

Abstract
The directed activation of carbon–hydrogen bonds (C–H) is important in the development of synthetically useful reactions, owing to the proximity-induced reactivity and selectivity that is enabled by coordinating functional groups1,2,3,4,5,6. Palladium-catalysed non-directed C–H activation could potentially enable further useful reactions, because it can reach more distant sites and be applied to substrates that do not contain appropriate directing groups; however, its development has faced substantial challenges associated with the lack of sufficiently active palladium catalysts7,8. Currently used palladium catalysts are reactive only with electron-rich arenes, unless an excess of arene is used9,10,11,12,13,14,15,16,17,18, which limits synthetic applications. Here we report a 2-pyridone ligand that binds to palladium and accelerates non-directed C–H functionalization with arene as the limiting reagent. This protocol is compatible with a broad range of aromatic substrates and we demonstrate direct functionalization of advanced synthetic intermediates, drug molecules and natural products that cannot be used in excessive quantities. We also developed C–H olefination and carboxylation protocols, demonstrating the applicability of our methodology to other transformations. The site selectivity in these transformations is governed by a combination of steric and electronic effects, with the pyridone ligand enhancing the influence of sterics on the selectivity, thus providing complementary selectivity to directed C–H functionalization.
Similar content being viewed by others

Main
Extensive research on various directing-group designs1,2,3,4,5,6 and strategies19,20,21 for C–H functionalization, as well as the development of ligands that accelerate it22, has greatly improved the practicality and utility of this approach. Ligand-induced acceleration has enabled palladium catalysts to functionalize remote C–H bonds of aromatic substrates by ‘recognizing’ distance and geometry23,24 (Fig. 1a). Despite the potential of directed C–H activation reactions where proximal or distal site selectivity can be controlled, non-directed C–H functionalization can reach sites that are not currently accessible by a directed approach, as demonstrated by Ir(iii)-catalysed C–H borylation chemistry25,26. Furthermore, substrates that do not contain appropriate directing groups can be functionalized only when using a non-directed approach. The challenges involved in achieving Pd(ii)-catalysed non-directed C–H activation reactions are best illustrated by the lack of progress over the past 60 years in the Fujiwara–Moritani olefination reaction of arenes, for which a large excess of arene is still required to achieve sufficient reactivity with palladium catalysts. Furthermore, such electrophilic palladation reactions, which are analogous to electrophilic aromatic substitution reactions, are limited to electron-rich arenes27,28.

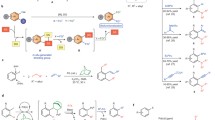
a, Directed (left) and non-directed (right) palladium-catalysed C(sp2)–H functionalization. DG, directing group. The current limitations in non-directed C–H activation are that excess substrate is required and regioselectivity is poor. b, Ligand-accelerated Pd-catalysed C–H functionalization (left). L, Ligand; FG, functional group. The DFT-optimized C–H activation transition state at the M06/SDD,6-311+G(d,p)(SMD)//B3LYP/LANL2DZ,6-31G(d) level of theory is shown on the right; the double dagger represents transition state. c, Crystal structures of the palladium–L69 complexes. d, Ligand effects on reactivity and site selectivity for selected substrates. HFIP, hexafluoroisopropanol; CHCl3, chloroform. For 1m, 1o and 1u, HFIP was used; for 1r, 1t and 1y, CHCl3 was used. The yield and selectivity were determined by 1H NMR; the red colouring highlights the enhancement in the regioselectivity. m/p is the ratio of olefination products at the meta- and para- positions; b/a is the ratio of olefination products at the b- and a- positions.
We discovered an electron-deficient 2-pyridone ligand that enables palladium-catalysed non-directed C–H olefination and carboxylation of both electron-deficient and electron-rich arenes, allowing arene to be used as the limiting reagent (Fig. 1b). We characterize the structures of a pre-catalyst Pd3(μ2-OH)(L69)5 and of a 1,10-phenanthroline-stabilized Pd(Phen)(L69)2 complex (where L69 is the highly electron-deficient ligand 3,5-bis(trifluoromethyl)-pyridin-2(1H)-one) using X-ray crystallography (Fig. 1c), which provide insight into the coordination mode of these ligands with a palladium catalyst. Kinetic and density functional theory (DFT) studies indicate that the pyridone ligand is likely to serve as an X-type ligand to palladium and can also act as an internal base to cleave the C–H bond via the concerted metalation deprotonation mechanism (Fig. 1b). Simple and heterocyclic arenes are both compatible with this protocol. We also apply these reactions to the late-stage modification of amino acids, dyes and pharmaceuticals. Although site selectivity is predominantly dictated by the intrinsic electronic and steric bias of the arene substrates, the presence of the ligand leads to unprecedented enhancement in selectivity with a few classes of arenes (Fig. 1d).
Guided by recent success in developing ligand-accelerated directed C–H activation22, we set out to develop highly active ligands for Pd(ii) to achieve non-directed C–H activation with arene as the limiting reagent (for details see Methods section ‘Ligand optimization for non-directed C–H activation of simple arenes’ and Supplementary Information, Schemes S1–S5). In short, we evaluated the ligands with 1,2-dichlorobenzene (1u) as a model substrate because it displayed poor reactivity and low β/α selectivity under previously reported conditions13. After evaluating various mono-protected 3-amino-2-hydroxypyridine ligands and 2-pyridone ligands, we found that L69 promoted the reaction, allowing formation of the olefinated product (3u) in 70% yield with improved site selectivity (β/α = 3.0/1.0). The yield was further improved to 85% (82% isolated yield) by using 30 mol% L69 and 2.0 equiv. ethyl acrylate.
To elucidate the role of L69 and the origin of this unprecedented reactivity when using arene as the limiting reagent, we carried out structural characterization, kinetic studies and DFT studies. We first obtained and characterized Pd(Phen)(L69)2 and the trimeric Pd3(μ2-OH)(L69)5 complexes using X-ray crystallography (Fig. 1c). The coordination mode of L69 in these complexes is shown to be analogous to that found with carboxylates29 and consistent with the most favourable transition state proposed by our DFT studies (Fig. 1b). The kinetic-isotope-effect experiments (kH/kD = 2.9) using benzene as a model substrate with ethyl acrylate indicate that the C–H bond cleavage is the rate-limiting step. Further kinetic investigation shows that L69 increases the initial rate by a factor of 1.4 (see Supplementary Fig. S5). In addition, the reaction profile using 1u as the substrate indicates that the ligand has a dual role in this reaction: it not only accelerates the initial rate of the reaction by a factor of three, but also prevents catalyst decomposition by forming a more stable complex with palladium. Such a stabilizing effect is crucial for maintaining the catalyst efficiency in this non-directed C–H activation reaction with arene as the limiting reagent.
Interestingly, electron-deficient arenes are nearly unreactive in the absence of L69. With C–H cleavage being the rate-limiting step in this reaction, it is plausible that the observed rate enhancement is due to the involvement of pyridone ligands in the C–H cleavage step. To obtain support for this hypothesis, we performed DFT studies to calculate the transition-state energies for the C–H cleavage step using palladium complexes containing two acetates, one acetate and one pyridone, or two pyridone ligands. We observed that with each successive replacement of an acetate with a pyridone ligand, the relative Gibbs free energy and the Gibbs free energy of activation for the C–H activation transition state decrease. The most favourable transition state (as shown in Fig. 1b) consists of palladium coordinated to two pyridone ligands, one of which serves as an X-type ligand and binds in a κ-2 fashion. The second pyridone ligand coordinates to palladium through the nitrogen centre and serves as an internal base to cleave the C–H bond via the concerted metalation deprotonation mechanism (see Supplementary Information section 3.17). The Gibbs free energy of activation for this transition state is 4.8 kcal mol−1 lower than that for the corresponding transition state with two acetates as the ligand. Although our preliminary DFT studies overestimate the rate enhancement of relatively reactive benzene by the new ligand, the rate enhancement observed with electron-deficient 1u is more marked. Detailed studies are underway to help to understand the effect of hexafluoroisopropanol (HFIP) and silver acetate on the reaction rate to obtain a more quantitative prediction, which could then be used for further catalyst design and development. Finally, the compelling site selectivity that we observed with the naphthalene contrasts the α-selectivity of the conventional Friedel–Crafts-type palladation pathway by favouring the β-position instead (see Supplementary Information, Scheme S6).
Using these optimal conditions, we next examined the generality of the substrate by using ethyl acrylate as the coupling partner. As summarized in Fig. 2, electron-rich and electron-deficient arenes are both suitable substrates, delivering the mono-olefinated products in moderate to high yields (3a–3ak). Di-olefinated products were also formed as minor products when using highly reactive substrates (see Supplementary Information section 3.4). The use of chloroform as the solvent reduces the amount of di-olefination products; for example, benzene afforded the mono-olefinated product 3a in 64% yield and a mixture of di-olefinated products in 18% yield in CHCl3 (compared with 45% mono-olefinated product and 36% di-olefinated products in HFIP). Mono-alkylated arenes were olefinated at the less sterically hindered meta- and para-positions in good yields (3b–3e). Anisole provided the ortho- and para-isomers as the main products, owing to electronic effects (3f), whereas the bulkier silyl-protected phenol gave the para-olefinated product as the main isomer (3g and 3h).

TBS, tert-butyldimethylsilyl; TIPS, triisopropylsilyl; Pin, pinacolate; Ts, 4-toluenesulfonyl. The values under each structure indicate isolated yields. Reaction conditions: Pd(OAc)2 (10 mol%), L69 (30 mol%), AgOAc (3.0 equiv.), HFIP (0.5 ml), 100 °C, 24 h; for 3a, 3r, 3t, 3w, 3y, 3ai, 3aj, 3ak, 3an, 3ao, 3ay, 3az and 3bb, CHCl3 (0.5 ml) was used instead of HFIP; for 3j and 3k, 2,2,2-trifluoro-N-(2-hydroxy-5-(trifluoromethyl)pyridin-3-yl)acetamide (L31; 20 mol%) was used instead of L69; for 3l and 3ab, Pd(OAc)2 (20 mol%), L69 (60 mol%) and ethyl acrylate (1.2 equiv.) were used; for 3as–3ax, the reaction was conducted at 90 °C; for 3al, 3am, 3aq and 3ar, Ag2CO3 (1.5 equiv.) was used instead of AgOAc; for 3am, the reaction time was shortened to 12 h; for 3ap, the reaction was conducted at 60 °C; for 3ar, the reaction time was shortened to 8 h; for 3bd, substrate (0.2 mmol), ethyl acrylate (0.1 mmol) were used in CHCl3. The red bonds signify the site for C–H activation corresponding to the major product.
Less-reactive electron-deficient arenes are also olefinated using this catalytic system, highlighting the importance of the ligand acceleration. Subjecting substrates with strongly electron-withdrawing groups, such as nitro, trifluoromethyl, aldehyde, ester, ketone and nitrile, to these olefination conditions afforded the corresponding products in moderate to good yields with synthetically useful meta-selectivity (3l–3q and 3af). Naphthalene also provided the mono-olefinated product (3r) in 68% yield, with a 13.5/1.0 selectivity ratio (β/α). The improvement in the regioselectivity by the ligand is particularly noteworthy (Fig. 1d); it seems that the ligand enhances the contribution of sterics in determining the selectivity. We also examined di-substituted and tri-substituted aromatics (3s–3ag and 3ah–3ak, respectively). The selectivity of symmetric 1,2-disubstituted arenes and of 1,3-disubstituted arenes is primarily governed by sterics; for example, 1,2,3,4-tetrahydronaphthalene, o-xylene and m-xylene afforded the β-olefinated products (3s, 3t and 3y, respectively) in excellent selectivity. Substrates with less sterically hindered substituents such as 1,2-dichlorobenzene and 1,2-difluorobenzene reacted to provide a mixture of α- and β-olefinated products (3u and 3v), although the β-olefinated products predominate as the major isomer. Methyl 3-(trifluoromethyl)benzoate was transformed to the meta-product (3ab) as a single isomer, owing to a combination of electronic and steric influence. Symmetric 1,4-disubstituted aromatics are also suitable substrates for this reaction and generally undergo mono-functionalization in high yields (3ac and 3ad). 4-substituted anisoles are olefinated exclusively at the ortho-position, owing to electronic effects (3ae–3ag). 1,3,5- and 1,2,3-trisubstituted arenes also react smoothly, providing the desired products in high yields (3ah–3ak). 2,6-Disubstituted aryl boronate ester is also compatible under the conditions (3ak).
We then investigated various heterocyclic aromatics. Olefination of thiophene, furan, benzothiophene and benzofuran occurred at the electron-rich positions to give the desired products selectively (3al–3ar). Use of pyrrole led to a mixture of 2- and 3-olefinated products (3ap), probably owing its high reactivity. Indolines, 1,2,3,4-tetrahydroquinoline, isoindoline, carbazole, dibenzofuran and indazole are also compatible with this non-directed C–H activation, generating the desired products in moderate to good yields (3as–3ba). Furthermore, vinylic C–H bonds in cyclic alkenes were olefinated to provide the corresponding diene products (3bb–3bd).
We evaluated the scope of the olefin coupling partners by using o-xylene as the model substrate (Fig. 3a). α,β-Unsaturated olefins served as particularly effective coupling partners for this palladium-catalysed olefination reaction (4a–4r); for example, the reactions with acrylate derivatives proceeded in 55%–85% yields (4a–4d). Other electron-withdrawing groups attached to the olefins, including carboxylic acid, amide, aldehyde, ketone, nitrile, sulfone, sulfonamide and phosphonate, are also compatible with these reaction conditions, providing the desired olefinated products in moderate to excellent yields (4e–4o). We note that acrylic acid, acrylamide and acrolein are often incompatible with palladium-catalysed C–H olefination reactions. 1,2-Disubstituted α,β-unsaturated olefins including crotonate (4p), maleate (4q) and cyclic tri-substituted olefin (4r) were also found to be suitable coupling partners. Although both the aryl C–H bonds and vinylic C–H bonds of styrenes are reactive under these conditions, electron-deficient styrene derivatives can be used as coupling partners (4s and 4t). We also performed a gram-scale C–H olefination of o-xylene with acrolein using 5 mol% Pd(OAc)2 and 15 mol% L69, which provided the desired product in 83% yield; L69 was recovered in 87% yield.

a, Scope of olefin coupling partners. HFIP, hexafluoroisopropanol. Reaction conditions: o-xylene (0.1 mmol), olefin (2.0 equiv.), Pd(OAc)2 (10 mol%), L69 (30 mol%), AgOAc (3.0 equiv.), HFIP (0.5 ml), 100 °C, 24 h; for 4a–4d, 4i, 4l and 4p, the reaction was conducted in CHCl3; for 4c, the reaction time was shortened to 16 h. b, Carboxylation of simple arenes. Phth, phthaloyl. Reaction conditions: substrate (0.2 mmol), CO (1 atm), Pd(OAc)2 (10 mol%), L69 (30 mol%), AgOAc (3.0 equiv.), HFIP (2.0 ml), 100 °C, 24 h; then NaOH (1.5 ml, 2 M), MeOH (2.0 ml), 12 h; for 5d and 5e, substrate (0.1 mmol) was used; for 5c and 5d, the products were isolated before hydrolysis.
To demonstrate the potential generality of ligand-accelerated non-directed C–H activation to provide a wide range of transformations, we have also developed a non-directed C–H carboxylation reaction (Fig. 3b). We found that L69 can promote the C–H carboxylation of arenes with arene as the limiting reagent under an atmosphere of carbon monoxide. The reaction produces a mixture of the mono-HFIP ester and phthalic anhydride derivatives. The phthalic anhydride derivatives arise from an ortho-C–H carboxylation reaction directed by the product of the first carboxylation. A mixture of mono-acid and phthalic acid derivatives can be obtained after treatment with an aqueous NaOH solution in methanol. Benzene and o-xylene were tested under the standard conditions and transformed to the mono- and di-acids in 79% and 85% combined yield, respectively (5a and 5b). Direct carboxylation of 1,3,5-trimethoxybenzene gave the corresponding HFIP-ester in 92% yield (5c). Bioactive molecules O-methyl-tyrosine derivative (5d) and estrone 3-methyl ether (5e) were also subjected to the carboxylation procedure, affording the desired products in moderate yields.
The development of ligand-accelerated, non-directed C–H functionalization with one equivalent of arene presents an opportunity for late-stage functionalization of C–H bonds that are not accessible by directing group strategies, owing to distance, geometry or compatibility (Fig. 4). C–H olefination of amino-acid derivatives proceeded smoothly to give the desired products in 68% and 52% yield (7a and 7b, respectively). [2.2]Paracyclophane, a commonly used ligand scaffold, was olefinated in 45% yield (7c). Fluorescein derivative (6d), a well-known dye and fluorescence probe, was olefinated in 53% yield (7d). Interestingly, the most active site for 6d is the α-position of the carbonyl group adjacent to the oxo-bridge. In addition, natural products including caffeine, estrone, podocarpic-acid derivatives and camptothecin were tested under the standard conditions, delivering the targets in moderate yields (7e–7h). Various pharmaceuticals including fenofibrate, gemfibrozil, diflunisal, viloxazine and betaxolol were also olefinated to give the products in 47%–91% yield (7i–7m).

Phth, phthaloyl; Ts, 4-toluenesulfonyl; HFIP, hexafluoroisopropanol; Phe, phenylalanine; Tyr, tyrosine. The values under each structure indicate isolated yields. Reaction conditions: substrate (0.1 mmol), ethyl acrylate (0.2 mmol), Pd(OAc)2 (10 mol%), L69 (30 mol%), AgOAc (3.0 equiv.), HFIP (0.5 ml), 100 °C, 24 h; for 7d, 7f and 7i, the reaction time was shortened to 16 h; for 7g, chloroform was used instead of HFIP.
In summary, the 2-pyridone ligand scaffold affords a much more reactive and stable Pd(ii) catalyst for non-directed C–H functionalization. Through the development of the ligand L69, we achieved non-directed C–H olefination and carboxylation with arene as the limiting reagent. We also observed that the ligand has a substantial influence on site selectivity in a few classes of arene substrates. Owing to the compatibility of both electron-rich and electron-deficient arenes, this reaction provides a way to diversify advanced synthetic intermediates, natural products, dyes and drug molecules.
Methods
Ligand optimization for non-directed C–H activation of simple arenes
To best evaluate the ligand effects on reactivity and site selectivity, we selected 1,2-dichlorobenzene (1u) as a model substrate because it displayed poor reactivity and low β/α selectivity under previously reported conditions13. In the absence of ligand, olefination of 1u was found to proceed in the presence of a catalytic amount of Pd(OAc)2 and 3.0 equiv. silver acetate in HFIP to provide the olefinated products in 8% yield with poor site selectivity (β/α = 1.0/1.0). A wide range of ligands that are commonly used in catalysis were then evaluated. Phosphine ligands, N-heterocyclic carbene ligands, oxazoline ligands, pyridine ligands, phenanthroline and 1,10-phenanthrolin-2-ol did not display an improvement in reactivity for this reaction (see Supplementary Information, Scheme S1). The first noticeable ligand enhancement was observed with mono-protected amino acid ligands, which improved the combined yield to 16%. The improved selectivity (α/β = 1.6/1.0) is evidence of ligand involvement in the C–H activation step. Although further screening of other mono-protected amino acid ligands did not improve the yield, we found that the ligand 3-acetylamino-2-hydroxypyridine (L19), previously disclosed for meta-C–H functionalization30, provided an improvement of the reactivity by a factor of nearly three (21% versus 8% yield). Following this lead, we evaluated various mono-protected 3-amino-2-hydroxypyridine ligands (L20–L50) and found that trifluoroacetyl-protected 3-amino-5-(trifluoromethyl)pyridin-2-ol (L31) afforded a substantial improvement in yield (62%) and selectivity (β/α = 2.3/1.0). Interestingly, L51 (5-trifluoromethyl-2-pyridone), which is devoid of the trifluoroacetyl-protected amino moiety, also imparts reactivity to the palladium catalyst, albeit less substantially than does L31 (24% yield). This finding is in line with a previous report on the use of 3-acetylamino-2-hydroxypyridine ligands, wherein it was shown that the 3-acetylamino group has a positive influence on the reaction, but the pyridone moiety is crucial to the reactivity30. This prompted us to perform further ligand screening with a focus on evaluating a diverse range of substituted 2-pyridones. We found that the highly electron-deficient 3,5-bis(trifluoromethyl)-pyridin-2(1H)-one (L69) greatly promoted the reaction, allowing formation of 3u in 70% yield with improved site selectivity (β/α = 3.0/1.0). The yield was further improved to 85% (82% isolated yield) by using 30 mol% L69 and 2.0 equiv. ethyl acrylate.
General procedure for the palladium/pyridone-promoted non-directed C–H activation of simple arenes
Substrate (0.1 mmol), ethyl acrylate (0.2 mmol), Pd(OAc)2 (2.2 mg, 10 mol%), L69 (3.0 mg, 20 mol%), AgOAc (50.1 mg, 0.3 mmol) and HFIP or CHCl3 (0.5 ml) were added to a 2-dram vial. The vial was capped and closed tightly, then the reaction mixture was stirred at 100 °C for 24 h. After cooling to room temperature, the mixture was filtered through a pad of Celite and washed with dichloromethane as the eluent to remove the insoluble precipitate. The resulting solution was concentrated and purified by preparative thin-layer chromatography to afford the desired arylated product. Full experimental details and characterization of new compounds are provided in Supplementary Information.
Data availability
The data that support the findings of this study are available within the paper and its Supplementary Information. Metrical parameters for the structures of Pd(Phen)(L69)2, the Pd3(μ2-OH)(L69)5 complex and 7d are available free of charge from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/) under reference numbers CCDC 1552055, CCDC 1538821 and CCDC 1538820, respectively.
References
Whisler, M. C., MacNeil, S., Snieckus, V. & Beak, P. Beyond thermodynamic acidity: a perspective on the complex-induced proximity effect (CIPE) in deprotonation reactions. Angew. Chem. Int. Ed. 43, 2206–2225 (2004)
Kakiuchi, F. et al. Catalytic addition of aromatic carbon–hydrogen bonds to olefins with the aid of ruthenium complexes. Bull. Chem. Soc. Jpn 68, 62–83 (1995)
Daugulis, O., Do, H.-Q. & Shabashov, D. Palladium- and copper-catalyzed arylation of carbon–hydrogen bonds. Acc. Chem. Res. 42, 1074–1086 (2009)
Engle, K. M., Mei, T.-S., Wasa, M. & Yu, J.-Q. Weak coordination as powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res. 45, 788–802 (2012)
Lyons, T. W. & Sanford, M. S. Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem. Rev. 110, 1147–1169 (2010)
Colby, D. A., Bergman, R. G. & Ellman, J. A. Rhodium-catalyzed C–C bond formation via heteroatom-directed C–H bond activation. Chem. Rev. 110, 624–655 (2010)
Kuhl, N., Hopkinson, M. N., Wencel-Delord, J. & Glorius, F. Beyond directing groups: transition-metal-catalyzed C–H activation of simple arenes. Angew. Chem. Int. Ed. 51, 10236–10254 (2012)
Hartwig, J. F. & Larsen, M. A. Undirected, homogeneous C–H bond functionalization: challenges and opportunities. ACS Cent. Sci. 2, 281–292 (2016)
Moritani, I. & Fujiwara, Y. Aromatic substitution of styrene-palladium chloride complex. Tetrahedr. Lett. 8, 1119–1122 (1967)
Dams, M., De Vos, D. E., Celen, S. & Jacobs, P. A. Toward waste-free production of Heck products with a catalytic palladium system under oxygen. Angew. Chem. Int. Ed. 42, 3512–3515 (2003)
Yokota, T., Tani, M., Sakaguchi, S. & Ishii, Y. Direct coupling of benzene with olefin catalyzed by Pd(OAc)2 combined with heteropolyoxometalate under dioxygen. J. Am. Chem. Soc. 125, 1476–1477 (2003)
Zhang, Y.-H., Shi, B.-F. & Yu, J.-Q. Pd(II)-catalyzed olefination of electron-deficient arenes using 2,6-dialkylpyridine ligands. J. Am. Chem. Soc. 131, 5072–5074 (2009)
Kubota, A., Emmert, M. H. & Sanford, M. S. Pyridine ligands as promoters in PdII/0-catalyzed C–H olefination reactions. Org. Lett. 14, 1760–1763 (2012)
Ying, C.-H., Yan, S.-B. & Duan, W.-L. 2-Hydroxy-1,10-phenanthroline vs 1,10-phenanthroline: significant ligand acceleration effects in the palladium-catalyzed oxidative Heck reaction of arenes. Org. Lett. 16, 500–503 (2014)
Li, R., Jiang, L. & Lu, W. Intermolecular cross-coupling of simple arenes via C–H activation by tuning concentrations of arenes and TFA. Organometallics 25, 5973–5975 (2006)
Shrestha, R., Mukherjee, P., Tan, Y., Litman, Z. C. & Hartwig, J. F. Sterically controlled, palladium-catalyzed intermolecular amination of arenes. J. Am. Chem. Soc. 135, 8480–8483 (2013)
Fujiwara, Y., Taniguchi, H. & Taniguchi, H. Palladium-promoted one-step carboxylation of aromatic compounds with carbon monoxide. J. Chem. Soc. Chem. Commun. 220–221 (1980)
Yoneyama, T. & Crabtree, R. H. Pd(II) catalyzed acetoxylation of arenes with iodosyl acetate. J. Mol. Catal. A 108, 35–40 (1996)
Huang, C., Chattopadhyay, B. & Gevorgyan, V. Silanol: a traceless directing group for Pd-catalyzed o-alkenylation of phenols. J. Am. Chem. Soc. 133, 12406–12409 (2011)
Bedford, R. B., Coles, S. J., Hursthouse, M. B. & Limmert, M. E. The catalytic intermolecular orthoarylation of phenols. Angew. Chem. Int. Ed. 42, 112–114 (2003)
Zhang, F.-L., Hong, K., Li, T.-J., Park, H. & Yu, J.-Q. Functionalization of C(sp3)–H bonds using a transient directing group. Science 351, 252–256 (2016)
Engle, K. M. & Yu, J.-Q. Developing ligands for palladium(II)-catalyzed C–H functionalization: intimate dialogue between ligand and substrate. J. Org. Chem. 78, 8927–8955 (2013)
Leow, D., Li, G., Mei, T.-S. & Yu, J.-Q. Activation of remote meta-C–H bond assisted by an end-on template. Nature 486, 518–522 (2012)
Chu, L. et al. Remote meta-C–H activation using a pyridine-based template: achieving site-selectivity via the recognition of distance and geometry. ACS Cent. Sci. 1, 394–399 (2015)
Mkhalid, I. A. I., Barnard, J. H., Marder, T. B., Murphy, J. M. & Hartwig, J. F. C–H activation for the construction of C–B bonds. Chem. Rev. 110, 890–931 (2010)
Cho, J.-Y., Tse, M. K., Holmes, D., Maleczka, R. E. Jr & Smith, M. R. III. Remarkably selective iridium catalysts for the elaboration of aromatic C–H bonds. Science 295, 305–308 (2002)
Grimster, N. P., Gauntlett, C., Godfrey, C. R. A. & Gaunt, M. J. Palladium-catalyzed intermolecular alkenylation of indoles by solvent-controlled regioselective C–H functionalization. Angew. Chem. Int. Ed. 44, 3125–3129 (2005)
Ueda, K., Yanagisawa, S., Yamaguchi, J. & Itami, K. A general catalyst for the β-selective C–H Bond arylation of thiophenes with iodoarenes. Angew. Chem. Int. Ed. 49, 8946–8949 (2010)
Bedford, R. B. et al. Facile hydrolysis and alcoholysis of palladium acetate. Angew. Chem. Int. Ed. 54, 6591–6594 (2015)
Wang, P. et al. Ligand-promoted meta-C–H arylation of anilines, phenols, and heterocycles. J. Am. Chem. Soc. 138, 9269–9276 (2016)
Acknowledgements
We acknowledge The Scripps Research Institute, the NIH (NIGMS, 2R01 GM102265), Bristol-Myers Squibb and Shanghai RAAS Blood Products Co., Ltd. for their financial support. We also thank Novartis for providing the drug molecules.
Author information
Authors and Affiliations
Contributions
P.W. developed the ligands and the reactions. P.V. performed the DFT calculations. G.X. performed the kinetic study. J.S., S.T. and P.T.W.C. separated the isomers using preparative HPLC. J.X.Q. and M.A.P. participated in the screening of acrylamide-derived coupling partners and investigation of the C–H olefination reaction for amino acid substrates. M.E.F. performed preliminary studies on 2-hydroxypyridine ligands. K.-S.Y. helped with the screening of sulphonamide-derived coupling partners. J.-Q.Y. conceived the concept and prepared the manuscript with feedback from P.W., P.V. and G.X.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks J. de Vries and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains Supplementary Information schemes S1-S5. (PDF 76894 kb)
Supplementary Data
This zip file contains the cif files and CheckCIF documents for X-ray structures. (ZIP 2016 kb)
Rights and permissions
About this article

Cite this article
Wang, P., Verma, P., Xia, G. et al. Ligand-accelerated non-directed C–H functionalization of arenes. Nature 551, 489–493 (2017). https://doi.org/10.1038/nature24632
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature24632
- Springer Nature Limited
This article is cited by
-
Electrocatalyzed direct arene alkenylations without directing groups for selective late-stage drug diversification
Nature Communications (2023)
-
Transannular C–H functionalization of cycloalkane carboxylic acids
Nature (2023)
-
Synergistic silver-mediated and palladium-catalyzed nondirected olefination of aryl C–H bond: quick access to multi-substituted aryl olefins
Science China Chemistry (2023)
-
Photoredox C–H functionalization leads the site-selective phenylalanine bioconjugation
Scientific Reports (2022)
-
Facile synthesis of 6-organyl-4-(trifluoromethyl)pyridin-2(1H)-ones and their polyfluoroalkyl-containing analogs
Russian Chemical Bulletin (2022)