachs, P., Iskratsch, T. & Sheetz, M. P. Finding the weakest link—exploring integrin-mediated mechanical molecular pathways. J. Cell Sci. 125, 3025–3038 (2012)
Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitiveness (grants BFU2016-79916-P and BFU2014-52586-REDT to P.R.-C.; BFU2015-65074-P to X.T.; DPI2015-64221-C2-1-R to J.M.G.-A.; PI14/00280 to D.N.; SAF2016-75241-R (MINECO-FEDER) to L.A.), the European Commission (grant agreement SEP-210342844 to X.T. and P.R.-C.), the Generalitat de Catalunya (grant 2014-SGR-927), the European Research Council (CoG-616480 to X.T. and StG 306571 to J.M.G.-A.), Obra Social ‘La Caixa’, Fundació la Marató de TV3 (project 20133330 to P.R.-C.), the German Science Foundation (DFG SFB1129 P15 to E.A.C.-A.), and the EMBO Young Investigator Programme. A.E.-A., R.O., and L.A. were supported respectively by a Juan de la Cierva Fellowship (Spanish Ministry of Economy and Competitiveness, fellowship number IJCI-2014-19156), an FI fellowship (Generalitat de Catalunya), and a Ramon y Cajal Fellowship (Spanish Ministry of Economy and Competitiveness). The support of the Max Planck Society and the Alexander von Humboldt foundation (to I.P.) is acknowledged. We thank P. Oakes, J. Spatz, J. L. Jones, M. D. Allen and the members of the P.R.-C. and X.T. laboratories for technical assistance and discussions.
Author information
Authors and Affiliations
Contributions
R.O. and P.R.-C. conceived the study; R.O., L.A., D.N., X.T., E.A.C.-A. and P.R.-C. designed the experiments; R.O., T.W., A.E.-A., J.J.U., I.P. and P.D. performed the experiments; J.E., C.M.-P., J.M.G.-A. and P.R.-C. carried out the theoretical modelling; and R.O. and P.R-C. wrote the manuscript. All authors commented on the manuscript and contributed to it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks M. Mrksich and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Nanopattern swelling on gels.
a, Scanning electron micrograph of a quasi-hexagonal 100-nm ordered pattern on a glass surface (from one of two independent experiments). b, Scanning electron micrograph of a quasi-hexagonal 100-nm ordered pattern on a polyacrylamide gel. c, Corresponding histograms showing the distribution of distances between nanodots and their first-order neighbours on glass and polyacrylamide substrates of rigidity 30 kPa (300 particles; two independent experiments). d, Corresponding quantification of mean distance between nanodots on polyacrylamide gels as a function of rigidity (n = 300 particles per condition; two independent experiments). Scale bar, 200 nm.
Extended Data Figure 2 Cell binding to nanopatterned substrates is specific to α5β1 integrins, cRGD, and nanodots.
a, Images showing breast myoepithelial cells plated on 30 kPa substrates with ligand spacing of 50 nm, under conditions that either allow integrin-mediated cell binding (cRGD + nanodots; top left) or do not (the remaining three images). RGE is a peptide with low affinity for α5β1 integrin; abα5β1 is an antibody that blocks α5β1 integrin. b, Corresponding quantification of the percentage spread of cells (n = 30/30/30/22 fields of view; three independent experiments). Scale bar, 100 μm; ***, P < 0.001. Error bars, mean ± s.e.m.
Extended Data Figure 3 Further characterization of cell response to rigidity and nanodot spacing.
a, Quantification of the fluorescence intensity of staining for phosphorylated paxillin (phospho-paxillin) at the cell edge (two different regions per cell; n = 10/10/11, 10/10/11, 10/11/11, 10/10/11, 10/10/11, 10/10/11, 10/10/11, 10/10/11 cells on 50/100/200-nm-spaced substrates and increasing rigidity; two independent experiments). The effect of both ligand spacing and rigidity was significant (P < 0.05; two-way ANOVA). Rather than measuring focal adhesions, this complementary measurement integrates phospho-paxillin recruitment in both adhesions and surrounding areas. The same trends were observed in Fig. 1e. b, Cell-spreading area (n = 13/13/11, 11/16/11, 11/19/11, 16/13/11, 13/16/11, 13/14/11, 13/13/11 cells on 50/100/200-nm-spaced substrates as rigidity increases; two independent experiments). Although nanodot spacing did affect cell spreading, we note that, on the 50-nm- and 100-nm-spaced substrates, the rigidities inducing adhesion formation and collapse are not associated with changes in cell spreading. c, Examples of cells transfected with GFP−paxillin, seeded on 30 kPa and 150 kPa substrates, with nanodots spaced 50 nm and 100 nm apart. The right-hand images of each pair correspond to rectangles marked in red in the left-hand image. d, Corresponding quantification of focal-adhesion length (ten adhesions per cell; n = 11/11, 10/10 cells for 50/100-nm-spaced substrates as rigidity increases; two independent experiments) **, P < 0.005; ***, P < 0.001, two-way ANOVA. Scale bar, 20 μm. Error bars, mean ± s.e.m.
Extended Data Figure 4 Effect of rigidity and nanodot spacing on different cell types and ligands.
a, Staining of phospho-paxillin-containing adhesions in myoepithelial cells, seeded on polyacrylamide substrates of different rigidities or on glass, with either 50-nm- or 100-nm-spaced nanodots coated with the collagen-mimicking GFOGER peptide. Right-hand images are magnifications of the red rectangular regions in the left-hand images. b, Corresponding quantification of focal-adhesion length (mean of at least three adhesions per cell for n = 15 cells per condition; two independent experiments). c–h, As for panels a and b, but for different cell types (HUVECs, MEFs or MCF 10As) seeded on cRGD-coated nanodots. At least three focal adhesions were analysed per cell. For 50/100-nm-spaced substrates and increasing rigidity, n = 16/16, 16/15, 15/16, 15/15 cells (HUVECs), n = 15/15, 16/15, 15/15, 14/14 cells (MEFs), and n = 15/15, 15/15, 15/15, 10/10 cells (MCF 10As); two independent experiments. Scale bars, 20 μm. Error bars, mean ± s.e.m.
Extended Data Figure 5 Adhesion collapse is not associated with changes in nanodot spacing between paxillin clusters.
a, STORM super-resolution images of phospho-paxillin stainings in cells seeded on 100-nm-spaced patterns on 30 kPa or 150 kPa gels. Left, overview images of different focal adhesions; scale bar, 5 μm. Right, magnified images; scale bar, 300 nm. White circles show examples of phospho-paxillin clusters. Two independent experiments. b, Pair-correlation functions (g(r)) of phospho-paxillin clusters as a function of distance in different adhesions (marked with different colours). In all cases, a first peak is observed at around 100 nm, indicating the periodicity of the cluster pattern. c, Histogram showing the distances between neighbouring phospho-paxillin clusters (n = 409 and 197 clusters for 30 kPa and 150 kPa substrates respectively; two independent experiments). No significant differences were observed.
Extended Data Figure 6 Spatial distribution of ordered and disordered nanopatterns.
a, b, Scanning electron micrographs of ordered and disordered nanopatterns on glass for average nanodot spacings of 50 nm (a) and 100 nm (b). Two independent experiments. c, d, Histograms showing the distribution of interparticle distances for ordered and disordered patterns with spacings of 50 nm (c) and 100 nm (d) (n = 300 particles for all the conditions measured in two independent experiments). Scale bar, 100 nm.
Supplementary information
Rights and permissions
About this article

Cite this article
Oria, R., Wiegand, T., Escribano, J. et al. Force loading explains spatial sensing of ligands by cells. Nature 552, 219–224 (2017). https://doi.org/10.1038/nature24662
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature24662
- Springer Nature Limited
This article is cited by
-
How multiscale curvature couples forces to cellular functions
Nature Reviews Physics (2024)
-
Physical biology of cell–substrate interactions under cyclic stretch
Biomechanics and Modeling in Mechanobiology (2024)
-
Glioblastoma Cells Use an Integrin- and CD44-Mediated Motor-Clutch Mode of Migration in Brain Tissue
Cellular and Molecular Bioengineering (2024)
-
Can a bulky glycocalyx promote catch bonding in early integrin adhesion? Perhaps a bit
Biomechanics and Modeling in Mechanobiology (2024)
-
Organization, dynamics and mechanoregulation of integrin-mediated cell–ECM adhesions
Nature Reviews Molecular Cell Biology (2023)