

Abstract
Cpf1 is an RNA-guided endonuclease that is emerging as a powerful genome-editing tool1,2. Here we provide insight into its DNA-targeting mechanism by determining the structure of Francisella novicida Cpf1 with the triple-stranded R-loop generated after DNA cleavage. The structure reveals the machinery involved in DNA unwinding to form a CRISPR RNA (crRNA)–DNA hybrid and a displaced DNA strand. The protospacer adjacent motif (PAM) is recognized by the PAM-interacting domain. The loop-lysine helix–loop motif in this domain contains three conserved lysine residues that are inserted in a dentate manner into the double-stranded DNA. Unzipping of the double-stranded DNA occurs in a cleft arranged by acidic and hydrophobic residues facilitating the crRNA–DNA hybrid formation. The PAM single-stranded DNA is funnelled towards the nuclease site through a mixed hydrophobic and basic cavity. In this catalytic conformation, the PAM-interacting domain and the helix–loop–helix motif in the REC1 domain adopt a ‘rail’ shape and ‘flap-on’ conformations, respectively, channelling the PAM strand into the cavity. A steric barrier between the RuvC-II and REC1 domains forms the ‘septum’, separating the displaced PAM strand and the crRNA–DNA hybrid, avoiding DNA re-annealing. Mutations in key residues reveal a mechanism linking the PAM and DNA nuclease sites. Analysis of the Cpf1 structures3,4 proposes a singular working model of RNA-guided DNA cleavage, suggesting new avenues for redesign of Cpf1.
Similar content being viewed by others
Main
Adaptive immunity against invading genetic elements in prokaryotes is accomplished by complexes composed of CRISPR-associated proteins (Cas)5,6,7 and crRNAs8. The resulting ribonucleoproteins are guided by the crRNA to recognize and cleave complementary DNA (or RNA) for interference9,10,11. The redesign of the guide RNA, to target specific DNA sites, is a powerful method for genome modification in multiple biomedical and biotechnological applications. The best-characterized CRISPR system is the Cas9 endonuclease, categorized as class 2 type II12,13,14. Another class 2 CRISPR system assigned to type V, which contains a large protein termed Cpf1 (CRISPR from Prevotella and Francisella; Fig. 1a, Extended Data Fig. 1) has been identified in bacterial genomes1,2,15. Cpf1 CRISPR arrays are processed into mature crRNAs without an additional trans-activating crRNA16 by Cpf1 (ref. 17). The Cpf1–crRNA complex efficiently cleaves a target double-stranded DNA (dsDNA) containing a short T-rich PAM motif on the 5′ end of the non-target strand, in contrast to the Cas9 which cleaves a target dsDNA containing a G-rich PAM motif located on the 3′ end of the non-target strand. Moreover, Cpf1 introduces a staggered double-stranded DNA break2,17 instead of the blunt end cut generated by Cas9 (refs 12, 13, 14) (Fig. 1b, c), which could potentially expand genome modification capabilities2. Recently, Cpf1 enzymes have been shown to mediate robust and precise genome editing in mammalian18,19,20 and plant21 cells, expanding our genome manipulation capabilities.

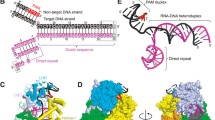
a, Domain organization of FnCpf1 (ref. 4). b, Schematic diagram of the R-loop formed by the crRNA and the target DNA. The orange triangles represent phosphodiester cleavage positions in the t- and nt-strands; the boxed light blue nucleotides represents a section of the cleaved target DNA not present in the structure. c, Urea gel depicting the length of the cleavage products of the target DNA. The markers indicate the position of the cleaved phosphodiester following Fig. 1b numeration (see Extended Data Table 2). The products show a cut at position −14 of the nt-strand, while the t-strand is cleaved at position 21 or 22. d, Overview of the FnCpf1–RNA–target-DNA ternary complex. e, View of the FnCpf1–R-loop structure (FnCpf1 polypeptide omitted for clarity). An omit map for the nucleotides of the nt-strand after the PAM is shown at 1σ level. BH, bridge helix domain; HLH, helix–loop–helix; PI, PAM-interacting domain.
The structure of Lachnospiraceae bacterium Cpf1 (LbCpf1)3 and Acidaminococcus sp. Cpf1 (AsCpf1)4 have been solved, and these structures provide catalytically inactive snapshots. Hence, questions regarding DNA recognition, unzipping and subsequent cleavage remained unanswered. We present the crystal structure of a catalytically active Francisella novicida Cpf1 (FnCpf1)–R-loop complex (Fig. 1d, e), revealing the critical residues that unzip and cleave the target DNA. To explore target DNA processing, we reconstituted a functional FnCpf1–crRNA, which was then associated with target DNA (Extended Data Fig. 2, Methods).
The FnCpf1–R-loop structure represents a snapshot of the endonuclease–product complex after substrate cleavage. The protein displays an oval ‘sea conch’ structure with a bilobal cavity formed by the Nuc and the REC lobes (Fig. 1d, Supplementary Video 1, Extended Data Table 1). The target DNA is cleaved yielding a triple-stranded R-loop (Fig. 1e, Extended Data Fig. 3) with the target (t)-strand hybridized on the crRNA, while the dissociated PAM non-target (nt)-strand is stabilized and funnelled towards the DNA nuclease site (Fig. 2a). Unambiguous electron density was observed for seven nucleotides of the nt-strand upstream of the PAM sequence. The lack of density for the nucleotides −8 to −14 in the nt-strand indicates the high mobility on the distal end of the PAM strand, as shown for Cas9 (ref. 22) (Fig. 1e, Extended Data Fig. 3a).

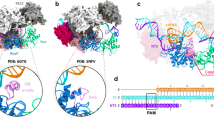
a, Surface representation of the FnCpf1–R-loop complex. The transparent surface permits visualization of the triple helix (crRNA–DNA hybrid and the nt-strand). The green dots show the predicted path of the nt-strand to the DNA nuclease site after dT–7. b, Schematic showing FnCpf1–R-loop interactions. For clarity, part of the crRNA–DNA hybrid is omitted. Polar contacts of the nucleic acids with the protein side and main chain (in brackets) are indicated. c, Detailed view of the PAM recognition and the dsDNA unwinding depicting the conserved K667, M668, K671 and K677 residues (side chains in orange). d, Zoom of the dG−1/dC+1 pair uncoupling. The 2mFo − DFc electron density refined map is contoured at 1.2σ. e, Urea gel showing the cleavage activity and pattern of the LKL mutants. Each experiment was repeated four times. WT, wild type. Error bars, s.d.
The RNA nuclease site in the WED-III subdomain is located at the back of the protein (Fig. 1d), where key residues involved in RNA processing embrace the 5′ end of the RNA handle (Extended Data Fig. 4a).
DNA nuclease activity takes place in a pocket at the interface between the RuvC and Nuc domains (Extended Data Fig. 4b), in which the E1006 and R1218 residues have an important role in catalysis. These residues are 11 Å apart and their alanine mutations suggested that the Nuc and RuvC domains cleave the t-strand and nt-strand in AsCpf1 (ref. 4). The E1006A and R1218A mutants did not show this selective cleavage in FnCpf1 (Extended Data Fig. 4c). The distances of dG at position +20 in the t-strand and dT at position −7 in the nt-strand to the R1218 and the E1006 is 20 Å and 28 Å, respectively. Although the central site of the RuvC–Nuc pocket is 25.5 Å away from the first nucleotide visualized in the nt-strand (dT−7), this distance can be covered by the extra seven nucleotides in the R-loop, which could not be modelled, in agreement with our cleavage analysis in the −15 position (Fig. 1c). However, the RuvC–Nuc pocket is around 32 Å away from the t-strand, implying that the enzyme undergoes a large conformational change to hydrolyse the phosphodiester in positions 21 or 22 of the t-strand (Fig. 1c). The high flexibility shown by the REC2 and Nuc domains and the differences between the apo and PAM-bound Cpf1 structures23 suggests a conformational change approximates the t-strand to the nuclease site.
PAM recognition is a critical aspect of DNA targeting by RNA-guided nucleases, as it is a prerequisite for ATP-independent DNA strand separation and crRNA–target-DNA heteroduplex formation24. In contrast with Cas9, the PAM-binding site in FnCpf1 is around 70 Å away from the nuclease active site (Fig. 2a). FnCpf1 recognizes a 5′-YTN-3′ PAM sequence2 by residues in the WED-II-III, REC1 and PAM-interacting domains (Fig. 2a, b).
The loop-lysine helix–loop (LKL) region (L662 to I679) in the PAM-interacting domain contains the conserved K667, K671 and K677 residues and has an essential role in recognizing PAM and promoting strand separation (Fig. 2c). Only K671 interacts with the bases in the PAM region, suggesting that shape might have a more important role than direct base readout in PAM recognition4. Our structure shows that the helix in the LKL region has a key role in dsDNA unzipping by displacing the PAM nt-strand (Fig. 2c). The helix is inserted at an angle of 45° with respect to the dsDNA longitudinal axis, working as a dentate plough unwinding the helical dsDNA (Extended Data Fig. 4d). The LKL element inserts its three-conserved lysine ‘pins’ (K667, K671 and K677) in a staggered manner on the DNA (Fig. 2c, Extended Data Fig. 4d). K677 is inserted in the major groove interacting with the backbone of 3′-dG-5′ in the t-strand, while K671 is inserted in the minor groove contacting the C2 carbonyl in dT+2, the N3 of dA−3 in the PAM sequence and the oxygen of the deoxyribose of dC−4 (Fig. 2b). Finally, the side chain of K667 disrupts the Watson–Crick interaction of the dG–1/dC+1 pair after the PAM (Fig. 2d). The uncoupling of the first base pair of the target DNA initiates the unzipping of the dsDNA, facilitating the hybridization of the t-strand with the crRNA. The conserved P663 and P670, collaborate to insert the LKL plough and unzip the target DNA. P663 positions the helix in the correct orientation for its insertion in the dsDNA; accordingly, the P663A mutation reduces FnCpf1 cleavage activity (Fig. 2e, Extended Data Fig. 5). P670 favours the insertion of K671 in the PAM and the conserved M668 also disrupts the base pairing of the double helix, facilitating the uncoupling of dG–1 by the invariant K667 (Fig. 2d). As in AsCpf1 (ref. 4), the K671A mutation in FnCpf1 causes a large reduction in activity. Remarkably, we found that the K667A and K677A mutants also reduce FnCpf1 DNA cleavage activity by around 70%, indicating the pivotal role of the lysines in PAM recognition and DNA unzipping (Fig. 2e). In addition, although the K667A mutant displays DNA cleavage products similar to the wild type, K677A and P663A generate a different pattern, in which the nt-strand is cleaved into shorter fragments than those produced by the wild type (Figs 1c, 2e). None of the lysine mutants recognizes a different PAM sequence (Extended Data Fig. 5c).
This suggests that P663 and K677 initiate the positioning of the LKL helix to facilitate the insertion of K671 in the PAM, placing K667 to disrupt the Watson–Crick pair after the PAM to determine the length of the cleaved product of the target DNA. The activity reduction and different cleavage pattern exhibited by the K671A mutant (Fig. 2e) underlines its key role to position the target DNA for cleavage. Our analysis suggests that the LKL would function as the marker of a molecular ruler linking the 70 Å distant PAM and DNA nuclease sites, and thereby recognition and catalysis, to hydrolyse the correct phosphodiester bond.
Overlapping with the region in which the first base pair of the target DNA is uncoupled, the WED-I-II-III, REC1 and RuvC-I domains build the unzipping cleft (Figs 2a, 3a) composed by the well-conserved E184, P883, I884, G826, A828, E827, E829, F831, H881, G608, L661, L662 and P663 residues (Extended Data Fig. 1, Extended Data Fig. 6a). The arrangement of the acidic and hydrophobic residues in the cleft facilitates the coupling of the dC+1 and G+1 (Fig. 3a, Extended Data Fig. 6a). E184 and E827, behind the t-strand, facilitate the scanning of the complementary base and the formation of the crRNA–DNA hybrid by electrostatic repulsion of the phosphate backbone of dC+1 and dT+2 (Extended Data Fig. 6a). The phosphate of dT−1 of the t-strand is inverted by the interaction with K823, Y659 and G826 and N825, which also house the dT−1 phosphate. We found that mutations in alanine and glutamate in the neighbouring G608, which contacts the PAM dA–2 phosphate, severely affect activity (Fig. 3b, Extended Data Fig. 5), as observed for G826 (ref. 4). Therefore, these two invariant glycine residues are essential for housing the target DNA.

a, Surface representation of the unzipping cleft formed by the WED, REC1 and RuvC domains, see Extended Data Fig. 6a. b, Urea gels showing the cleavage activity and pattern of mutations in the cleft, the PAM-interacting domain and the septum. Each experiment was repeated four times. Error bars, s.d. c, R692, N666 and T696 interaction with the nt-strand in the PAM-interacting domain rail conformation. Dashed lines show the polar contacts. d, Magnified view of the septum displaying the K1065 and Y201 interaction, separating the paths towards the DNA nuclease site for the crRNA–DNA and the nt-strand. e, View of the ‘septum’ showing the displaced nt-strand separated from the hybrid crRNA–DNA.
A comparison of the LbCpf1 apo3 and FnCpf1–R-loop structures indicates that Cpf1 undergoes a large conformational rearrangement in the PAM-interacting domain (Extended Data Fig. 7) and the REC lobe (Extended Data Fig. 8). The PAM-interacting domain in the FnCpf1–R-loop complex readjusts to accommodate the nt-strand. The nucleotides of the nt-strand exhibit an extended distorted helical conformation, and the phosphate backbone displays a number of contacts with residues of the PAM-interacting, RuvC-I and-II domains (Fig. 2b). The residues (P670 to E715) in the loop of the PAM-interacting domain adopt a ‘rail shape’, embracing the nt-strand into a hydrophobic cavity with basic patches and guiding it to the active site (Fig. 3c, Extended Data Figs 6b, c, 7).
R692, N666 and T696 interact with the phosphate backbone of the nt-strand (Fig. 3c), funnelling the ssDNA to the RuvC–Nuc pocket. The R692A mutation reduces the endonuclease activity of FnCpf1 by 70%, suggesting that this first residue in the ‘rail’ has a key role interacting with the uncoupled dG–1 and directing the nt-strand to the DNA nuclease site. Although the N666A and H694A mutants produce only a small activity reduction (5–15%), their cleaved nt-strand products are two bases shorter than the wild type, indicating that in addition to the LKL region, the residues in this loop of the PAM-interacting domain are also involved in the molecular ruler determining the nt-strand cleavage length (Fig. 3b).
The conformational differences observed between the apo3 and the R-loop complex structures suggests that changes in the HLH segment of REC1 (F61–F123) could be induced by the binding of the PAM to protect the nt-strand after unzipping (Extended Data Fig. 8b). The HLH segment in the apo conformation would be in contact with the conserved T197–S220 and F264–G277 helices in REC1, like in the LbCpf1 apo structure3, aligned with the REC lobe (Extended Data Figs 1, 8b). The binding of the target DNA induces a closure of the protein, positioning the Nuc and REC lobes closer23. W971 in the bridge helix domain would be anchored in the REC2 hydrophobic pocket, as observed in AsCpf1 (ref. 4), stopping the movement and locking the closed active conformation. Hence, the FnCpf1–R-loop structure suggests that PAM recognition and nt-strand displacement induce a HLH around 45° rotation mimicking a ‘flap-on’ movement (Supplementary Video 2). The HLH ‘flap-on’ and the PAM-interacting ‘rail’ conformations would protect the nt-strand by partially occluding the cleft, directing it to the DNA nuclease site (Extended Data Figs 7, 8).
The structure shows two paths in its bilobal architecture. The crRNA–t-strand hybrid spans all the length of the central cavity between the REC and Nuc lobes, while the nt-strand runs along a trail separated from the previous one by contacts between the conserved REC1 (T197–V204) and RuvC (F1061–Q1070) segments. K1065 and Y201 form a thin barrier at the level of the dT−7 (the ‘septum’) avoiding DNA re-annealing (Fig. 3d, e). The double mutant K1065A/K1066A showed 52% activity, revealing the importance of the septum (Fig. 3b). Remarkably, the cleavage product of the nt-strand is six nucleotides shorter, whereas the t-strand is cut as the wild type, indicating that the different regions building the path leading the nt-strand to the DNA nuclease site are essential for correct cleavage (Fig. 3b).
Our findings suggest a working mechanism for Cpf1 (Fig. 4). The apo Cpf1–RNA complex undergoes a conformational change from a relaxed open conformation to a compact DNA-bound conformation to scan target DNAs. The recognition of the PAM promotes the ‘flap-on’ conformation of the HLH motif. After PAM recognition, the LKL helix is inserted in the dsDNA, starting the melting of the bases proximal to the PAM. K671 and K677 interact with the PAM, and K667 promotes the uncoupling of the first Watson–Crick base pair after the PAM (Fig. 4b), the phosphate in position −1 of the t-strand is inverted and the dsDNA unzips in the cleft, allowing the pairing of the initial uncoupled bases of the t-strand with the crRNA. The electrostatic environment in the cleft favours the pairing of the uncoupled DNA nucleotides with the crRNA. If a mismatch occurs and the energetics of the process do not favour the crRNA–DNA coupling, the LKL helix is removed, the DNA re-hybridizes, and Cpf1 continues searching for a PAM containing the complementary target, as has been proposed for Cas9 (refs 25, 26). In case of complementarity after the PAM, the PAM-interacting domain adopts the ‘rail’ conformation while the nt-strand is displaced and the ‘septum’ is formed. Consequently, propagation will result in the crRNA–DNA hybrid residing in the main cavity generating a 5′ overhang after cleavage. Mutations in the RuvC-II–Nuc pocket (Extended Data Fig. 4c) do not suggest a selective cleavage of each strand to generate the double-stranded break (Extended Data Fig. 4b, c), similarly to C2c1 (ref. 27). The FnCpf1–R-loop structure exhibits a different pathway for each strand to reach the RuvC–Nuc pocket, determining the length of the overhang. Further studies are needed to provide the details of phosphodiester hydrolysis.

a, Cpf1 is in an open conformation (I), the presence of target DNA induces a compact conformation allowing scanning of PAM (II). Binding to PAM fosters insertion of the LKL helix and the ‘flap-on’ conformation of the HLH motif (III). If coupling between the t-strand and the crRNA occurs, the PAM-interacting domain adopts the ‘rail’ conformation, the nt-strand is displaced and the ‘septum’ is formed (IV). Finally, catalysis proceeds generating an overhang (V). b, The LKL scans the PAM (I). K677 contacts the phosphate before the first base in the PAM site in the t-strand, allowing K671 to test binding of the PAM sequence contacting the bases in positions dA−3 and dT+2 (II). Once K671 is properly positioned, the LKL helix is inserted in the dsDNA. The K667 uncouples the base pair after PAM and the inversion of the phosphate in the −1 base by K823 and Y659 in the t-strand starts unzipping to allow hybridization. The PAM-interacting domain adopts the rail conformation and R692 and N666 stabilize the nt-strand (III).
Our findings provide a detailed understanding into Cpf1 endonucleases, which can be harnessed to gene targeting19,20,21,28 or adapted for other applications for example, base-editing29 or large-scale genetic studies30 widening the CRISPR–Cas repertoire.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Protein expression and purification
The gene encoding Cpf1 from Francisella novicida U112 (FnCpf1) was obtained from Addgene (item ID: 69975, UniProt entry A0Q7Q2). The open reading frame was amplified by PCR using the primers FnCpf1 forward and FnCpf1 reverse (Extended Data Table 2) and cloned into pET21a. The obtained construct was transformed in Escherichia coli BL21 star (DE3) cells containing the pRare2 plasmid. Protein expression was induced with 1 mM IPTG for 3 h. For the preparation of selenomethionine-substituted protein, the cells were grown in SelenoMethionine Medium Complete (Molecular Dimensions) including 40 μg ml−1 selenomethionine and the expression was induced as described above. Cells were resuspended in lysis buffer (50 mM Bicine (pH 8.0), 150 mM KCl, 1 tablet per 50 ml Complete Inhibitor cocktail EDTA Free (Roche), 50 U ml−1 Benzonase, 1 mg ml−1 lysozyme and 0.5 mM TCEP). After cell disruption by French press, cell debris and insoluble particles were removed by centrifugation at 10,000g at 4 °C. The supernatant was loaded onto a HisTrap column (GE Healthcare) equilibrated in buffer A (50 mM Bicine (pH 8.0), 150 mM KCl and 0.5 mM TCEP). After the sample was loaded, the column was washed with buffer A containing 5 mM imidazole to prevent non-specific binding of contaminants to the resin. The sample was eluted with 50 mM Bicine (pH 8.0), 150 mM KCl, 0.5 mM TCEP and 250 mM imidazole. Enriched protein fractions were pooled together and applied onto a HiTrap Heparin HP column (GE Healthcare) equilibrated with buffer A. The protein was eluted with a linear gradient of 0–100% buffer H (50 mM Bicine (pH 8.0), 1 M KCl and 0.5 mM TCEP). Protein-rich fractions were collected and concentrated (using 100 kDa MWCO Centriprep Amicon Ultra devices) and subsequently loaded onto a HiLoad 16/60 200 Superdex column (GE Healthcare) equilibrated in buffer A. The protein peaks were concentrated (using 100 kDa MWCO Centriprep Amicon Ultra devices), flash-frozen in liquid nitrogen and stored at −80 °C. The protein concentration was determined using the theoretical molar extinction coefficient at 280 nm calculated from the amino-acid composition. An overloaded SDS–PAGE gel stained with SimplyBlue (Invitrogen) displayed a highly pure protein preparation.
Mutagenesis
Mutagenesis was performed using the Q5 site direct mutagenesis kit (NEB). The primers were designed according to the NEBaseChanger (see Extended Data Table 2). The FnCpf1 mutants were expressed and purified as described above. The purity of mutant proteins was analysed by SDS–PAGE stained with SimplyBlue (Invitrogen; Extended Data Fig. 5).
RNA production and purification
DNA oligonucleotides corresponding to the reverse complemented sequence of the target site (RNA template) and a short T7 priming sequence (T7 primer) were purchased from Integrated DNA Technologies (IDT; Extended Data Table 2). The oligonucleotides were annealed at a final concentration of 20 μM in annealing buffer containing 150 mM KCl by heating the mixture up to 95 °C for 10 min followed by a cool ramp to 4 °C over 10 min. This partial DNA duplex was used as template in the transcription reaction carried out with HiScribe T7 Quick High Yield RNA Synthesis (NEB). The reaction was stopped using 2× stop solution (50 mM EDTA, 20 mM Tris-HCl pH 8.0 and 8 M urea) and the RNA was denatured at 95 °C for 10 min. The transcription product was purified by preparative electrophoresis with a Bio-Rad Model 491 PrepCell apparatus equipped with the 37 mm i.d. gel tube using a 9-cm 1× TBE, 15% (19:1) polyacrylamide/7 M urea gel at room temperature.
FnCpf1–R-loop complex formation
The 31-nucleotide DNA template was annealed using a single-stranded oligonucleotide, according to the protocol above for the preparation of the transcription template. The complex was assembled in reconstitution buffer consisting of 150 mM KCl, 50 mM Bicine (pH 8.0), 5 mM MgCl2 at a molar ratio of protein:RNA:DNA 1:1.3:1.7. The FnCpf1 protein was mixed with crRNA and incubated for 30 min at 25 °C before adding the 31 bp target DNA duplex (nt-strand and t-strand; Extended Data Table 2) followed by one-hour incubation at 25 °C. The reconstituted FnCpf1–R-loop complex was further purified by preparative electrophoresis with a Bio-Rad Model 491 PrepCell apparatus using a 5% (w/v) non-denaturing polyacrylamide gel at 4 °C. The highly pure and homogeneous complex was separated from free DNA and high-molecular weight aggregates and immediately concentrated to 7 mg ml−1 using a Vivaspin 20 50000 MWCO.
In vitro cleavage assay
The fluorescently labelled DNA substrate was prepared by annealing the t-strand–6FAM-A and nt-strand–6FAM-A (DNA1A) under the same condition described above (Extended Data Table 2). To identify which strands were cleaved the DNA substrate was prepared by annealing nt-strand–6FAM-A with t-strand-A (DNA2A) and t-strand–6FAM-A with t-strand-A (DNA3A) (Extended Data Table 2). The RNA–protein complexes were formed in 20 mM Bicine-HCl (pH 8), 150 mM KCl, 0.5 mM TCPE (pH 8) and 5 mM MgCl using 6 nmoles of purified RNA and 4 nmoles of purified protein. The complex was incubated at 25 °C for 30 min before adding 2 nmoles of DNA substrate. The mixture was incubated at 25 °C for 1 h and the reaction was stopped by adding equal volume of stop solution (8 M urea and 100 mM EDTA at pH 8) followed by incubation at 95 °C for 5 min. The samples were loaded on 15% Novex TBE-Urea Gels (Invitrogen) and run according to the manufacturer’s instructions. The gel was visualized using an Odyssey FC Imaging System (Li-Cor) and the intensity (I) of the DNA bands was quantified using ImageStudio. The percentage of DNA cleavage was calculated using the following formula: I(t-strand product) + I(nt-products) / I(total DNA). The total DNA was calculated as the sum of I(duplex) + I(single strands) + I(t-strand products) + I(nt-products). The presented data are representative of four experiments performed with two independent FnCpf1–RNA preparations.
In vitro saturation assay
The DNA substrate was prepared by annealing the oligonucleotide t-strand–6FAM and nt-strand–6FAM (DNA1) under the same condition describe above. To identify which strand was cleaved the DNA substrate was prepared by annealing nt-strand–6FAM with t-strand (DNA2) and t-strand–6FAM with t-strand (DNA3) (Extended Data Table 2 and Extended Data Fig. 2). The RNA–FnCpf1 complex was prepared as described above and incubated with increasing amounts of DNA1 from 60 pmoles to 60 nmoles. The reactions were incubated at 25 °C for 3 h, and then stopped and loaded on the gels as described above. The presented data are representative of four experiments performed with two independent FnCpf1 preparations.
Cleavage and activity analysis
The ribonucleoprotein complex was assembled with a target dsDNA of 31 base pairs and then purified for crystallization (Fig. 1b, see Methods). An in vitro analysis of the reconstituted R-loop, formed when the crRNA associated with the target DNA, revealed that the DNA overhang produced by FnCpf1 is 7–8 nucleotides (Fig. 1c), which is 2–3 nucleotides wider than previously reported2,17. In addition, FnCpf1 showed a dependence of Mg2+ for catalysis (Extended Data Fig. 2a, b).
The target (complementary) DNA strand (t-strand) shows two cleavage sites at position 21 and 22 (Fig. 1b, c). Previous reports have shown that the t-strand is cleaved at position 22 (ref. 17) (in this case the crRNA used in the experiment was 2 bases shorter on the 3′) or 23 (ref. 2), and an in vivo analysis suggested that at least 19 bases of crRNA must be coupled to the target DNA to support Cpf1 activity20. Thereby, both in vitro and in vivo data indicate that cleavage on the t-strand occurs on the nucleotides not coupled to the crRNA after position 20. In the non-target (not complementary) DNA strand (nt-strand), our R-loop shows a cleavage site at position −15 (Fig. 1b, c), thus forming a 5′ overhang of 7−8 nucleotides. Earlier reports have shown that the nt-strand is cleaved at position −19 (ref. 2) or at position −18 (ref. 17), thus generating a staggered cut with 4–5 nucleotides. The difference observed in the length of the 5′ overhangs may depend on the size of the DNA substrate containing the target site.
A time course cleavage assay shows that the reaction reaches a plateau in 60 min (Extended Data Fig. 2c). To test the efficiency of the endonuclease, we performed a saturation assay by increasing the concentrations of the substrate target DNA while keeping the amount of endonuclease constant. We observed saturation of the cleavage activity near the equimolar ratio of endonuclease/substrate DNA (3–4 nmoles), suggesting a slow dissociation of the enzyme from the cleaved product (Extended Data Fig. 2d). A similar enzymatic behaviour has been observed for Cas9 (ref. 31).
Crystallization
Initial crystallization screening was performed at 20 °C using the sitting-drop vapour-diffusion method and testing a collection of commercially available crystallization screens. After five days of incubation, the extensive initial screening rendered plate-like crystals in 0.35 M sodium thiocyanate, 20% w/v PEG 3350. Following the initial hit identification, crystal growth was optimized using a Dragonfly screen optimizer. Selenomethionine-modified crystals were obtained under the same conditions.
Structure determination, model building and refinement
The structure of FnCpf1–R-loop complex was determined by combining a molecular replacement solution, using AsCpf1 (ref. 4) as a search model in a native dataset (λ = 1.00), and a FnCpf1 selenium derivative crystal, where a single-wavelength anomalous diffraction (SAD) dataset was collected at the peak of Se K absorption edge (λ = 0.978). Both the native and the SAD data were collected from frozen crystals at 100 K using an EIGER detector at the PXI-XS06 beamline (Swiss Light Source Villigen). Data processing and scaling were accomplished with XDS32 and AIMLESS33 as implemented in autoPROC34 (Extended Data Table 1). All methionines were substituted by selenomethionine and 10 out of the 13 possible Se sites were identified using SHELX package35. Initial phases were calculated at 3.7 Å resolution PHASER as implemented in the PHENIX suite36,37. These initial phases were extended to 3.0 Å resolution using the native dataset with the PHENIX Autobuild routine. The initial Cα model was remodelled manually with Coot38 and O39,40 and refined initially using PHENIX36. Se anomalous maps were used as a guide during model building. Several rounds of manual building and refinement using BUSTER41 and REFMAC42 yielded the refinement and data collection statistics summarized in the Extended Data Table 1. Resolution cut-offs were determined using correlation-coefficient based criteria43. The identification and analysis of the protein–DNA–RNA hydrogen bonds and van der Waals contacts were performed with the Protein Interfaces, Surfaces and Assemblies service (PISA) as implemented in the CCP4 suite44 and Coot38. Unambiguous electron density was observed for seven nucleotides of the nt-strand upstream of the PAM sequence (Fig. 1e, Extended Data Fig. 3a). The figures for the manuscript have been produced with PyMOL (The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC). The final model has an Rwork/Rfree of 23/26% with good stereochemistry according to MolProbity (Extended Data Table 1) and only 0.7% of the residues in disallowed regions of the Ramachandran plot.
Data availability
The atomic coordinates and structure factors have been deposited in the Protein Data Bank under the accession code 5MGA. All other data are available from the corresponding authors upon request.
Accession codes
References
Vestergaard, G., Garrett, R. A. & Shah, S. A. CRISPR adaptive immune systems of Archaea. RNA Biol. 11, 156–167 (2014)
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell 163, 759–771 (2015)
Dong, D. et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 532, 522–526 (2016)
Yamano, T. et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell 165, 949–962 (2016)
Marraffini, L. A. CRISPR–Cas immunity in prokaryotes. Nature 526, 55–61 (2015)
Mohanraju, P. et al. Diverse evolutionary roots and mechanistic variations of the CRISPR–Cas systems. Science 353, aad5147 (2016)
Mojica, F. J., Díez-Villaseñor, C., García-Martínez, J. & Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60, 174–182 (2005)
Makarova, K. S. et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 13, 722–736 (2015)
Abudayyeh, O. O. et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 (2016)
Horvath, P. & Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170 (2010)
Sorek, R., Lawrence, C. M. & Wiedenheft, B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 82, 237–266 (2013)
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013)
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012)
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013)
Makarova, K. S. & Koonin, E. V. Annotation and classification of CRISPR–Cas systems. Methods Mol. Biol. 1311, 47–75 (2015)
Deltcheva, E. et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607 (2011)
Fonfara, I., Richter, H., Bratovicˇ, M., Le Rhun, A. & Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521 (2016)
Fagerlund, R. D., Staals, R. H. & Fineran, P. C. The Cpf1 CRISPR–Cas protein expands genome-editing tools. Genome Biol. 16, 251 (2015)
Kim, Y. et al. Generation of knockout mice by Cpf1-mediated gene targeting. Nat. Biotechnol. 34, 808–810 (2016)
Kleinstiver, B. P. et al. Genome-wide specificities of CRISPR–Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 34, 869–874 (2016)
Endo, A., Masafumi, M., Kaya, H. & Toki, S. Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci. Rep. 6, 38169 (2016)
Jiang, F. et al. Structures of a CRISPR–Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 (2016)
Gao, P., Yang, H., Rajashankar, K. R., Huang, Z. & Patel, D. J. Type V CRISPR–Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell Res. 26, 901–913 (2016)
Anders, C., Niewoehner, O., Duerst, A. & Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573 (2014)
Jiang, F., Zhou, K., Ma, L., Gressel, S. & Doudna, J. A. Structural biology. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481 (2015)
Szczelkun, M. D. et al. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl Acad. Sci. USA 111, 9798–9803 (2014)
Yang, H., Gao, P., Rajashankar, K. R. & Patel, D. J. PAM-dependent target DNA recognition and cleavage by C2c1 CRISPR–Cas endonuclease. Cell 167, 1814–1828 (2016)
Kim, D. et al. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 34, 863–868 (2016)
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016)
Wang, T. et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015)
Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C. & Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014)
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010)
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D 69, 1204–1214 (2013)
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D 67, 293–302 (2011)
Sheldrick, G. M. A short history of SHELX. Acta Crystallogr. A 64, 112–122 (2008)
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010)
Adams, P. D. et al. The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 (2011)
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004)
Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 (1991)
Kleywegt, G. J. & Jones, T. A. Model building and refinement practice. Methods Enzymol. 277, 208–230 (1997)
Bricogne G. et al. BUSTER version 2.10.3 (2016)
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255 (1997)
Diederichs, K. & Karplus, P. A. Better models by discarding data? Acta Crystallogr. D 69, 1215–1222 (2013)
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 (2011)
Acknowledgements
The Novo Nordisk Foundation Center for Protein Research is supported financially by the Novo Nordisk Foundation (Grant NNF14CC0001). This work was also supported by a grant from the Danish Cancer Society to G.M. We thank the beamline staff of X06SA at the Swiss Light Source (PSI, Switzerland) and the EMBL beamline P14 at DESY (Germany) for support during data collection. Data processing has been performed in the Computerome, the Danish National Computer for Life Sciences. We also thank H. Koc and M. Melynis from the Protein Production Facility Platform at CPR for excellent technical assistance.
Author information
Authors and Affiliations
Contributions
S.S. expressed and purified the Cpf1–R-loop complex with the help of P.A. S.S. and P.A. prepared the crystals and P.A. performed the cleavage and saturation experiments designed by S.S. All authors collected diffraction data. G.M. performed data processing and crystallographic analysis. All authors were involved in model building and refinement. All authors discussed the data, and helped G.M. to write the manuscript and prepare the figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks J. Doudna, D. Liu and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Sequence alignment of representative members of the Cpf1 family.
The amino acid sequences of Cpf1 from Francisella novicida (FnCpf1), Acidaminococcus sp. (AsCpf1), Lachnospiraceae bacterium (LbCpf1), Francisella tularensis (FtCpf1) and Porphyromonas crevioricans (PcCpf1) were aligned by Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo). The figure was prepared with ESPript (http://espript.ibcp.fr). Residue numbers are labelled according to the FnCpf1 sequence. Similar residues are shown in red and identical residues in white over red background. Blue frames indicate homologous regions. The different structural domains of FnCpf1 and their amino acid composition are depicted as boxes above the sequences and labelled with the same names and colour code as in Fig. 1a. In addition, thin boxes above the domains, following the same colour code, mark the positions of relevant regions mentioned in the manuscript. The mutations performed in key residues are marked according to their region: PI rail conformation (square), catalytic (circle), unzipping cleft (triangle), LKL (star) and ‘septum’ (cross). The protein sequence identity and similarity (in percentage) of FnCpf1 (1,300 amino acids) with AsCpf1, LbCpf1, FtCpf1 and PcCpf1 are FnCpf1:AsCpf1, 36:42; FnCpf1:LbCpf1, 43:49; FnCpf1:FtCpf1, 99:100; and FnCpf1:PcCpf1, 43:52.
Extended Data Figure 2 DNA saturation cleavage assay.
a, Scheme of the oligonucleotides used in the assay. In DNA1 both strands are labelled with 6-FAM in order to visualize the cleaved products of both non-target and target DNA. In DNA2 the non-target strand is labelled with 6-FAM in order to visualize the cleaved products from the non-target DNA. In DNA3 the target strand is labelled with 6-FAM to visualize the cleaved products from the target strand. b, Target DNA cleavage depends on Mg2+. Depletion of the cation by EDTA abrogates phosphodiester hydrolysis. c, Time course of the cleavage assay shows that FnCpf1 endonuclease completes the reaction in approximately 60 min. Quantification is shown in the graph below, representing the intensity of the corresponding bands (AU, arbitrary units) at different time points. d, Cleavage assay using DNA1, DNA2 and DNA3, shows the cleavage products of the different strands. For the saturation assay an increasing amount of DNA1 was used. Quantification of the cleaved and non-cleaved DNA1 is shown in the chart below (see Methods). The chart represents the intensity of the corresponding bands (AU) at increasing concentrations of the DNA1 substrate. The activity curve shows an asymptotic behaviour after 3 nmol of substrate. Each experiment was repeated 4 times. Error bars, s.d.
Extended Data Figure 3 The crRNA–DNA hybrid and the displaced non-target strand.
a, Detailed view of the 2mFo − DFc map of the R-loop inside FnCpf1 contoured at 1σ. b, Superposition of the FnCpf1–R-loop with the crRNA–DNA hybrid containing the PAM sequence of AsCpf1.
Extended Data Figure 4 The RNAase site and the LKL helix insertion.
a, Inset of the 5′ RNA region. Cpf1 can process its own crRNA. The RNA catalysis involves residues F873, H843, K869 and K852. 2mFo−DFc sigmaA map contoured at 1σ is displayed on the RNA molecule. The dashed lines indicate the flexible loop not built in the model. H843 is included in this highly mobile six residues loop and cannot be observed in our structure, showing the high flexibility of this region after RNA processing. The alanine mutants of these residues abolished RNA nuclease activity17. b, Overall view of the sliced FnCpf1 from the RuvC–Nuc interface, the key residues in each domain are surrounded by a pink and cyan oval. The dashed lines depict the distances between E1006 and R1218, and the dT–7 of the nt-strand and the dG+20 the t-strand to the RuvC and Nuc centres, respectively. The lower panel shows a detailed view of the RuvC–Nuc interface where the catalysis of the nt-strand and the t-strand occur after DNA unzipping. c, Cleavage assay of the E1006A and R1218A FnCpf1 mutants. Both mutations abrogate DNA cleavage. The experiment was repeated 4 times. d, Detailed view of the LKL helix insertion in the dsDNA. The dashed red lines show the 45° between the longitudinal DNA axis and the helix axis.
Extended Data Figure 5 Cleavage assay of FnCpf1 Mutants.
a, SDS–PAGE gel of the purified proteins used in the assay. b, Substrate target DNA oligonucleotide used in this assay. In DNA1A both strands are labelled with 6FAM in order to visualize the cleaved products of both the non-target and the target DNA strands in Figs 2e, 3b and Extended Data Fig. 5c. The encircled region indicates the PAM sequence c, Wild-type FnCpf1 and K671A, K667A and K677A mutants cleavage with different PAM sequences. The PAMs are indicated in bold above the gels. No cleavage can be observed for the wild-type FnCpf1 or mutants with PAMs different than the TTA. Neither FnCpf1 nor the LKL mutants exhibit PAM promiscuity in this context. Each experiment was repeated 4 times.
Extended Data Figure 6 Electrostatic potential map of FnCpf1.
a, The close-up of the unzipping cleft shows the disposition of the hydrophobic and acidic amino acids to facilitate the formation of the initial hybridization between the target DNA and the crRNA. b, c, Two different views of the cavity where the displaced nt-DNA resides after target cleavage. The cavity consists of hydrophobic and basic residues that direct the nt-DNA to the DNA nuclease site. The PAM, and DNA nuclease site and the septum are encircled in a dashed yellow line.
Extended Data Figure 7 PAM-interacting domain conformation.
a, The PI domains of LbCpf1, AsCpf1 and FnCpf1 structure have been superimposed using the Lsq routine in O36 to observe the different conformations depending on the presence or absence of the non-target DNA strand. The dashed black line represents the path of the nt-DNA. b, View of the non-target PAM strand embraced by the ‘rail’ conformation of the PI domain. The AsCpf1 PI domain is superimposed for comparison. The electron density map (2mFo − DFc map) displayed on the non-target strand inside FnCpf1 is contoured at 1σ.
Extended Data Figure 8 Comparison of Cpf1 conformations upon target DNA binding.
a, The figure displays surface models after superimposition of LbCpf1, AsCpf1 and FnCpf1 conformations representative of different states. The comparison shows the differences in the Cpf1 structure from the less to the more compact conformation. Nucleic acids are not shown for clarity. b, The figure depicts the differences in the HLH section between the LbCpf1 apo structure and the FnCpf1–R-loop structure. The HLH domain adopts the ‘flap-on’ conformation when the PAM sequence is bound.
Supplementary information
Supplementary Figure
This file contains uncropped scans of all the UREA-PAGE gels in the main and in the Extended Data Figures. (PDF 8001 kb)
Overview of the FnCpf1 R-loop crystal structure
The protein-crRNA-target DNA ternary complex is rotated 180º around its longitudinal axis. The polypeptide is composed by the WED (I, II and II subdomains), REC1, REC2, PI, BH, RuvC (I, II and III subdomains) and Nuc domains. The colours assigned to FnCpf1 domains are the same in Fig. 1a. (MPG 16085 kb)
Morphing between the apo and R-loop structures reflecting conformational change for the HLH motif in the REC1 domain between these two states
The video shows the morphing between the apo and R-loop structures reflecting conformational change for the HLH motif in the REC1 domain between these two states (Flap-on conformation). (MOV 3930 kb)
Rights and permissions
About this article

Cite this article
Stella, S., Alcón, P. & Montoya, G. Structure of the Cpf1 endonuclease R-loop complex after target DNA cleavage. Nature 546, 559–563 (2017). https://doi.org/10.1038/nature22398
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature22398
- Springer Nature Limited
This article is cited by
-
Development of a CRISPR/Cas12a-based fluorescent detection method of Senecavirus A
BMC Veterinary Research (2024)
-
An alpha-helical lid guides the target DNA toward catalysis in CRISPR-Cas12a
Nature Communications (2024)
-
Targeting miRNA by CRISPR/Cas in cancer: advantages and challenges
Military Medical Research (2023)
-
CRISPR applications in cancer diagnosis and treatment
Cellular & Molecular Biology Letters (2023)
-
CRISPR/Cas9 therapeutics: progress and prospects
Signal Transduction and Targeted Therapy (2023)