

Abstract
Singly or doubly bonded polynitrogen compounds can decompose to dinitrogen (N2) with an extremely large energy release. This makes them attractive as potential explosives or propellants1,2,3, but also challenging to produce in a stable form. Polynitrogen materials containing nitrogen as the only element exist in the form of high-pressure polymeric phases4,5,6, but under ambient conditions even metastability is realized only in the presence of other elements that provide stabilization. An early example is the molecule phenylpentazole, with a five-membered all-nitrogen ring, which was first reported in the 1900s7 and characterized in the 1950s8,9. Salts containing the azide anion (N3−)10,11,12 or pentazenium cation (N5+)13 are also known, with compounds containing the pentazole anion, cyclo-N5−, a more recent addition14,15,16. Very recently, a bulk material containing this species was reported17 and then used to prepare the first example of a solid-state metal–N5 complex18. Here we report the synthesis and characterization of five metal pentazolate hydrate complexes [Na(H2O)(N5)]·2H2O, [M(H2O)4(N5)2]·4H2O (M = Mn, Fe and Co) and [Mg(H2O)6(N5)2]·4H2O that, with the exception of the Co complex, exhibit good thermal stability with onset decomposition temperatures greater than 100 °C. For this series we find that the N5− ion can coordinate to the metal cation through either ionic or covalent interactions, and is stabilized through hydrogen-bonding interactions with water. Given their energetic properties and stability, pentazole–metal complexes might potentially serve as a new class of high-energy density materials19 or enable the development of such materials containing only nitrogen20,21,22,23. We also anticipate that the adaptability of the N5− ion in terms of its bonding interactions will enable the exploration of inorganic nitrogen analogues of metallocenes24 and other unusual polynitrogen complexes.
Similar content being viewed by others
Main
We hypothesized that metal–N5 complexes could be prepared through cation exchange of cyclo-N5− precursor salts in solution. Adapting the procedure17 for C–N bond cleavage gave an air-stable, off-white NaN5 hydrate (1), which was recrystallized to produce [Na(H2O)(N5)]·2H2O (2) (Fig. 1a) and then characterized using high-resolution mass spectrometry (HRMS) and 15N NMR spectroscopy (Extended Data Fig. 1). The only peak in the 15N NMR spectrum, with a chemical shift (δ) of −5.7 p.p.m., was assigned to N5−, consistent with the computed chemical shifts ranging from −6.5 to 0 p.p.m. (refs 23, 24, 25). When preparing singly-15N-labelled N5− (using Na15NO2 in the synthetic process) and examining the crude reaction mixture by 15N NMR (Extended Data Fig. 1c), a signal for the 15N-labelled N5− ring was detected at −3.0 p.p.m. (in CD3OD); in contrast, the 15N nitrate anion signal (oxidized from the nitrite anion15NO2−) was observed at −12.4 p.p.m., in agreement with published data (−10 ± 2 p.p.m.)21,26. When dissolving the appropriate metal salts into ethanol solutions containing N5−, [Mn(H2O)4(N5)2]·4H2O (3), [Fe(H2O)4(N5)2]·4H2O (4), [Co(H2O)4(N5)2]·4H2O (5) and [Mg(H2O)6(N5)2]·4H2O (6) formed via metathesis reactions between NaN5 and MnCl2, FeCl2, Co(NO3)2 and MgCl2, respectively (Fig. 1b).

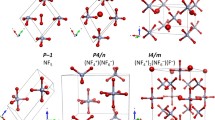
Ellipsoids represent the 50% probability level. a, Synthetic route to 2. b, Preparation of 3–6 by ion metathesis of 2. c–e, Extended molecular view of 2 normal to (100) (c), (001) (d) and (100) (e). f–i, Molecular structures of 3–6, respectively, in their crystalline states, shown by ORTEP representations.
The single-crystal X-ray structures determined in this study for complexes 2–6 (Extended Data Table 1) differ from all the configurations predicted in previous theoretical studies that explored metal pentazolates22,24,27,28. As depicted in Fig. 1c–e and Extended Data Fig. 2a, b, the sodium complex 2 crystallized in solution to form a two-dimensional porous metal–inorganic framework with orthorhombic Cmcm symmetry, with the pentagonal N5− units adopting a perfectly planar arrangement and a perfectly co-planar arrangement with adjacent sodium atoms (as evidenced by a Na–N1–N2–N3 torsion angle of 180°). The three complexes 3–5 all crystallized in the orthorhombic Fmmm space group, with almost identical unit cell arrangements (Fig. 1f–h and Extended Data Fig. 2c–k). The N5 rings bind to the metal atoms (Mn, Fe, or Co) through η1-coordination and appear perfectly coplanar (Extended Data Fig. 2c–e). The ball-and-stick packing diagrams of 2–6 at the (100) surfaces show the metal–N5 fragments all stacked face-to-face, with an interlayer distance in 2 of 3.453 Å (a/2) and interlayer distances in 3, 4 and 5 of 3.269, 3.285 and 3.233 Å (all c/2), respectively. The crystal structure of 6 is characterized by triclinic (P1) symmetry, with no coordination bonds between the N5 rings and magnesium atoms (Fig. 1i). Extended Data Fig. 2l–n demonstrates that the N5 ring in 6 lacks perfect planarity, with N1–N2–N3–N4 and N1–N5–N4–N3 torsion angles of 0.85° and 0.14°, respectively.
The N–N bond lengths of the N5 rings in complexes 2–6 are not identical, with average values found to be 1.316, 1.320, 1.329, 1.314 and 1.316 Å, respectively. Moreover, even in a given ring, the N–N bonds differ as indicated by X-ray photoelectron spectroscopy (XPS; see Extended Data Fig. 3): except for complex 1, three distinct N 1s peaks are observed at 398.2, 400.3 and 402.5 eV. Considering the similar electronegativity values of C and N, the three peaks are suggested to arise from pyridinic N, pyrrolic N and quaternary N, respectively. Bond critical point data, based on experimental electron density analysis29, indicate that the bonding between the metal and the N5 ring differs in complexes 2–6 (Extended Data Fig. 4a–e and Extended Data Table 2): whereas 2 and 6 can be considered as typical ionic compounds with no obvious electron transfer between N in the charged N5 ring and the Na/Mg ion, complexes 3–5 exhibit some covalent-type bonding, as evidenced by electron transfer from lone pair electrons in the N5 ring to the metal.
We note that in all complexes, water plays an important role in crystallization (Extended Data Table 3). In complex 2, hydrogen bonding between H2O and cyclo-N5− (O–H···N) acts as an important bridge during the formation of the metal–inorganic framework (Extended Data Fig. 2a). In complex 6, each N atom in the N5− ring forms a hydrogen bond with H2O (Extended Data Fig. 2l), with the surrounding H2O molecules providing a hydrophilic and polar environment that helps to stabilize and protect the N5− rings in the crystal. For 3–5, the stabilizing effect of the coordinated water is evident when considering the Kohn–Sham orbitals30. The M(η1-N5)2 mode, away from the lowest point on the potential energy surface, was found to be unstable in previous studies24,27. But in complexes 3–5, the participation of H2O in the coordination process gives rise to new orbitals and thus stabilizes both the metal and the N5− ring (Extended Data Fig. 4f–k). We note, however, that water also gives rise to steric hindrance that limits the direction of coordination of N5−.
The geometric parameters of the complexes (Fig. 2a, b) indicate that cyclo-N5− is unable to maintain D5h symmetry ( ) upon coordination. Instead, it exhibits C2v symmetry and is predicted to have nine fundamentals classified as A1 + (A1 + B2) + 2(A1 + B2) + (A2 + B1). The (A2 + B1) modes derived from
) upon coordination. Instead, it exhibits C2v symmetry and is predicted to have nine fundamentals classified as A1 + (A1 + B2) + 2(A1 + B2) + (A2 + B1). The (A2 + B1) modes derived from  are neither infrared (IR)-active nor Raman-active, whereas the (A1 + B2) modes derived from
are neither infrared (IR)-active nor Raman-active, whereas the (A1 + B2) modes derived from  are IR-active and give rise to experimentally observed signals in the ranges from 1,244 to 1,256 cm−1 (B2) and 1,219 to 1,236 cm−1 (A1) (Fig. 2c). The two vibrations are assigned to two strong N–N stretches, in good agreement with the theoretical frequency for these modes19. The A1 and 2(A1 + B2) modes evolved from
are IR-active and give rise to experimentally observed signals in the ranges from 1,244 to 1,256 cm−1 (B2) and 1,219 to 1,236 cm−1 (A1) (Fig. 2c). The two vibrations are assigned to two strong N–N stretches, in good agreement with the theoretical frequency for these modes19. The A1 and 2(A1 + B2) modes evolved from  and
and  are Raman-active, with five theoretical peaks. One strong peak observed at 1,188–1,199 cm−1 is attributed to the A1 mode, whereas the other four peaks expected, associated with the 2(A1 + B2) modes, are of low intensity such that only three of them are seen in the spectra, at 1,234–1,239, 1,120–1,124 and 1,005–1,037 cm−1 (Fig. 2d).
are Raman-active, with five theoretical peaks. One strong peak observed at 1,188–1,199 cm−1 is attributed to the A1 mode, whereas the other four peaks expected, associated with the 2(A1 + B2) modes, are of low intensity such that only three of them are seen in the spectra, at 1,234–1,239, 1,120–1,124 and 1,005–1,037 cm−1 (Fig. 2d).

a, Change of cyclo-N5− symmetry from D5h to C2v. b, Geometric parameters of cyclo-N5− in 2–5. Bond lengths in ångströms; bond angles in degrees. c, Infrared spectra of 2–6 and H2O. d, Raman spectra of 2–6.
The decomposition behaviour of the N5 complexes was examined using differential scanning calorimetry (DSC) and thermogravimetric (TG) analysis (Fig. 3a–e). For 2–5, only one endothermic process occurred at about 50 °C, whereas for compound 6, three endothermic peaks were observed below 100 °C. All complexes exhibited two exothermic stages, except for complex 5. The first exothermic peaks were similar for each of complexes 2–4 and 6, occurring at 111.3, 104.1, 114.7 and 103.5 °C, respectively, but the position of the second exothermic peak varied greatly. Further DSC measurement of decomposition kinetics showed that the apparent activation energies of the first exothermic peaks were 98.4, 106.6, 108.4 and 118.2 kJ mol−1, for 2–4 and 6, respectively (Extended Data Fig. 5).

For DSC measurements, about 0.5 mg of the sample was placed in a perforated stainless steel container. a–e, TG and DSC curves of 2–6. All temperature points in the stability study were onset temperatures. The decomposition energies of cyclo-N5− are calculated by integration of the curves, shown hatched. exo, exothermic. f, Chromogenic experiments of complexes 2–6 after heating at 60 °C (2′–6′; top row) and 110 °C (2″–6″; bottom row). The almost colourless solution containing N3− ions became red upon the addition of two drops of a yellow FeCl3 solution (0.1 g ml−1).
As representatives of the three different crystal structures, complexes 2, 5 and 6 were further analysed with TG–DSC–MS (Fig. 4a and Extended Data Fig. 6). Mass spectra collected for 5 and 6 indicated that the marked weight loss during the endothermic process generated a peak at an m/z value of 18 and hence was related to dehydration. To further probe the endothermic process, complexes 2–6 were maintained at 60 °C for 0.5 h and the products, denoted as 2′–6′, were then analysed by chromogenic experiments (Fig. 3f), IR spectrophotometry (Extended Data Fig. 7a). No decomposition of cyclo-N5− was found in products 2′–4′ and 6′, and temperature-dependent IR indicated some H2O remaining after the endothermic process. Heating to a higher temperature removed the remaining H2O and resulted in decomposition of cyclo-N5− (Extended Data Fig. 7c, e). In product 5′, water loss was accompanied by loss of N2 and azide formation (Extended Data Fig. 7d). We attribute this to the Co–N bond (2.122 Å) being the shortest among the metal–N5 bonds, so the strained N5− ring decomposes more easily owing to its closer distance to Co2+ after the loss of water. These observations, overall, also illustrate that water helps to stabilize these complex systems.

a, TG–DSC–MS analysis of 2 under argon. The signals at m/z 14, 18 and 28 in the mass spectra are shown. During the vacuum period at 40 °C (the exchange of air with argon), H2O was lost before measurement. b, c, IR (b) and Raman (c) spectra of 2, 2″ and NaN3.
During the first exothermic process, MS peaks at m/z values of 14 and 28 for 2 and 6 indicate the generation of N and N2 during the decomposition process. The exothermic process was also probed further by maintaining 2–6 at 110 °C for 0.5 h, and the residues, marked as 2″–6″, were then analysed with the same methods as before (Figs 3f, 4b, c and Extended Data Fig. 7b). It was found that all of the cyclo-N5− rings decomposed into N3− during this step. Because N3− had already formed from complex 5 during the earlier dehydration step, the single exothermic peak at 149 °C is attributed to the decomposition of N3− in the residue.
The series of energetic metal–N5 complexes we have developed illustrates the adaptability of the cyclo-N5− unit in terms of its ability to take part in ionic, coordination and hydrogen-bonding interactions. We anticipate that this feature will enable the development of other complexes, such as novel nitrogen-based analogues of metallocenes, complexes in which non-metallic elements stabilize the cyclo-N5−, or even polynitrogen energetic materials.
Methods
Safety precautions
Caution. NaN5 and other metal–N5 complexes are, in part, extremely energetic compounds with increased sensitivities towards various stimuli, therefore proper protective measures (safety glasses, face shield, leather coat, earthen equipment and shoes, Kevlar gloves and ear plugs) should be used. All compounds should be stored in explosive cases as they can explode spontaneously.
General methods
All reagents and solvents were purchased from Sigma-Aldrich, Aladdin, and Energy Chemical as analytical grade and were used as received. The filtration and storage of the intermediate product were performed in a Coolingway DW-86W58 cryopreservation box. A Bruker AVANCE 500 nuclear magnetic resonance spectrometer operating at 50.69 MHz was used to collect 15N spectral data. DMSO-d6 and CD3OD were employed as the solvent and locking solvent, respectively. Chemical shifts are given relative to MeNO2 for 15N NMR. High-resolution mass spectra (electrospray ionization) were recorded on a Waters Q-TOF MicroTM high-resolution mass spectrometer operated in the splitless mode. The samples were dissolved in methanol/H2O (70/30 by volume), and introduced via a syringe pump at 5 μl min−1. The instrument was run in the negative ion mode with a capillary voltage of 2,500 V. DSC plots were acquired on a differential scanning calorimeter (Mettler Toledo DSC-1) at a scan rate of 5 °C min−1 in perforated stainless steel containers under a nitrogen flow of 50 ml min−1. TG analysis was also performed at a heating rate of 5 °C min−1 on a Mettler Toledo TGA/SDTA851e instrument. X-ray photoelectron spectroscopy (XPS, XPS Microprobe, PHI Quantera II) was performed using Al Kα as a monochromatic radiation source (hν = 1,486.7 eV) at a power of 240 W (12 kV × 20 mA) at 25 °C. The total pressure in the main vacuum chamber during analysis was typically 5 × 10−8 Pa. The pass energy was set to 160 eV (energy step 0.5 eV) for recording survey spectra and 20 eV (energy step 0.05 eV) for high-resolution spectra. The carbon peak at 285.0 eV was used as a reference to correct for charging effects. IR spectra were recorded on a Thermo Nicolet IS10 instrument. Raman spectra were collected using a Horiba-Jobin Yvon Labram HR800 Raman spectrometer with a 514.532 nm Ar+ laser. A 50× objective was used to focus the laser beam. TG–DSC–MS was performed on a Netzsch STA 449 F3 Jupiter and QMS 403C at a heating rate of 5 °C min−1 under an argon atmosphere.
[Na(H2O)(N5)]·2H2O (2)
To a magnetically stirred H2O/tetrahydrofuran mixed solution (20 ml/20 ml) containing 4-hydroxy-3,5-dimethylbenzenaminium chloride31 (6 g, 34.58 mmol) and hydrochloric acid (36%, 3.025 ml, 36.31 mmol), sodium nitrite (2.505 g, 36.31 mmol) in 5 ml water was added dropwise at −3 to 0 °C. After 45 min, 45 ml methanol and 45 ml THF were added and the reaction system was cooled to −38 °C, after which sodium azide (2.360 g, 36.31 mmol) in methanol/H2O (22 ml, 1/1 by volume) was added dropwise. The resulting mixture was stirred at −38 °C for 1.5 h. The solution was removed by filtration at −60 °C (in a cryopreservation box), and the filter residue was washed with acetone (10 ml × 4). The off-white solid was collected and used in the next step without further purification after freeze-drying.
A solution of the intermediate product sodium salt of arylpentazole (5.000 g, 21.64 mmol) and ferrous glycinate (8.600 g, 42.16 mmol) in a mixed solution of 100 ml methyl alcohol and 100 ml acetonitrile was stirred at −47 °C. After 30 min, meta-chloroperbenzoic acid (85%, 19.25 g, 94.82 mmol) was added in portions. The reaction mixture was maintained at –43 °C for 24 h. The precipitate was removed by filtration, and the filtrate was evaporated under reduced pressure. The residue was suspended in 200 ml of water, after which the precipitate was filtered off and washed with 50 ml water. The filtrate was concentrated, and after removing the solvent under vacuum, the residue was purified by chromatography with ethyl alcohol/ethyl acetate (1/10–1/3 gradient elution). Crude NaN5 hydrate was obtained as an off-white product (427 mg, 19.56%). Colourless crystals of 2 were obtained by maintaining alcohol (95%) solutions at ambient temperature for several days. Decomposition point (onset): 111.3 °C; 15N NMR (50.69 MHz, DMSO-d6): δ − 5.7 p.p.m. (s); IR (neat): νmax 3,491, 3,354, 3,293, 2,166, 1,651, 1,246, 1,219 cm−1; Raman (neat): νmax 1,188, 1,120, 1,005, 112 cm−1; HRMS (m/z): [M]− calcd for N5, 70.0154; found, 70.0156; analysis (% calcd, % found for NaH6N5O3): H (4.08, 4.13), N (47.62, 47.74).
General procedures for the preparation of complexes 3–6
1.0 mmol of 2 (147 mg) was added to 0.5 mmol MnCl2 (FeCl2, Co(NO3)2, MgCl2) dissolved in 15 ml of ethanol (95%). Single crystals of 3–6 were obtained by maintaining alcohol solutions at ambient temperature for several days. The crystals were used for DSC, TG, IR, Raman and XPS analysis.
[Mn(H2O)4(N5)2]·4H2O (3)
Colourless crystals; decomposition point (onset): 104.1 °C; IR (neat): νmax 3,453, 3,330, 3,258, 2,162, 1,709, 1,663, 1,252, 1,232 cm−1; Raman (neat): νmax 1,234, 1,197, 1,121, 1,022, 151 cm−1; analysis (% calcd, % found for MnH16N10O8): H (4.76, 4.72), N (41.30, 41.24).
[Fe(H2O)4(N5)2]·4H2O (4)
Pale green crystals; decomposition point (onset): 114.7 °C; IR (neat): νmax 3,442, 3,335, 3,251, 2,162, 1,712, 1,668, 1,255, 1,232 cm−1; Raman (neat): νmax 1,197, 1,124, 156 cm−1; analysis (% calcd, % found for FeH16N10O8): H (4.74, 4.59), N (41.19, 41.08).
[Co(H2O)4(N5)2]·4H2O (5)
Bright orange crystals; decomposition point (onset): 58.9 °C; IR (neat): νmax 3,435, 3,331, 3,243, 1,713, 1,669, 1,256, 1,236 cm−1; Raman (neat): νmax 1,239, 1,199, 1,122, 1,029, 169, 134 cm−1; analysis (% calcd, % found for CoH16N10O8): H (4.70, 4.65), N (40.82, 40.73).
[Mg(H2O)6(N5)2]·4H2O (6)
Colourless crystals; decomposition point (onset): 103.5 °C; IR (neat): νmax 3,523, 3,209, 2,062, 1,693, 1,648, 1,244, 1,236 cm−1. Raman (neat): νmax 1,238, 1,192, 1,122, 1,037, 184, 127 cm−1; analysis (% calcd, % found for MgH20N10O10): H (5.85, 5.89), N (40.66, 40.72).
Computational methods
All computations were performed using the Gaussian 09 program32 invoking hybrid HF-DFT calculations with the three-parameter gradient-corrected exchange potential of Becke and the gradient-corrected correlation potential of Lee, Yang and Parr (B3LYP)33,34. The elements H, Na, Mg, N and O were examined using the cc-pVTZ basis set. Transition metals invoking effective core potentials on the heavier elements used the double-ζ valence basis set of Hay and Wadt, denoted as LANL2DZ35. The structures were optimized for energy minimization before spectra acquisition and frequency calculations. The wavefunctions of complexes generated from Gaussian were imported into the software Multiwfn36 to perform Kohn–Sham orbital prediction29, bond critical point analysis30, charge decomposition analysis and localized orbital locator prediction37.
X-ray diffraction studies
The single-crystal X-ray diffraction measurements for 2–6 were conducted on a Bruker Smart Apex II diffractometer using Mo-Kα radiation (λ = 0.71073 Å) with a graphite monochromator at 170 K or 205 K. An Oxford Cobra low-temperature device was used to maintain the temperature. Integration and scaling of intensity data were accomplished using the SAINT program38. The structures were solved by intrinsic methods using SHELXT2014 and refinement was carried out by a full-matrix least-squares technique using SHELXT201439. The hydrogen atoms were refined isotropically, and the heavy atoms were refined anisotropically. N–H and O–H hydrogens were located from different electron density maps, and C–H hydrogens were placed in calculated positions and refined with a riding model. Data were corrected for the effects of absorption using SADABS40.
Data availability
The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 1527747, [Na(H2O)(N5)]·2H2O; 1527748, [Mn(H2O)4(N5)2]·4H2O; 1527749, [Fe(H2O)4(N5)2]·4H2O; 1527750, [Co(H2O)4(N5)2]·4H2O; 1527751, [Mg(H2O)6(N5)2]·4H2O. The source crystallographic data can be found in Supplementary Information and also obtained free of charge from the CCDC. The source data that support the plots in this Letter and the other findings of this study are available from the corresponding authors upon reasonable request.
Change history
23 May 2018
In this Letter, under Methods subsection '[Na(H2O)(N5)]·2H2O (2)', the description "the intermediate product arylpentazole (5.000 g, 26.18 mmol)" should read "the intermediate product sodium salt of arylpentazole (5.000 g, 21.64 mmol)". In the legend of Fig. 3, we add that "All temperature points in the stability study were onset temperatures." to avoid misunderstanding. These corrections have been made online.
References
Christe, K. O. Polynitrogen chemistry enters the ring. Science 355, 351–353 (2017)
Hirshberg, B., Gerber, R. B. & Krylov, A. I. Calculations predict a stable molecular crystal of N8 . Nat. Chem. 6, 52–56 (2014)
Christe, K. O. Recent advances in the chemistry of N5+, N5− and high-oxygen compounds. Propellants Explos. Pyrotech. 32, 194–204 (2007)
Eremets, M. I., Gavriliuk, A. G., Trojan, I. A., Dzivenko, D. A. & Boehler, R. Single-bonded cubic form of nitrogen. Nat. Mater. 3, 558–563 (2004)
Steele, B. A. & Oleynik, I. I. Sodium pentazolate: a nitrogen rich high energy density material. Chem. Phys. Lett. 643, 21–26 (2016)
Steele, B. A. et al. High-pressure synthesis of a pentazolate salt. Chem. Mater. 29, 735–741 (2017)
Curtius, T., Darapsky, A. & Müller, E. Die sogenannten Pentazol-Verbindungen von J. Lifschitz. Ber. Dtsch. Chem. Ges. 48, 1614–1634 (1915)
Huisgen, R. & Ugi, I. Zur Lösung eines klassischen Problems der organischen Stickstoff-Chemie. Angew. Chem. 68, 705–706 (1956)
Huisgen, R. & Ugi, I. Pentazole, I. Die Lösung Eines Klassischen Problems der Organischen Stickstoffchemie. Chem. Ber. 90, 2914–2927 (1957)
Fehlhammer, W. P. & Beck, W. Azide chemistry – an inorganic perspective, Part I metal azides: overview, general trends and recent developments. Z. Anorg. Allg. Chem. 639, 1053–1082 (2013)
Haiges, R. et al. Polyazide chemistry: preparation and characterization of Te(N3)4 and [P(C6H5)4]2[Te(N3)6] and evidence for [N(CH3)4][Te(N3)5]. Angew. Chem. Int. Ed. 42, 5847–5851 (2003)
Crawford, M. J., Ellern, A. & Mayer, P. UN213−: a structurally characterized binary actinide heptaazide anion. Angew. Chem. Int. Ed. 44, 7874–7878 (2005)
Christe, K. O., Wilson, W. W., Sheehy, J. A. & Boatz, J. A. N5+: a novel homoleptic polynitrogen ion as a high energy density material. Angew. Chem. Int. Ed. 38, 2004–2009 (1999)
Vij, A., Pavlovich, J. G., Wilson, W. W., Vij, V. & Christe, K. O. Experimental detection of the pentaazacyclopentadienide (pentazolate) anion, cyclo-N5−. Angew. Chem. Int. Ed. 41, 3051–3054 (2002)
Östmark, H. et al. Detection of pentazolate anion (cyclo-N5−) from laser ionization and decomposition of solid p-dimethylaminophenylpentazole. Chem. Phys. Lett. 379, 539–546 (2003)
Bazanov, B. et al. Detection of cyclo-N5− in THF solution. Angew. Chem. Int. Ed. 55, 13233–13235 (2016)
Zhang, C., Sun, C., Hu, B., Yu, C. & Lu, M. Synthesis and characterization of the pentazolate anion cyclo-N5− in (N5)6(H3O)3(NH4)4Cl. Science 355, 374–376 (2017)
Zhang, C. et al. A symmetric Co(N5)2(H2O)4·4H2O high-nitrogen compound formed by cobalt(II) cation trapping of a cyclo-N5− anion. Angew. Chem. Int. Ed. 56, 4512–4514 (2017)
Klapötke, T. M. & Hammerl, A. in Comprehensive Heterocyclic Chemistry III (eds Katritzky, A. R., Ramsden, C. A., Scriven, E. F. V. & Taylor, R. J. K. ) 739–757 (Elsevier, 2008)
Butler, R. N., Stephens, J. C. & Burke, L. A. First generation of pentazole (HN5, pentazolic acid), the final azole, and a zinc pentazolate salt in solution: A new N-dearylation of 1-(p-methoxyphenyl) pyrazoles, a 2-(p-methoxyphenyl) tetrazole and application of the methodology to 1-(p-methoxyphenyl) pentazole. Chem. Commun. 1016–1017 (2003)
Schroer, T., Haiges, R., Schneider, S. & Christe, K. O. The race for the first generation of the pentazolate anion in solution is far from over. Chem. Commun. 1607–1609 (2005)
Choi, C., Yoo, H. W., Goh, E. M., Cho, S. G. & Jung, Y. Ti(N5)4 as a potential nitrogen-rich stable high-energy density material. J. Phys. Chem. A 120, 4249–4255 (2016)
Burke, L. A., Butler, R. N. & Stephens, J. C. Theoretical characterization of pentazole anion with metal counter ions. Calculated and experimental 15N shifts of aryldiazonium, -azide and -pentazole systems. J. Chem. Soc., Perkin Trans. 2 1679–1684 (2001)
Tsipis, A. C. & Chaviara, A. T. Structure, energetics, and bonding of first row transition metal pentazolato complexes: a DFT study. Inorg. Chem. 43, 1273–1286 (2004)
Perera, S. A., Gregusová, A. & Bartlett, R. J. First calculations of 15N–15N J values and new calculations of chemical shifts for high nitrogen systems: a comment on the long search for HN5 and its pentazole anion. J. Phys. Chem. A 113, 3197–3201 (2009)
Crawford, M. J. & Mayer, P. Structurally characterized ternary U–O–N compound, UN4O12: UO2(NO3)2·N2O4 or NO+UO2(NO3)3−? Inorg. Chem. 44, 8481–8485 (2005)
Malar, E. J. P. Do penta- and decaphospha analogues of lithocene anion and beryllocene exist? Analysis of stability, structure, and bonding by hybrid density functional study. Inorg. Chem. 42, 3873–3883 (2003)
Zhang, X., Yang, J., Lu, M. & Gong, X. Structure, stability and intramolecular interaction of M(N5)2 (M = Mg, Ca, Sr and Ba): a theoretical study. RSC Adv. 5, 21823–21830 (2015)
Bianchi, R., Gervasio, G. & Marabello, D. Experimental electron density analysis of Mn2(CO)10: metal–metal and metal–ligand bond characterization. Inorg. Chem. 39, 2360–2366 (2000)
Stowasser, R. & Hoffmann, R. What do the Kohn–Sham orbitals and eigenvalues mean? J. Am. Chem. Soc. 121, 3414–3420 (1999)
Zhang, C., Sun, C., Hu, B. & Lu, M. Investigation on the stability of multisubstituted arylpentazoles and the influence on the generation of pentazolate anion. J. Energ. Mater. 34, 103–111 (2016)
Frisch, M. J. et al. Gaussian 09, Revision A.02 (Gaussian, Inc., 2009)
Becke, A. D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993)
Lee, C., Yang, W. & Parr, R. G. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988)
Hay, P. J. & Wadt, W. R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 299–310 (1985)
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012)
Cremer, D. & Kraka, E. Chemical bonds without bonding electron density — does the difference electron-density analysis suffice for a description of the chemical bond? Angew. Chem. Int. Edn 23, 627–628 (1984)
SAINT v7.68A (Bruker AXS Inc., 2009)
Sheldrick, G. M. SHELXL-2014/7 (Univ. Göttingen, 2014)
SADABS v2008/1 (Bruker AXS Inc., 2008)
Acknowledgements
This work was supported by the NSAF (U1530101) and the National Natural Science Foundation of China (51374131). We thank C. Zhang and B. Hu for co-exploring the rupture of C–N bonds in phenylpentazole at the beginning of the project, Z. Zhang for analysis of the crystal structures, L. Lu for analysis of Raman and NMR spectra, and L. Cheng for DSC measurements of decomposition kinetics.
Author information
Authors and Affiliations
Contributions
Y.X., P.W. and M.L. conceived and designed the experiments. C.S. and Q.L. prepared N5− solid. Y.X. and Q.W. performed the crystal experiments. Y.X., Q.W. and P.W. performed the measurements and analysed the data. P.W. performed the DFT calculations. Y.X., P.W. and M.L. co-wrote the manuscript. All authors contributed to the overall scientific interpretation and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks K. O. Christe, T. M. Klapötke and H. Östmark for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 High-resolution mass spectrum and 15N NMR spectra of 2.
a, Single mass and formula analysis of 2. b, 15N NMR spectra of 2 (in DMSO-d6, MeNO2 as external standard). c, 15N NMR spectra of NaN5 (15N labelled on N2) before column chromatography (in CD3OD, MeNO2 as external standard); inset, synthetic scheme for the preparation of 15N-labelled N5−.
Extended Data Figure 2 Molecular structures of 2–6 shown by ORTEP representations.
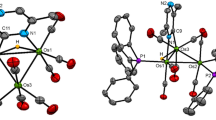
a, b, Ball-and-stick packing diagrams of 2, viewed normal to (001) with hydrogen bonds (a), and normal to (010). (b). c–e, Molecular structures of 3–5, respectively, viewed normal to (100), shown by ORTEP representations. f–k, Ball-and-stick packing diagrams of 3–5, viewed normal to (001) (f–h), and normal to (100) (i–k). l, Hydrogen bonds in the packing of 6, and unit cell parameters. m, A unit cell of 6 viewed normal to (100), shown by ORTEP representation. n, Ball-and-stick packing diagram of 6 viewed normal to (001).
Extended Data Figure 3 XPS spectra of complexes 1–6.
a–f, Survey spectra of 1–6, respectively. g–l, Narrow scan of the N 1s peak of 1 (g), 2 (h), 3 (j), 4 (k), 5 (l) and 6 (i). The experimental and fitting curves are shown in black and red, with the pyrrolic (N1), pyridinic (N2) and quaternary (N3) nitrogen curves shown in blue, pink and green, respectively.
Extended Data Figure 4 Theoretical simulation of complexes 2–6.
a–e, Model deformation density maps of 2 (a), 3 (c), 4 (d), 5 (e) and 6 (b) in the plane defined by the N5 rings. Scale in a.u. The lone pair of electrons on N is attracted by H+, Mn2+, Fe2+ and Co2+. f–k, Molecular orbital correlation diagrams for interactions between M2+ (M = Mn, Fe and Co) and N5− with and without H2O in complexes: Mn(N5)2 and Mn(H2O)4(N5)2 (f, g); Fe(N5)2 and Fe(H2O)4(N5)2. (h, i); Co(N5)2 and Co(H2O)4(N5)2. (j, k) The positive and negative phases of the molecular orbitals are shown in deep red and green; hydrogen, nitrogen, oxygen and metal atoms are shown in white, blue, red and brown respectively.
Extended Data Figure 5 DSC measurement of the decomposition kinetics of complexes 2–4 and 6, and their apparent activation energies.
a–d, DSC curves of complexes at heating rates of 2, 5, 8 and 10 °C min–1: 2 (a), 3 (b), 4 (c), and 6 (d). e, Apparent activation energy Ea of the first exothermic peak of complexes 2–4 and 6, calculated using the Kissinger method. b, heating rate; Tp, peak temperature.
Extended Data Figure 6 TG–DSC–MS measurements of complexes 5 and 6.
a, b, TG–DSC–MS curves of 5 (a) and 6 (b); ions of m/z 14, 18, 28 and 42 are selected. Note explosion in a.
Extended Data Figure 7 IR spectra of complexes 3–6 after heating at different temperatures.
a, Complexes 3–6 after heating at 60 °C for 0.5 h, denoted as 3′–6′, respectively. cyclo-N5– remains after the partial loss of water, as shown in the highlighted regions. b, Complexes 3–6 heated at 110 °C for 0.5 h, denoted as 3″–6″, respectively. The signal for N3– indicates that cyclo-N5– decomposed under these conditions. c–e, Temperature-dependent IR spectra of 4–6 in air. Significant decomposition of cyclo-N5– occurs at 95, 65 and 100 °C, respectively.
Supplementary information
Supplementary Tables
This file contains Supplementary Tables 1-4. (PDF 163 kb)
Supplementary Data
This file contains crystallographic data for [Na(H2O)(N5)]•2H2O. (CIF 84 kb)
Supplementary Data
This file contains crystallographic data for [Mn(H2O)4(N5)2]•4H2O. (CIF 67 kb)
Supplementary Data
This file contains crystallographic data for [Fe(H2O)4(N5)2]•4H2O. (CIF 84 kb)
Supplementary Data
This file contains crystallographic data for [Co(H2O)4(N5)2]•4H2O. (CIF 11 kb)
Supplementary Data
This file contains crystallographic data for [Mg(H2O)6(N5)2]•4H2O. (CIF 12 kb)
Rights and permissions
About this article

Cite this article
Xu, Y., Wang, Q., Shen, C. et al. A series of energetic metal pentazolate hydrates. Nature 549, 78–81 (2017). https://doi.org/10.1038/nature23662
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature23662
- Springer Nature Limited
This article is cited by
-
Experimental detection of the diamino-pentazolium cation and theoretical exploration of derived high energy materials
Scientific Reports (2024)
-
Stabilization of N6 and N8 anionic units and 2D polynitrogen layers in high-pressure scandium polynitrides
Nature Communications (2024)
-
Taming cyclo-pentazolate anions with a hydrogen-bonded organic framework
Communications Materials (2024)
-
Prediction of impact sensitivity and electrostatic spark sensitivity for energetic compounds by machine learning and density functional theory
Journal of Materials Science (2024)
-
A physical organic strategy to predict and interpret stabilities of chemical bonds in energetic compounds for the discovery of thermal-resistant properties
Journal of Molecular Modeling (2024)