

Abstract
Many animals keep track of their angular heading over time while navigating through their environment. However, a neural-circuit architecture for computing heading has not been experimentally defined in any species. Here we describe a set of clockwise- and anticlockwise-shifting neurons in the Drosophila central complex whose wiring and physiology provide a means to rotate an angular heading estimate based on the fly’s angular velocity. We show that each class of shifting neurons exists in two subtypes, with spatiotemporal activity profiles that suggest different roles for each subtype at the start and end of tethered-walking turns. Shifting neurons are required for the heading system to properly track the fly’s heading in the dark, and stimulation of these neurons induces predictable shifts in the heading signal. The central features of this biological circuit are analogous to those of computational models proposed for head-direction cells in rodents and may shed light on how neural systems, in general, perform integration.
Similar content being viewed by others
Main
Many animals, including birds1, mammals1,2,3,4 and insects5,6,7, navigate to specific locations in their environment, such as their home or a source of food. To do so, they often use an internal sense of heading, which can persist even without visual landmarks3,4,5,6. Neurons that keep track of heading or head-direction were first discovered in rodents8,9 and have more recently been found in other animals10,11,12,13, including Drosophila14. Whereas elegant computational models have been proposed to account for the firing properties of such neurons15,16,17,18,19,20, a biological circuit that computes an animal’s heading remains unknown in any species. Here we describe a neural shifting mechanism in the Drosophila central complex, akin to models proposed for rodent head direction cells15,16,17,18,19, which allows flies to integrate their turning velocity quantitatively in order to update an internal heading estimate over time.
Heading signals in the central complex
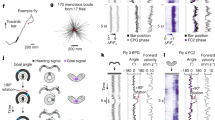
We focus on two cell types that make direct connections between the protocerebral bridge and the ellipsoid body in the fly central complex: E-PGs (ellipsoid body-protocerebral bridge-gall neurons, also PBG1–8.b-EBw.s-D/Vgall.b21 or EIP22; Fig. 1a, b) and P-ENs (protocerebral bridge-ellipsoid body-noduli neurons, also PBG2–9.s-EBt.b-NO1.b21, PEN22, or ‘shifting neurons’ in this paper; Fig. 1a, b). Each cell type tiles both the protocerebral bridge and the ellipsoid body. To assess the role of E-PGs and P-ENs in building an internal heading signal, we first measured calcium levels in each cell type separately using the genetically encoded calcium indicator GCaMP6m23 under two-photon excitation. We imaged P-ENs expressing Gal4 under two independent drivers (VT032906–Gal4, which we call P-EN1, and VT020739–Gal4, which we call P-EN2) because we observed differences in their physiology in later experiments (see below). In all figures, the fly brain is viewed from the posterior side, such that the left bridge is displayed on the left and the right bridge on the right (except Supplementary Videos; see Video Legends). In all experiments, the fly was tethered24 and walking on an air-cushioned ball25,26 at the centre of a cylindrical LED arena27 (Fig. 1c, see Methods). The fly viewed either a dark screen or a bright vertical bar that rotated in closed loop with the fly’s behaviour, simulating a fixed, distant landmark (Fig. 1c).

a, Protocerebral bridge and ellipsoid body in the fly brain. b, Example E-PG and P-EN neurons. Each cell type tiles the bridge and ellipsoid body. c, Imaging neural activity in a fly walking on a ball with an LED arena. d–f, Z-projected bridge volumes of GCaMP6m over time for each cell type. Scale bars, 20 μm. g–i, Left, bridge activity as the fly walks with a bar in closed-loop; right, phase of the bridge activity and bar position. The 90° gap in the back of the arena is highlighted in grey. j–l, Correlations between phase position and ball position as well as phase velocity and ball velocity. Each circle represents one fly. The mean and s.d. across flies are shown. P-ENs are in orange and E-PGs in blue throughout.
In both conditions, we observed two or three periodic peaks of activity in the bridge for each cell type (Fig. 1d–i and Extended Data Figs 1a–l, 2). In all cell types, these peaks moved in unison to the left or right along the bridge as the fly turned right or left, respectively. The position of these peaks in the bridge—which we will call the E-PG or P-EN ‘phase’—quantitatively tracks the virtual heading of the fly (Fig. 1g–l, Supplementary Videos 1–7). Given a bar in closed loop, the phase tracks the bar’s position with an offset that is typically constant for many minutes, but differs from fly to fly (Extended Data Fig. 1m–o). In the dark, however, the phase tracks the fly’s heading with an error that accumulates over time, consistent with a system that integrates self-motion inputs (Extended Data Fig. 2). These properties, measured for three cell types in the bridge, are very similar to those previously observed for rodent head-direction cells8,9,41,42 (Extended Data Fig. 3) and E-PGs in the ellipsoid body14.
A model for angular integration
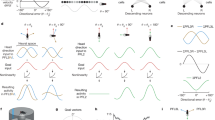
Do P-ENs and E-PGs interact in a circuit to track the fly’s heading? Our first clue came from examining a previous anatomical study21 that characterized how each cell type projects between the bridge, a linear array of 18 glomeruli, and the ellipsoid body, a circular array of 8 tiles21 (Fig. 2a). In linking this anatomical work to our physiological observations, we noticed that left- and right-bridge P-ENs project clockwise and anticlockwise, respectively, to the ellipsoid body (Fig. 2a (orange arrows), Extended Data Fig. 4 and Supplementary Table 1). E-PGs, on the other hand, project without shifting from the ellipsoid body to the bridge (Fig. 2a, blue arrows). Although the full connectome for this circuit will surely reveal more complexity, this coarse level of description already suggests a path by which an activity peak could propagate clockwise or anticlockwise around this circuit when the fly turns. For example, assuming that there are reciprocally excitatory interactions between E-PGs and P-ENs, E-PG activity in tile 5 of the ellipsoid body would activate P-EN cells in glomerulus 5 in both the left and right bridge (Fig. 2a, using a modified numbering scheme for functional clarity; see Extended Data Fig. 4a–d). If, when the fly turns right, the right-bridge P-ENs become more active than their left-bridge counterparts (Fig. 2b), these right-bridge P-ENs would drive the E-PGs in tile 4 in the ellipsoid body, shifting the E-PG activity peak anticlockwise by one tile (Fig. 2c). The E-PG activity in tile 4 in the ellipsoid body would then reverberate back to activate P-ENs in glomeruli 4 of the left and right bridge, shifting P-ENs to the left in the bridge in unison with E-PGs (Fig. 2d). If the fly continued to turn right, the E-PG and P-EN activities would continue to propagate leftward in the bridge and anticlockwise in the ellipsoid body, with stronger turning driving larger asymmetries in P-EN activity and faster propagation of the E-PG peak. The opposite sequence of events would cause the E-PG peak to rotate clockwise in the ellipsoid body if the fly turned left. In this way, an asymmetry in the activity of the right versus left P-ENs could cause the E-PG signal to rotate in one direction or the other in response to the fly turning.

a–d, How an asymmetry in left and right P-EN neurons could rotate the E-PG phase (see text). e–g, Bridge activity and accumulated phase in constant darkness for each cell type. Arrows highlight right–left asymmetries when the fly turns. h–j, Right–left bridge activity (bottom) triggered on the onset of left or right turns (top). The mean and s.e.m. across turns are shown. k–m, Right–left bridge activity versus turning velocity. Thin lines represent single flies. Thick lines represent the mean across flies. h–m, Averaged over bar and dark conditions.
Turning velocity signals in P-ENs
Consistent with this model, in addition to the GCaMP peaks shifting along the bridge (Fig. 1), we observed an asymmetry in the amplitude of the left and right P-EN peaks, but not of the E-PG peaks, when the fly turned (Fig. 2e–g, white arrows). To quantify this asymmetry, we subtracted the GCaMP signal averaged across all glomeruli in the left bridge from that averaged across the right bridge, and computed the time course of this right–left GCaMP signal triggered on the start of left and right turns (Fig. 2h–j, see Methods). When the fly turned right, we observed a transient increase in activity on the right bridge relative to the left (hereafter referred to as the ‘bridge asymmetry’) for both P-EN lines, and vice versa when the fly turned left (Fig. 2h, i). We observed this asymmetry when the fly turned—both in the dark and with a closed-loop bar (Extended Data Fig. 5)—as well as when the fly was not turning, but was viewing panoramic visual motion (that is, optic flow) that it would normally experience during turning (Extended Data Fig. 6). These results indicate that both visual optic-flow inputs and non-visual inputs (such as proprioceptive or efference copy signals) that carry information about the fly’s angular velocity contribute to the bridge asymmetry. Moreover, we found a quantitative, positive relationship between right–left GCaMP and the fly’s turning velocity for both P-EN lines (Fig. 2k, l), but not for E-PGs (Fig. 2m). This bridge asymmetry between left and right P-ENs could provide the quantitative signal necessary for integrating the fly’s turns over time.
Spatiotemporal relationships across cells
A second prediction of the anatomical model in Fig. 2a–d is that E-PG and P-EN activity peaks should occupy similar positions in the bridge. We therefore imaged E-PGs simultaneously with either P-EN1s or P-EN2s, with E-PGs expressing GCaMP6f23 and P-ENs expressing jRGECO1a, a red-shifted calcium indicator28 (Fig. 3). In the bridge, the calcium peaks from both P-EN lines shifted in unison with those of E-PGs (Fig. 3a–d). However, the two P-EN lines differed greatly in that P-EN1 and E-PG peaks were in phase with each other (Fig. 3a, c), whereas the P-EN2 and E-PG peaks were nearly in antiphase (Fig. 3b, d). Anatomical experiments suggested that we imaged genuine P-ENs in both Gal4 lines (Extended Data Fig. 4g–l, Supplementary Information Table 1), but that our P-EN1 and P-EN2 Gal4 lines target different subsets of P-ENs (Extended Data Fig. 7). Owing to these physiological and anatomical differences, we operationally defined two P-EN subtypes: P-EN1 and P-EN2.

a, b, Co-imaging of E-PGs (GCaMP6f) with P-EN1s (jRGECO1a) (a) or P-EN2s (jRGECO1a) (b) in the bridge in constant darkness. c, d, Phase-nulled signals in the bridge, averaged over time. e, f, Bridge data from c, d replotted onto the ellipsoid body using each cell type’s anatomical projection pattern21. g, h, Sum of the left- and right-bridge curves in e, f (scales adjusted). i, j, Co-imaging of E-PGs with P-EN1s or P-EN2s in the ellipsoid body. k–p, Phase-nulled signals in the ellipsoid body averaged over time when the fly was walking straight (k, l), turning left (m, n) or turning right (o, p). Arrows indicate the velocity of the peaks. The P-EN ellipsoid body asymmetries were significantly different during turning and walking straight (P < 0.02), and during turning left and right (P < 0.01) (Wilcoxon rank-sum test). The mean and s.e.m. across flies are shown. c–h, k–p, Averaged over bar and dark conditions.
What are the implications of these in-phase and nearly antiphase activity peaks in the bridge on the interactions between P-ENs and E-PGs? As P-ENs are likely to output (directly or indirectly) onto E-PGs in the ellipsoid body21,22 (Extended Data Fig. 4e, f), we replotted the mean GCaMP signal measured from each bridge glomerulus (Fig. 3c, d) over the appropriate tile in the ellipsoid body (Fig. 3e, f), using the anatomical mappings described above21 (Extended Data Fig. 4a–d). For E-PGs, the two peaks in the bridge map to a single peak in the ellipsoid body (Fig. 3e, f, blue curves), as expected14. However, the two P-EN peaks in the bridge map to either side of the single E-PG peak in the ellipsoid body (Fig. 3e, f, orange curves) because of their offset anatomy (Fig. 2a). Furthermore, the positions of these P-EN peaks are inverted across the two P-EN subtypes: the right bridge peak maps to the anticlockwise flank of the E-PG ellipsoid body-peak for P-EN1 (Fig. 3e, dashed orange curve) but to the clockwise flank for P-EN2 (Fig. 3f, dashed orange curve). The reverse is true for the left bridge peak in each cell type.
To evaluate whether these projected activity patterns match the actual patterns in the ellipsoid body, we performed dual imaging from E-PGs and either P-EN1s or P-EN2s in the ellipsoid body. With E-PGs, we observed a single, sharp peak in the ellipsoid body (Fig. 3i–l), as expected14. With P-ENs, we had the null expectation, based on summing the left and right bridge signals after projecting to the ellipsoid body, that we would observe a broad GCaMP peak for P-EN1 and a broad valley for P-EN2 at the position of the E-PG peak (Fig. 3g, h). Unexpectedly, however, P-ENs in both lines also showed a single, sharp peak in the ellipsoid body, which overlapped with the E-PG peak (Fig. 3i–l). These data suggest that the broad P-EN1 and P-EN2 calcium signals are reshaped by presynaptic modulation in the ellipsoid body, the mechanism for which should be investigated in future work. Despite this reshaping, we nevertheless observed an asymmetry in P-EN activity relative to the E-PG peak in the ellipsoid body (hereafter referred to as the ellipsoid body asymmetry) when the fly turned (Fig. 3m–p). Specifically, P-EN1 activity increased on the leading edge of the moving E-PG peak (Fig. 3m, o) and P-EN2 activity increased on the trailing edge (Fig. 3n, p), consistent with the sign of the bridge asymmetry during turns (Fig. 2h–i, k, l), the relative phase of P-EN1 and P-EN2 to E-PG activity in the bridge (Fig. 3c, d), and the known anatomical mappings of P-ENs and E-PGs between the bridge and ellipsoid body (Fig. 3e, f). Control experiments and analyses indicate that these results were not due to bleed-through between indicator channels, differences in indicator kinetics, or the specific lag times chosen (Extended Data Figs 8, 9).
Given that P-EN1 and P-EN2 have inverted spatial activity patterns, how are they coordinated in time? When the activity peaks moved in the bridge, the P-EN1 bridge asymmetry was evident early, before the onset of the movement of the peaks, whereas the P-EN2 bridge asymmetry came on late, after the peaks had begun to move, as measured by averaging these signals triggered on movements of the peaks (Fig. 4a–c). A cross-correlation analysis also showed this timing difference between P-EN1s and P-EN2s in the bridge (Fig. 4d) and the ellipsoid body (Extended Data Fig. 9c, d).

a, b, Bridge activity during phase shifts in constant darkness for P-EN1 (a) and P-EN2 (b). White arrows highlight right–left asymmetries. c, Right–left bridge activity (bottom) triggered on phase changes (top). The mean and s.e.m. across phase shifts are shown. d, Correlation between right–left bridge activity and phase velocity versus time lag between the two signals. Thin lines represent single flies. Thick lines represent the mean across flies. c, d, Traces are averaged over bar and dark conditions. e, f, E-PG activity in the bridge with P-EN1 cells expressing shibirets at 22 °C (e) or 32 °C (f). Arrows highlight atypical deviations in the E-PG phase from the ball’s heading at 32 °C. g, E-PG phase velocity versus ball velocity in a control fly and a P-EN1>shibirets fly (same as in e, f) in constant darkness. R, Pearson correlation coefficient. h, Phase velocity versus ball velocity correlations for three P-EN-Gal4 lines. Each circle represents one fly. The mean and s.e.m. across flies are shown. The cold-to-hot changes in correlation are significantly different between P-EN>shibirets and control groups (P < 0.01) (Wilcoxon rank-sum test). Note the different rotation scales in a, b and e, f.
Blocking P-ENs impairs integration
If P-EN neurons serve an important role in moving the E-PG peak, then impeding P-EN synaptic output should impair the ability of the E-PG peak to properly update its position when the fly turns. We expressed shibirets, which prevents synaptic vesicle recycling in a temperature-dependent manner29, in each P-EN line, and measured E-PG activity in the bridge using GCaMP6f (Fig. 4e–h). When we impeded P-EN synaptic output at 32 °C, the E-PG signal failed to consistently track the dynamics of the fly’s heading in the dark (Fig. 4f (black arrows), g, h). We note that at 34 °C, the E-PG signal appeared dim or blurred, indicating a stronger effect (and suggesting that P-ENs contribute to the overall magnitude, or general stability, of the E-PG peak), but this made it difficult to properly estimate the E-PG phase. We therefore performed our experiments at 32 °C, expecting that P-ENs would be only partially impaired. Moreover, we did not observe a consistent impairment in the ability of the E-PG phase to track a closed-loop bar in the same flies (Extended Data Fig. 10a–d). These experiments argue that proper synaptic transmission in P-ENs is necessary for this circuit to integrate the fly’s heading without a visual landmark.
Activating P-ENs shifts E-PG activity
If P-ENs contribute to the rotation of the E-PG activity peaks, then experimental stimulation of P-ENs should drive specific changes in E-PG activity. We stimulated P-ENs, with 1–2-glomeruli resolution, on the left or right bridge by expressing the ATP-gated cation channel P2X230 and locally releasing ATP from a pipette (see Methods), while measuring E-PG activity in the bridge using GCaMP6f (Fig. 5a). When we locally excited P-ENs in the left bridge, the E-PG peaks appeared (on both sides of the bridge) to the right of the stimulated glomerulus, and vice versa when P-ENs were excited on the right bridge (Fig. 5b–g), for both P-EN1 and P-EN2 Gal4 lines. These data are consistent with an excitatory relationship (direct or indirect) between P-ENs and E-PGs. Control experiments without ATP and without Gal4 indicate that we specifically stimulated P-ENs (Extended Data Fig. 10e–j). Notably, the flies occasionally performed a behavioural turn immediately after the local release of ATP, in a direction that would return the E-PG peaks to their position just before stimulation (for example, Fig. 5b–e, asterisks)—an effect that we do not pursue further here. At the neural level, these experiments demonstrate that experimentally activating P-ENs induces the E-PG activity peaks to relocate to the expected positions in the bridge relative to the stimulated glomerulus. We note that the effects we observed in our perturbation experiments across P-EN1 and P-EN2 Gal4 lines could, formally, have been due to perturbing one P-EN subtype alone since all of our Gal4 lines contain a mixture of both subtypes but with different ratios (Extended Data Fig. 7). Regardless, these data support a role for P-ENs of at least one subtype in the shift-circuit hypothesis for angular integration.

a, Stimulating P2X2-expressing P-ENs in the bridge with ATP is expected to excite E-PGs in the medial neighbouring glomerulus. b, Left, ATP (Alexa594) signal in the bridge; right, E-PG bridge activity while stimulating P-EN1 neurons in the left bridge. c, Same as b, except for stimulating P-EN1s in the right bridge. d, e, Same as b, c but for stimulating P-EN2 neurons. Asterisks highlight events when the fly turned following stimulation in a direction that would return the E-PG phase to its pre-stimulus position. f, g, Phase-nulled E-PG activity and ATP (Alexa594) signal after P-EN1 (f) or P-EN2 (g) stimulation. Thin lines represent the mean response in each fly. Thick lines represent the mean across flies. h, Summary model. P-EN and E-PG interactions projected onto a single ring. Only ellipsoid body tiles are represented for clarity. i, Model proposed in ref. 15 for rat head-direction cells.
Discussion
These anatomical and physiological data support a model in which an asymmetry in P-EN activity rotates the E-PG activity peak in response to the fly turning (Figs 2a–d, 5h). This model also requires additional inhibitory circuitry to maintain the width of the E-PG peak in the face of spreading P-EN excitation. A second, parallel study employing electrophysiological measurements and calcium imaging provides further evidence that P-ENs serve a role in angular integration31.
Whereas previous models of angular integration15,16,17,18,19 have required only P-EN1-like neurons, we observe a second set of asymmetrically active neurons (P-EN2) whose activity is biased to the trailing edge of the ellipsoid body E-PG peak and whose asymmetry arises later during tethered-walking turns. One interpretation of this discovery is that, given their relative timing, the early P-EN1 cells start the movement of the E-PG peak, and the late P-EN2 cells stop its movement. This model implies that a moving E-PG peak would otherwise continue rotating—a property not featured to date in models of head direction15,16,17,18,19. Another interpretation is that P-EN1s are the main drivers of angular integration, whereas P-EN2s serve a different role (see Supplementary Discussion). Further work will be needed to rigorously test such functional hypotheses.
The physiology of E-PGs and P-ENs resembles that of rodent head-direction cells in the presubiculum (and other areas)8,32 and the lateral mammillary nucleus33,34, respectively. Moreover, the models proposed to account for these physiological properties in Drosophila (here) and rodents15,16,17,18,19 are remarkably similar (Fig. 5h, i), suggesting that insects and mammals may use common circuit architectures to update their sense of heading. Although apparently hard-wired to integrate turning velocities, the essential features of this circuit are general enough to integrate other variables (including variables in two or more dimensions19) over time, and may also therefore appear in other integrating neural systems, such as the mammalian grid cell system19,35,36, among others37,38,39,40.
Methods
Fly stocks
Flies were raised with a 12-h light, 12-h dark cycle at 25 °C. All physiological experiments were performed with 1–3-day-old females with at least one wild-type white allele. Flies were selected randomly for all experiments. We were not blinded to the flies’ genotypes. For experiments imaging one cell type (Figs 1, 2, 4a–d; Extended Data Figs 1, 2, 3, 5, 6, 8a–f), we used + (Canton S, Heisenberg Laboratory)/w; UAS-GCaMP6m (Bloomington Drosophila Stock Center, BDSC #42748); VT032906–Gal4 (Vienna Drosophila Resource Center, VDRC #202537) flies for P-EN1; +/w; 60D05-LexA (BDSC #52867)/LexAop-tdTomato (Ruta Laboratory); VT020739–Gal4 (VDRC #201501)/UAS–GCaMP6m (BDSC #42750) flies for P-EN2; and +/w; +; 60D05–Gal4 (BDSC #39247)/UAS–GCaMP6m flies for E-PG. The same flies were analysed in Figs 1, 2, 4a–d, Extended Data Figs 1, 2, 3, 5. For imaging P-EN1 or P-EN2 simultaneously with E-PGs (Fig. 3, Extended Data Fig. 9), we used +/w; 60D05-LexA/LexAop–GCaMP6f (BDSC #44277); VT032906–Gal4/UAS–jRGECO1a (BDSC #63794), and +/w; 60D05-LexA/LexAop–GCaMP6f; VT020739–Gal4/UAS–jRGECO1a flies, respectively. As a control for differences in calcium indicator kinetics, we imaged GCaMP6f and jRGECO1a in the same cell type (Extended Data Fig. 8g–j), E-PGs, using +/w; UAS–GCaMP6f (BDSC #42747)/+; 60D05–Gal4/UAS–jRGECO1a flies. For the shibirets experiments (Fig. 4e–h, Extended Data Fig. 10a–d), we used three Gal4 lines to drive shibirets in P-ENs: VT032906–Gal4 (P-EN1), VT020739–Gal4 (P-EN2), and 12D09–Gal4 (P-EN2, BDSC #48503). We used pJFRC99-20XUAS-IVS-Syn21-Shibire-ts1-p10 inserted at VK00005 (referred to here as UAS-shibirets) to drive shibirets (Rubin Laboratory). For each X-Gal4 line, we used +/w; 60D05-LexA/LexAop–GCaMP6f; X-Gal4/UAS-shibirets flies with +/w; 60D05-LexA/LexAop–GC6aMPf; X-Gal4/+ flies as a control. We also used +/w; 60D05-LexA/LexAop–GCaMP6f; +/UAS-shibirets flies as a control without Gal4. For P-EN stimulation (Fig. 5, Extended Data Fig. 10e–j), we used +/w; 60D05-LexA/LexAop–GCaMP6f; X-Gal4/UAS–P2X2 flies (Ruta Laboratory), where X was either VT032906 (P-EN1) or VT020739 (P-EN2). We used +/w; 60D05-LexA/LexAop–GCaMP6f; +/UAS–P2X2 flies as a control without Gal4. For multicolour flip-out experiments (Supplementary Information Table 1, Extended Data Fig. 4g–l), we used 57C10-FLP/+; +; 10×UAS–FRT.stop-myr::smGdP–HA, UAS–FRT.stop-myr::smGdP–V5-THS-10×UAS–FRT.stop-myr::smGdP–FLAG/X-Gal4 flies, where X was VT032906, VT020739 or 12D09. We used either 57C10-FLPL (BDSC #64087) or 57C10-FLPG5 (BDSC #64088) to label more or fewer neurons, respectively. To label putative axon terminals with synaptotagmin–GFP (Extended Data Fig. 4e, f), we used +/w; +/UAS-syt–GFP (BDSC #6925); X-Gal4/UAS–tdTomato (BDSC #32221) flies, where X was VT032906 or VT020739. To co-label different P-EN driver lines (Extended Data Fig. 7), we used +/w; 12D09-LexA (BDSC #54419)/LexAop–myrGFP (BDSC #32210);X-Gal4/UAS–tdTomato flies, where X was VT032906 or VT020739.
Immunohistochemistry
We dissected fly brains in S2 medium at room temperature and fixed them in 1% paraformaldehyde at 4 °C overnight. Fixed brains were washed 3 times for 30–60 min with PAT3 (0.5% Triton X-100 and 0.5% bovine serum albumin in phosphate buffered saline), then blocked with 3% NGS in PAT3 for 1.5 h at room temperature. We incubated brains with primary and secondary antibodies as previously described43, and mounted them in VectaShield (Vector Labs). For co-labelling tdTomato with GFP (Extended Data Figs 4e, f, 7), we used anti-DsRed antibody (Clontech) at 1:1,000. For the multicolour flip-out experiments (Extended Data Fig. 4g–l, Supplementary Information Table 1), we used antibodies as previously described43. We imaged the central complex using a 40 × 1.20 NA objective on a Zeiss LSM780 confocal microscope with 0.58 or 1.0 μm separating each optical slice.
Tethered walking setup
We shaped 6.35 mm (1/4 inch) diameter balls from Last-A-Foam FR-4618 (General Plastics)25. The balls had a mass ranging from 42 to 46 mg. To shape the ball from raw foam, we machined a steel concave file with the same diameter as the ball, with sharp edges to cut the foam25. The ball rested in an aluminium base with a concave hemisphere 6.75 mm (17/64 inch) in diameter. A 1-mm channel was drilled through the bottom of the hemisphere and connected to air flowing at approximately 260 ml per min.
Behavioural imaging
For all experiments, we imaged the fly and ball from the front under 850-nm illumination with a Prosilica GE680 camera (Allied Vision Technologies) externally triggered at 50 Hz, with a zoom lens (MLM3X-MP, Computar) set at 0.3×. The lens also held an OD4 875-nm shortpass filter (Edmund Optics) to block the two-photon excitation laser (925 nm or 1,035–1,040 nm). This camera was used both to position the fly and to track the ball.
Ball tracking and closed-loop experiments
We tracked the ball using FicTrac software26. FicTrac calculates the angular position of the ball for each frame, rather than integrating rotational velocities, which ensures no drift in the estimated ball position over time. The ball was marked with irregular black spots, which allowed a single camera (see Behavioural Imaging) facing the fly to track all three rotational axes of the ball in real time at 50 Hz. We verified the accuracy of the FicTrac software using a servo motor rotating a ball at known velocities. A plastic square was laser-cut with a hole at the centre so that it fit as a sleeve on the ball holder, under the ball. The plastic square was aligned to the fly, such that its side edges were parallel to the fly’s body axis. This plastic square was then used to calibrate the FicTrac tracking system to the fly’s frame of reference (in other words, to determine which rotational axes represented forward, heading, and sideways walking). We modified FicTrac to output analogue voltages corresponding to the angular position of the ball along each axis through a digital-to-analogue converter (USB-3101, Measurement Computing). In closed loop experiments, we used the heading axis voltage output to control the azimuthal position of a bar displayed on the LED arena. That is, when the fly turned left, the bar rotated right, and vice versa, simulating the natural visual input a turning fly would experience from a prominent, stationary, visual landmark at infinity.
LED arena and visual stimuli
We used a cylindrical LED arena27, spanning 270° in azimuth and 81° in height. Pixels were spaced by 1.875°. We positioned the empty quadrant of the arena directly behind the fly. We used blue LEDs (BM-10B88MD, Betlux Electronics), covered by five sheets of blue filter (Tokyo Blue, Rosco) to reduce detection of the blue LEDs by the two-photon’s photomultiplier tubes. For all experiments, the microscope was surrounded by a black shroud to block light from the monitors, and all light-emitting sources inside the shroud other than the LED arena were covered with black tape. For the bar stimulus, we presented a single bright bar, 6 pixels wide (11°) and spanning the height of the arena. The bar did not jump across the 90° gap in the arena behind the fly; rather, we kept track of the bar position behind the fly without displaying it. For the dark stimulus, all LEDs were turned off. For the moving dots stimulus (Extended Data Fig. 6), we generated a series of frames—one for each azimuthal pixel (spaced by 1.875°)—in which single-pixel (1.875° pitch) dots appeared at random locations, travelled for four pixels, and then reappeared at a new random location. We chose this stimulus to separate the contributions of position and velocity of a moving object (like a bar), because with this stimulus the fly cannot track the position of any single dot for more than 7.5° (four frames). We designed the stimulus such that the optic flow could proceed with the same number of dots disappearing and reappearing on each frame, uninterrupted, for an infinite number of rotations. The total number of dots (including those not visible, behind the fly) was kept constant on all frames. In Extended Data Fig. 6, we presented this stimulus in closed loop with the fly's rotational walking behaviour, which we interrupted with 1-s open-loop rotations of the moving dots stimulus (45°/s and 90°/s to the left or right) every 6 s.
Fly tethering and preparation
Flies were anaesthetized at 4 °C, and were tethered to a custom holder that was similar to that used in previous studies24, except that the back wall was pitched to 45° instead of 90°, allowing more light from the brain to reach the objective (Fig. 1c). We also modified the rear of the holders to ensure that the flies could see 280° around them. Flies were fixed to the holder by gluing the thorax and the front of the head between the eyes with glue cured by blue light (Bondic). Additional glue was applied to the posterior side of the head to stabilize the head for dissection. The head was pitched forward during tethering to provide a posterior view of the central complex. We cut a window in the cuticle immersed in saline at the centre of the posterior side of the head to gain optical and pipette access to the central complex. The holder to which each fly was tethered was placed in a base at the centre of the LED arena, under the objective. The holder was stabilized by magnets in the holder and the base. The ball holder was mounted on a manipulator to adjust the position of the ball under each fly.
Calcium imaging
We used a two-photon microscope with a movable objective (Bruker) and custom stage (ThorLabs, Siskiyou). For two-photon excitation we used a Chameleon Ultra II Ti:Sapphire femtosecond pulsed laser (Coherent). To image GCaMP6m or GCaMP6f alone, we tuned the laser to 925 nm, whereas to image GCaMP6f and jRGECO1a together, we used 1,035–1,040-nm light, to excite both fluorophores simultaneously. The emitted light was split by a 575-nm dichroic mirror. We used a 490–560-nm bandpass filter (Chroma) for the green channel in single indicator experiments (Figs 1, 2, 4; Extended Data Figs 1–3, 5, 6, 8a–f, 10a–d). For dual imaging of GCaMP6f and jRGECO1a (Fig. 3, Extended Data Figs 8g–j, 9) we used a 500–550-nm bandpass filter for the green channel and a 585–635-nm bandpass filter for the red channel, except during dual imaging of E-PGs and P-EN2 in the bridge (Fig. 3b, d), where we used a 490–560-nm bandpass filter for the green channel. We detected light signals with GaAsP detectors (Hamamatsu). We used a 40× 0.8 NA objective (Olympus) to image the brain. We perfused the brain with extracellular saline composed of, in mM: 103 NaCl, 3 KCl, 5 N-Tris(hydroxymethyl) methyl-2-aminoethanesulfonic acid (TES), 10 trehalose, 10 glucose, 2 sucrose, 26 NaHCO3, 1 NaH2PO4, 1.5 CaCl2, 4 MgCl2, and bubbled with 95% O2/5% CO2. The saline had a pH of 7.3–7.4, and an osmolarity of 280 ± 5 mOsm. The temperature of the bath was controlled by flowing the saline through a Peltier device, with feedback from a thermistor in the bath (Warner Instruments). This thermistor measurement was used to set the bath to 22 °C or 32 °C for the shibirets experiments. To image the protocerebral bridge, we selected a region framing the bridge, about 140 × 50 pixels in size, and scanned through 2 or 3 z-planes separated by 7–9 μm using a Piezo motor to achieve a volumetric scanning rate of 5–7 Hz. To image the ellipsoid body, we selected a region 64 × 64 pixels in size and scanned through 3 z-planes separated by 7–9 μm at 5–7 Hz.
Trial structure
For all experiments, unless noted, we interleaved two visual conditions: closed-loop bar (1.0× gain between angular rotations of the ball and bar) and dark screen. For GCaMP6m imaging of single cell types in the bridge (Figs 1, 2, 4a–d; Extended Data Figs 1–3, 5), we presented each fly with six 50-s trials of each visual condition. For GCaMP6m imaging of single cell types in the bridge and ellipsoid body (Extended Data Fig. 8a–f), we presented each fly with two 50-s trials of each visual condition. For GCaMP6f and jRGECO1a imaging (Fig. 3, Extended Data Figs 8g–j, 9), we presented each fly with one or two 20–30 s trials of each visual condition. For imaging E-PGs with P-EN-shibirets (Fig. 4e–h; Extended Data Fig. 10a–d), we presented each fly with two 50-s trials each of closed-loop bar and a dark screen (and additional stimuli not analysed here) for each temperature. For the optic flow stimuli (Extended Data Fig. 6), we presented 16 blocks of the four stimuli (−45°/s, +45°/s, −90°/s, +90°/s open loop optic flow) to each fly. The order of the four open loop stimuli was randomized within each block. Half of the 1-s open loop stimuli were separated by 5 s of 0.5× gain closed loop optic flow, and half by 5 s of 1.0× gain closed loop optic flow. The P2X2 experiments (Fig. 5; Extended Data Fig. 10e–j) were performed in constant darkness.
Data acquisition and alignment
All data were digitized at 10 kHz using a Digidata 1440 (Axon Instruments), except for the two-photon scanning images, which were acquired using PrairieView (Bruker). Behavioural, stimulus and two-photon scanning data were aligned using triggers acquired on the same Digidata 1440.
Comparing data acquired at different sampling rates
When comparing two-photon imaging (~5–7 Hz) and behavioural data (50 Hz) within a fly on a time-point-by-time-point basis (Figs 1j–l, 2k–m, 4g, h; Extended Data Figs 1m–o, 3, 9e, f, 10c, d), we subsampled behavioural data to the imaging frame rate by computing its mean during each volumetric imaging time point. Because different flies were imaged at slightly different frame rates depending on the size of the region of interest, when averaging across flies or across turns (Fig. 2h–j, bottom row, 4c, d, thick lines; Extended Data Figs 5, 6, 9a–d), we linearly interpolated each time series to a common time base of 10 Hz, and then averaged over these interpolated time series. Our conclusions are not altered if we interpolate to slower time bases.
Data analysis
Two photon images were first registered using Python 2.7 (see Image registration). These images were then manually parsed in Fiji44 (see Processing imaging data). All subsequent data analysis was performed in Python 2.7. We did not exclude flies from any analysis, except for the jRGECO1a experiments, where a few recordings were excluded because the red signal was too weak (0/20 flies for Figs 3c, 1/12 flies for Fig. 3d, 8/22 flies for Fig. 3k, m, o, 0/11 flies for Fig. 3l). In analysed jRGECO1a flies, we also sometimes excluded the second of two sets of stimuli because of jRGECO1a bleaching. If we do not exclude these data, our conclusions are unaltered. No statistical method was used to choose the sample size.
Image registration
Two-photon imaging frames were computationally registered by translating each frame in x and y to best match the time-averaged frame for each z-plane. We registered multiple recordings from the same fly to the same time-averaged template for each z-plane, unless a significant shift was introduced between recordings. Rarely, time points were discarded from analysis if the registration failed because the signal in a particular frame was too weak. We did not register dual cell type imaging data from the ellipsoid body, but rather analysed the raw data directly.
Processing imaging data
Regions of interest were manually defined in Fiji44. For the protocerebral bridge, we manually defined regions delineating each glomerulus from the registered time-average of each z-plane (Extended Data Fig. 1a). Note that E-PGs do not innervate the outer two glomeruli of the bridge, and P-ENs do not innervate the inner two glomeruli21, and thus no region was defined for these glomeruli for the respective cell type. In P-EN2 GCaMP experiments, tdTomato was expressed in E-PGs, which helped us to parse glomeruli. For P-EN1 GCaMP experiments, two copies of UAS-GCaMP and VT032906-Gal4 were required to produce enough signal, and therefore tdTomato was not used for parsing glomeruli. For the ellipsoid body recordings, we first smoothed the imaging data with a 2-pixel (~ 2-μm) Gaussian. We manually defined an ellipsoid body region of interest from the time-average of each z-plane. We then subdivided these regions into 16 equal wedges radiating from a manually selected centre, as described previously14. Note that while E-PGs tile the ellipsoid body in 16 wedges, and P-ENs tile the ellipsoid body in half the number of tiles21, we used the same 16-wedge analysis for both as an equal means of comparing the two signals. We calculated the mean pixel value for each glomerulus or wedge across z-planes for each time point, producing a matrix of raw mean intensity values for each region over time. We then calculated ∆F/F values for each glomerulus or wedge independently, by defining F as the mean of the lower 5% of raw values in a given glomerulus or wedge over time. We also normalized each glomerulus or wedge independently with a z-score, which measures how many standard deviations each imaging data point is from the mean. We used this metric to estimate bridge asymmetries (Figs 2, 4a–d; Extended Data Figs 5, 6), because it tended to normalize constant, absolute differences in intensity across glomeruli better than the ∆F/F approach. Constant, absolute differences in fluorescence across glomeruli might reflect functional differences among glomeruli or simply result from differences in the number of cells per glomerulus targeted by each Gal4 line, or the amount of GCaMP in each cell. We observed the same asymmetries in the bridge using the ∆F/F normalization approach, but with more variability (Extended Data Fig. 5).
Analysis of periodicity and phase
To analyse the protocerebral bridge signal, we started with a matrix of ∆F/F values, in which each row represents a time point, and each column represents a glomerulus. We took the Fourier transform of each row or time point independently, and observed a consistent peak at a periodicity of about eight glomeruli for each cell type (the peak periodicity of the power spectrum averaged over time is shown in Extended Data Fig. 1j–l for closed-loop bar and Extended Data Fig. 2g–i for dark conditions). Given that this periodicity was relatively constant over time (Extended Data Figs 1a–i, 2a–f), we extracted the phase from the Fourier component with a period of eight glomeruli for each time point independently. When overlaid on the protocerebral bridge GCaMP time series, this phase accurately tracked the shift in the protocerebral bridge over time (Extended Data Figs 1a–i, 2a–f). For the ellipsoid body, we computed the population vector average, as previously described14. For summary analyses, and for the dual-imaging sample traces (Fig. 3a, b, i, j), the phase was filtered with a three-point moving average. To calculate the offset between the phase and bar position (Extended Data Fig. 1m–o), we computed the circular mean of the difference between the phase and bar position during time points when the bar was visible to the fly. We shifted the phase by this constant offset in Fig. 1g–i and Extended Data Figs 1c, f, i and 10a, b. In Figs 2e–g and 4a, b, we nulled the accumulated phase and ball position at time zero, and applied a gain to the ball heading to best match the phase: 1.0 for Fig. 2e (P-EN1), 1.40 for Fig. 2f (P-EN2), 0.75 for Fig. 2g (E-PG), 1.0 for Fig. 4a (P-EN1) and 0.89 for Fig. 4b (P-EN2). We do not interpret these different gains measured in different flies to mean that the three cell types have phase signals that drift relative to each other; indeed, when we imaged E-PGs and P-ENs simultaneously in the same fly (Fig. 3), their peaks moved in unison (that is, with the same gain) along the bridge and ellipsoid body. The different gains measured in separate flies could be due to fly-to-fly or tethering variability14.
Correlation analysis
For closed-loop bar experiments, we computed the circular correlation45 between GCaMP phase and bar position (Fig. 1j–l, ‘position’). For experiments in the dark, we computed the Pearson correlation between GCaMP phase velocity and ball velocity (Fig. 1j–l, ‘velocity’ and Fig. 4g, h). We correlated velocities instead of position for dark screen data because the phase tended to drift away from the ball’s heading without a visual landmark. We calculated these correlations for different time lags between the phase and ball signals, and in each figure we report the correlation at the time lag for which the correlation was highest. Specifically, the phase was delayed by 300 ms relative to the ball for E-PG and P-EN2 neurons in Fig. 1j, l, and by 600 ms for P-EN1 neurons in Fig. 1k. The sign of these delays suggests that the heading system updates in response to the fly turning, not vice versa, although more experiments are needed (see Supplementary Discussion). The longer delay for P-EN1 compared to P-EN2 and E-PG was likely to be an artefact of overexpressing GCaMP6m in P-EN1 rather than to reflect a genuine biological difference among cell types (see Supplementary Discussion). For example, in other imaging experiments in which we measured P-EN1 activity side-by-side with E-PG activity, we observed that the P-EN1 peak actually led the E-PG peak (Fig. 3m, o). In Fig. 4g, h, the time lag for which the correlation was highest between E-PG GCaMP phase and ball velocity was 200 ms (rather than 300 ms), which was the time lag used for the correlation values reported in this Figure; this shorter delay was probably due to the fact that in Fig. 1 we used GCaMP6m, and in Fig. 4e–h we used GCaMP6f. For the P-EN>shibirets experiments (Fig. 4e–h), we computed the difference between the velocity correlations at 22 °C and 32 °C for each fly. We then used a two-sided Wilcoxon rank-sum test to reject the null hypothesis that this difference was the same in each of the three P-EN-Gal4, UAS-shibirets fly populations and in the respective P-EN-Gal4- or UAS-shibirets-only populations (P < 0.01 for all individual comparisons). For all phase correlations, we included only data during which the fly was walking with a speed of at least 1 mm/s. We also required the peak activity (the mean of the top two values in the bridge) to be greater than 0.8 ∆F/F, to ensure the phase was properly estimated.
Computing the bridge asymmetry as a function of turning velocity
For each time point, we subtracted the mean z-score-normalized signal in the left bridge from that in the right bridge (referred to as the bridge asymmetry). We binned these bridge asymmetries based on the turning velocity of the fly in 30°/s bins, and computed the mean asymmetry in each bin (Fig. 2k–m). We repeated this process for different time lags between the bridge asymmetry and the fly’s turning behaviour, to find the lag at which the slope (measured between −200°/s and +200°/s) was the steepest. The curve computed at this time lag (bridge asymmetry lagging by +400 ms with respect to behaviour for P-EN1 and P-EN2) is shown in Fig. 2k, l. (Many other lags, before and after +400 ms, also show a significant positive slope for P-EN1 and P-EN2.) The time lag chosen does not affect the E-PG curve, whose slope is always near zero.
Culling individual turns or phase shifts
To isolate individual turns (Fig. 2h–j) or phase shifts in the bridge (Fig. 4c), we detected peaks in the turning velocity or phase velocity signal, respectively. In both cases, we first smoothed the velocity signal by convolving it with a Gaussian (300 ms s.d.). From this smoothed velocity signal, we isolated peaks with a minimum peak height of 30°/s and a minimum peak width of 0.5 s for the turns and the phase shifts. We further required that each peak be isolated from other peaks by a minimum distance of 1.5 s. We aligned the turning velocity or phase velocity and bridge asymmetry signals to the start of each turn or phase shift.
Phase nulling
To compute the average GCaMP signal in the bridge or ellipsoid body independent of phase (Fig. 3c, d, k–p; Extended Data Figs 8, 9a, b), we computationally shifted the GCaMP signals at each time point so that the phase was the same across all time points. To achieve this phase nulling, we first interpolated the GCaMP signal at each time point to 1/10 of a glomerulus or wedge with a cubic spline. We then shifted this interpolated signal by the phase estimate at that time point. In the ellipsoid body, this shift is naturally circular. In the protocerebral bridge, we wrapped the signal around the same side of the bridge, such that values shifted past the left edge of the left bridge would return on the right edge of the left bridge, and so on, in order to preserve left and right asymmetries. This was possible because each side of the bridge innervated by a given cell type consisted of eight glomeruli, which matched the period of the signals. Once we nulled the phase, we computed the mean signal over time when the fly was walking straight (Fig. 3c, d, k, l, computed for 0 ± 30°/s). For the ellipsoid body, we also averaged the phase-nulled signal over times when the fly turned at 300 ± 30°/s to the left (negative) or right (positive). In Fig. 3k–p, we show these phase nulled plots at the time lag for which the P-EN1 or P-EN2 ellipsoid body asymmetry (see below) and the fly’s turning velocity showed a maximum correlation (Extended Data Fig. 9e, f, black arrows). In Fig. 3 we nulled both the P-EN and E-PG signals using the E-PG phase. In Extended Data Fig. 8a–f, we nulled both the bridge and ellipsoid body signals using the ellipsoid body phase. In Extended Data Fig. 8g–j, we nulled both the GCaMP6f and jRGECO1a signals using the GCaMP6f phase (analogous to Fig. 3). In Fig. 5f, g, and Extended Data Fig. 10e–j, we nulled both the GCaMP6f and Alexa594 signals using the position of the pipette.
Computing the ellipsoid body asymmetry in P-ENs
To compute the ellipsoid body asymmetry in P-ENs (Extended Data Fig. 9c–f), we integrated the P-EN signal 180° clockwise and anticlockwise from the E-PG phase, and subtracted the integrated anticlockwise (left on the linearized plots) signal from the integrated clockwise (right on the linearized plots) signal. We used a two-sided Wilcoxon rank-sum test to reject the null hypothesis that the ellipsoid body asymmetry in P-ENs was the same when the fly was turning to the left at –300°/s (Fig. 3m, n) as when it was walking straight (Fig. 3k, l), and the same when turning to the right at +300°/s (Fig. 3o, p) as when it was walking straight (Fig. 3k, l). All P values were <0.02 when analysing either z-score normalized data (not shown) or ∆F/F normalized data (Fig. 3k–p). The null hypothesis that the ellipsoid body asymmetry in P-ENs was the same when the fly turned left (Fig. 3m, n) as when it turned right (Fig. 3o, p) was rejected for both P-EN1 and P-EN2 (P < 0.01), using a two-sided Wilcoxon rank-sum test with either z-score or ∆F/F normalized data.
P-EN stimulation
To measure the effect of stimulation of P-ENs on E-PGs, we expressed GCaMP6f in E-PGs and the ATP-gated cation channel P2X2 in either P-EN1 or P-EN2 cells (see Fly stocks). Na2ATP (A7699, Sigma) was dissolved in extracellular solution (see Calcium imaging) at 1 mM, and stored in aliquots at −80 °C. Working solutions of 0.5 mM ATP for VT032906-Gal4 or 0.1 mM ATP for VT020739-Gal4 with 20 μM Alexa594 were prepared on the same day as the experiment and loaded into a pipette with a bore <1 μm in diameter. We adjusted the concentration of ATP to provide as gentle a perturbation as possible, as measured by the E-PG signal shapes remaining the same, and the E-PG phase returning to following the fly’s movements a few seconds after the stimulation. 0.5 mM ATP was used for controls without Gal4. The pressure in the pipette was controlled using a Pneumatic PicoPump (PV820, World Precision Instruments), and the pressure recorded using a Pressure Monitor (PM 015R, World Precision Instruments). We applied pressure pulses ranging from 5 to 20 psi with a 20-ms duration. The pipette was controlled using a PatchStar micromanipulator (Scientifica). To access the bridge, we locally applied 0.5 mg/ml collagenase type 4 (Worthington) through a pipette, while keeping the bath at ~30 °C, to breach the sheath above the bridge. For dual imaging of GCaMP6f and Alexa594, we used a 500–550-nm bandpass filter for the green channel, and a 585–635-nm bandpass filter for the red channel. We computed ∆F/F values for Alexa594 by defining the baseline, F0, as the mean of the lowest 5% of values in the entire bridge, rather than independently for each glomerulus, because the glomerulus-independent normalization is meant to compensate for varying GCaMP baselines across glomeruli, presumably owing to varying levels of GCaMP expression, or the amount of innervation within each glomerulus. In Fig. 5f, g and Extended Data Fig. 10e–j, both channels were nulled using the position of the pipette, which differed from fly to fly. In Fig. 5f, g, we averaged the E-PG signal 0.7–1.0 s after stimulation, and the Alexa594 (ATP) signal during the first frame after stimulation. In Extended Data Fig. 10e–j, we computed the change in each phase-nulled signal by subtracting the average over 0.3 s before stimulation from the averages in Fig. 5f, g for each channel. The four examples in Fig. 5b–e highlight trials in which we happened to stimulate P-ENs at roughly the glomeruli where we expected P-EN1 or P-EN2 activity peaks to already have been residing immediately before stimulation based on the measured E-PG phase (see Fig. 3) (that is, approximately in phase with E-PGs for P-EN1 and approximately anti-phase with E-PGs for P-EN2). All stimulation trials are included in the phase-nulled averages in Fig. 5f, g. All stimulation experiments were performed in the dark.
Data and code availability
All data and code are available on request from the authors.
References
O’Keefe, J . & Nadel, L. The Hippocampus as a Cognitive Map (Clarendon, 1978)
Tolman, E. C., Ritchie, B. F. & Kalish, D. Studies in spatial learning: orientation and the short-cut. J. Exp. Psychol. Gen. 36, 13–24 (1946)
Etienne, A. S. & Jeffery, K. J. Path integration in mammals. Hippocampus 14, 180–192 (2004)
Mittelstaedt, M. L. & Mittelstaedt, H. Homing by path integration in a mammal. Naturwissenschaften 67, 566–567 (1980)
Collett, T. S. & Collett, M. Path integration in insects. Curr. Opin. Neurobiol. 10, 757–762 (2000)
Wehner, R. & Srinivasan, M. in The Neurobiology of Spatial Behaviour (ed. Jeffery, K. J. ) Ch. 1 (Oxford Univ. Press, 2003)
Von Frisch, K. The Dance Language and Orientation of Bees (Harvard Univ. Press, 1967)
Taube, J. S., Muller, R. U. & Ranck, J. B. Jr. Head-direction cells recorded from the postsubiculum in freely moving rats. I. Description and quantitative analysis. J. Neurosci. 10, 420–435 (1990)
Taube, J. S., Muller, R. U. & Ranck, J. B. Jr. Head-direction cells recorded from the postsubiculum in freely moving rats. II. Effects of environmental manipulations. J. Neurosci. 10, 436–447 (1990)
Varga, A. G. & Ritzmann, R. E. Cellular basis of head direction and contextual cues in the insect brain. Curr. Biol. 26, 1816–1828 (2016)
Heinze, S. & Homberg, U. Maplike representation of celestial E-vector orientations in the brain of an insect. Science 315, 995–997 (2007)
Finkelstein, A. et al. Three-dimensional head-direction coding in the bat brain. Nature 517, 159–164 (2015)
Robertson, R. G., Rolls, E. T., Georges-François, P. & Panzeri, S. Head direction cells in the primate pre-subiculum. Hippocampus 9, 206–219 (1999)
Seelig, J. D. & Jayaraman, V. Neural dynamics for landmark orientation and angular path integration. Nature 521, 186–191 (2015)
Skaggs, W. E., Knierim, J. J., Kudrimoti, H. S. & McNaughton, B. L. A model of the neural basis of the rat’s sense of direction. Adv. Neural Inf. Process. Syst. 7, 173–180 (1995)
Zhang, K. Representation of spatial orientation by the intrinsic dynamics of the head-direction cell ensemble: a theory. J. Neurosci. 16, 2112–2126 (1996)
Redish, A. D., Elga, A. N. & Touretzky, D. S. A coupled attractor model of the rodent head direction system. Network 7, 671–685 (1996)
Sharp, P. E., Blair, H. T. & Brown, M. Neural network modeling of the hippocampal formation spatial signals and their possible role in navigation: a modular approach. Hippocampus 6, 720–734 (1996)
McNaughton, B. L., Battaglia, F. P., Jensen, O., Moser, E. I. & Moser, M.-B. Path integration and the neural basis of the ‘cognitive map’. Nat. Rev. Neurosci. 7, 663–678 (2006)
Kakaria, K. S. & De Bivort, B. L. Ring attractor dynamics emerge from a spiking model of the entire protocerebral bridge. Front. Behav. Neurosci. 11, 6706–6713 (2017)
Wolff, T., Iyer, N. A. & Rubin, G. M. Neuroarchitecture and neuroanatomy of the Drosophila central complex: A GAL4-based dissection of protocerebral bridge neurons and circuits. J. Comp. Neurol. 523, 997–1037 (2015)
Lin, C.-Y. et al. A comprehensive wiring diagram of the protocerebral bridge for visual information processing in the Drosophila brain. Cell Reports 3, 1739–1753 (2013)
Chen, T.-W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013)
Maimon, G., Straw, A. D. & Dickinson, M. H. Active flight increases the gain of visual motion processing in Drosophila. Nat. Neurosci. 13, 393–399 (2010)
Seelig, J. D. et al. Two-photon calcium imaging from head-fixed Drosophila during optomotor walking behavior. Nat. Methods 7, 535–540 (2010)
Moore, R. J. D. et al. FicTrac: a visual method for tracking spherical motion and generating fictive animal paths. J. Neurosci. Methods 225, 106–119 (2014)
Reiser, M. B. & Dickinson, M. H. A modular display system for insect behavioral neuroscience. J. Neurosci. Methods 167, 127–139 (2008)
Dana, H. et al. Sensitive red protein calcium indicators for imaging neural activity. eLife 5, 413 (2016)
Poodry, C. A. & Edgar, L. Reversible alteration in the neuromuscular junctions of Drosophila melanogaster bearing a temperature-sensitive mutation, shibire. J. Cell Biol. 81, 520–527 (1979)
Zemelman, B. V., Nesnas, N., Lee, G. A. & Miesenbock, G. Photochemical gating of heterologous ion channels: remote control over genetically designated populations of neurons. Proc. Natl Acad. Sci. USA 100, 1352–1357 (2003)
Turner-Evans, D. et al. Angular velocity integration in a fly heading circuit. eLife http://dx.doi.org/10.7554/eLife.23496 (2017)
Taube, J. S. The head direction signal: origins and sensory-motor integration. Annu. Rev. Neurosci. 30, 181–207 (2007)
Stackman, R. W. & Taube, J. S. Firing properties of rat lateral mammillary single units: head direction, head pitch, and angular head velocity. J. Neurosci. 18, 9020–9037 (1998)
Blair, H. T., Cho, J. & Sharp, P. E. Role of the lateral mammillary nucleus in the rat head direction circuit: a combined single unit recording and lesion study. Neuron 21, 1387–1397 (1998)
Hafting, T., Fyhn, M., Molden, S., Moser, M.-B. & Moser, E. I. Microstructure of a spatial map in the entorhinal cortex. Nature 436, 801–806 (2005)
Sargolini, F. et al. Conjunctive representation of position, direction, and velocity in entorhinal cortex. Science 312, 758–762 (2006)
Robinson, D. A. Integrating with neurons. Annu. Rev. Neurosci. 12, 33–45 (1989)
Major, G. & Tank, D. Persistent neural activity: prevalence and mechanisms. Curr. Opin. Neurobiol. 14, 675–684 (2004)
Pastor, A. M., De la Cruz, R. R. & Baker, R. Eye position and eye velocity integrators reside in separate brainstem nuclei. Proc. Natl Acad. Sci. USA 91, 807–811 (1994)
Aksay, E., Gamkrelidze, G., Seung, H. S., Baker, R. & Tank, D. W. In vivo intracellular recording and perturbation of persistent activity in a neural integrator. Nat. Neurosci. 4, 184–193 (2001)
Mizumori, S. J. & Williams, J. D. Directionally selective mnemonic properties of neurons in the lateral dorsal nucleus of the thalamus of rats. J. Neurosci. 13, 4015–4028 (1993)
McNaughton, B. L., Chen, L. L. & Markus, E. J. ‘Dead reckoning,’ landmark learning, and the sense of direction: a neurophysiological and computational hypothesis. J. Cog. Neurosci. 3, 190–202 (1991)
Nern, A., Pfeiffer, B. D. & Rubin, G. M. Optimized tools for multicolor stochastic labeling reveal diverse stereotyped cell arrangements in the fly visual system. Proc. Natl Acad. Sci. USA 112, E2967–E2976 (2015)
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012)
Fisher, N. I. & Lee, A. J. A correlation coefficient for circular data. Biometrika 70, 327–332 (1983)
Acknowledgements
We thank the Ruta and Rubin laboratories for fly stocks, and C. Kirst, C. Lyu, V. Vijayan, L. Fenk, A. Katsov, C. Bargmann, V. Ruta, S. Simon and members of the Maimon laboratory for discussions. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. Stocks obtained from the Vienna Drosophila Resource Center were used in this study. Research reported in this publication was supported by the New York Stem Cell Foundation (NYSCF-R-NI13), Searle Scholars Foundation (12-SSP-153), McKnight Foundation, and the National Institute on Drug Abuse of the NIH (DP2DA035148). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
J.G., A.A. and G.M. designed the experiments. J.G. performed, analysed and interpreted all physiological experiments, with input from G.M. A.A. performed and analysed all immunohistochemistry experiments. K.K.S. and J.G. fabricated parts for the experimental setup. J.D.H. developed the pipeline for image registration and rendered the sample videos. P.S.M. adapted FicTrac to our closed-loop setup. J.G. and G.M. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks A. Cheung, R. Ritzmann and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations
Extended data figures and tables
Extended Data Figure 1 Processing of protocerebral bridge signals from E-PG, P-EN1 and P-EN2 neurons in the presence of a closed-loop bar.
a, Each z slice, averaged over an entire E-PG>GCaMP6m recording, with glomeruli outlined. b, Processing of the EPG>GCaMP6m signal to generate the plot in Fig. 1g. From left to right: raw mean signal in each glomerulus over time, z-score normalization for each glomerulus independently, ∆F/F normalization for each glomerulus independently, power spectrum of the ∆F/F signal computed for each time point (row) independently. The E-PG phase extracted from the Fourier component with a period of eight glomeruli of the ∆F/F bridge signal is overlaid on each GCaMP plot in black. c, E-PG phase (blue) shifted with a constant offset to best match the bar position (dark grey). d, e, Same as a, b but for P-EN1 neurons originally plotted in Fig. 1h. That P-EN cells do not innervate the middle two glomeruli of the bridge slightly complicates the power spectrum analysis. Specifically, black arrows highlight transient peaks in the power spectrum at approximately 16 glomeruli, which are artefacts of the P-EN GCaMP peaks crossing the centre of the bridge. f, From left to right: P-EN1 ∆F/F signal with the middle two glomeruli filled in by averaging signals located one period (8 glomeruli) away, power spectrum of the ‘filled in’ ∆F/F bridge signal (note absence of artefactual peaks at 16 glomeruli), P-EN phase extracted from the Fourier component with a period of 8 glomeruli of the ‘filled in’ ∆F/F bridge signal (orange), shifted with a constant offset to best match the bar position (dark grey). g–i, Same as d–f but for P-EN2 neurons originally plotted in Fig. 1i. In all plots showing bar position over time, the gap in the arena where the bar is not displayed is shown in grey. j–l, Periodicity of the bridge signal at peak power in the power spectrum for each cell type. Each circle represents one fly. The mean and s.d. are shown. m–o, Offsets that minimize the distance between GCaMP phase and bar position for all 50-s trials for each cell type. Only data for which the bar was visible were included in computing the offsets. In o, fly 10 had only three bar trials. See Methods for details. a.u., arbitrary units; DFT, discrete Fourier transform.
Extended Data Figure 2 Processing of protocerebral bridge signals from E-PG, P-EN1 and P-EN2 neurons in the dark.
a, Processing of the E-PG>GCaMP6m signal to generate the plot in Fig. 2g. Bridge signals are plotted over time as in Extended Data Fig. 1b, but in the dark. b, Phase from the ∆F/F signal and ball position. Because the phase and ball position drift over time in the dark, we did not align the two signals by finding the best offset over the entire trial; rather, we nulled the offset between the GCaMP phase and ball heading at time zero, letting the signals drift naturally over time. For display purposes, we applied a constant gain to the ball position signal, which we determined from the slope of a linear regression between the GCaMP phase and ball velocity. c, d, P-EN1 signals (originally plotted in Fig. 4a) over time as in Extended Data Fig. 1e, f, but in the dark. e, f, Same as c, d, but for P-EN2 signals (originally plotted in Fig. 4b). The ball position gains are 0.75 for E-PG (b), 1.0 for P-EN1 (d) and 0.89 for P-EN2 (f). For P-EN1, the slope of the linear regression between phase and ball velocity was poorly estimated (see Supplementary Discussion) and thus we hand-picked the gain (1.0) in this case. That these gains are not all equal does not mean that each cell type has its own gain (see Supplementary Discussion). Note the different timescale compared to Extended Data Fig. 1. Also note that the time window was expanded in a, b, compared to Fig. 2g, to be the same length as in c–f. g–i, Periodicity of the bridge signal at peak power in the power spectrum for each cell type. Each circle represents one fly. The mean and s.d. are shown.
Extended Data Figure 3 Example visual tuning curves in E-PG, P-EN1 and P-EN2 neurons across glomeruli in the protocerebral bridge.
a–c, Tuning curves of GCaMP activity as a function of bar position for each glomerulus in a sample fly for E-PGs (a), P-EN1s (b) and P-EN2s (c). Data associated with bar positions in the 90° gap in the back of the arena (not visible) are not shown. The mean and s.d. across time points for each 22.5° bar position bin are shown.
Extended Data Figure 4 P-EN neuroanatomy: explanation for the numbering scheme, sytGFP localization, and multicolour single cell labelling.
a, Numbering used in the literature for the protocerebral bridge and ellipsoid body. b, Rearrangement of the left and right bridges and a linearized ellipsoid body that highlights the pattern of anatomical projections for E-PGs and P-ENs. Arrows indicate the expected direction of signalling (dendrite to axon) for each cell21,22 (also see e, f). The dashed line in a shows where the ellipsoid body is opened to display it linearly. Tile 1 is repeated as a visual aid, as the ellipsoid body is circular. c, d, Same as a, b but using a modified numbering scheme. In d, the numbers are constant along each column (with the exception that glomerulus 9 from either side of the bridge matches up with ellipsoid body tile 1), highlighting the fact that E-PGs project within the same column, whereas left-bridge P-ENs project to the right (+1, or clockwise) and right-bridge P-ENs project to the left (−1, or anticlockwise). e, f, Sample images of synaptotagmin–GFP (sytGFP, labelling putative axonal terminals) and tdTomato (labelling the entire cell) expressed in P-EN1 (e) and P-EN2 (f) neurons. These data are consistent with P-ENs having extensive presynaptic terminals in the ellipsoid body and noduli but few in the protocerebral bridge. g–l, Sample multicolour flip-out images for P-EN neurons driven by VT032906–Gal4 (P-EN1, g, h), VT020739–Gal4 (P-EN2, i, j), and 12D09–Gal4 (P-EN2, k, l). The multicolour flip-out method41 allows one to visualize single randomly selected cells from a Gal4 driver line (which might label a dense thicket of cells) in their entirety, like a multicoloured Golgi stain. The neuropil is shown in grey. Single neurons are coloured. Glomerulus numbers, including L for left and R for right, are shown in the bridge. After tracing each neuron from the bridge to the ellipsoid body, we labelled the terminals in the ellipsoid body with the bridge glomerulus from which they originated, using our revised numbering scheme (c, d). VT032906–Gal4 stains a neuron type that passes near the bridge, but does not innervate the bridge, ellipsoid body or noduli (for example, the green neuron in g). VT020739–Gal4 stains a neuron type that innervates the noduli, but not the ellipsoid body or bridge (for example, the blue neurons innervating the noduli from the sides in j). Virtually all neurons labelled in the bridge and ellipsoid body were consistent with P-ENs (see Supplementary Information Table 1). 12D09–Gal4 very rarely revealed flip-outs of protocerebral bridge local neurons, not shown here (see Supplementary Information Table 1). eb, ellipsoid body; no, noduli; pb, protocerebral bridge.
Extended Data Figure 5 P-EN1 and P-EN2 bridge asymmetry during turns in closed-loop bar and dark conditions, computed with z-score and ∆F/F normalization.
a–c, Right–left bridge activity (bottom) and the fly’s turning velocity (top), averaged over multiple turns, for P-EN1s (a), P-EN2s (b) and E-PGs (c), as in Fig. 2h–j, in closed-loop bar conditions. The right–left GCaMP signal is computed from z-score normalized data. d–f, Same as a–c but in constant darkness. g–l, Same as a–f, except that the right–left GCaMP signal is computed from ∆F/F normalized data. The mean and s.e.m. across turns are shown. Only data for which the bar was visible on the front 270° of the LED arena were included for closed-loop bar plots. See Methods for details.
Extended Data Figure 6 P-EN1 and P-EN2 asymmetries are driven in part by optic flow.
a, Sample trajectory of one of hundreds of dots used to create our optic flow stimulus. Each dot appeared at a random location, travelled 4 azimuthal pixels (7.5°), and then disappeared. The dashed circle is drawn as a point of reference, and was not presented on the screen. b, c, Right–left P-EN1 bridge activity during open-loop optic flow to the left (b) and to the right (c) at 45°/s (left column) and 90°/s (right column) during trials in which the fly did not, on average, turn ( ± 10°/s) in response to the optic flow stimulus. d, e, Same as b, c but for P-EN2 neurons. f–i, Same as b–e, except that trials were included only if the fly turned with the direction of optic flow (>10°/s in the direction of optic flow). The mean and s.e.m. across trials are shown. For display, the stimulus position was nulled at time zero to highlight the movement of the stimulus. In trials in which flies turned with the direction of optic flow, the direction of visual motion experienced on their retinas was opposite to that expected from their own turning behavior. That is, the visual optic flow inputs (presented in open loop) indicated an angular velocity with the opposite sign to that indicated by proprioceptive/efference-copy inputs. The fact that we observe a weaker asymmetry in f–i compared to b–e, argues that optic flow and proprioceptive/efference-copy inputs are combined to generate the P-EN bridge asymmetry.
Extended Data Figure 7 Co-labelling of P-EN1 and P-EN2 driver lines.
a–c, Maximum z-projection of a brain with 12D09-driven neurons expressing GFP and VT032906-driven neurons expressing tdTomato. a, GFP (12D09) signal. b, tdTomato (VT032906) signal. c, Composite of a and b. Physiological experiments suggest that VT032906 primarily labels P-EN1 neurons, whereas 12D09 primarily labels P-EN2 neurons (Fig. 3, Extended Data Fig. 8b, d). As expected, most P-EN neurons are primarily labelled by one of the two drivers, but some neurons are labelled by both (examples denoted with asterisks). d–f, Same as a–c but with VT020739-driven neurons expressing tdTomato. Physiological experiments suggest that both 12D09 and VT020739 primarily label P-EN2 neurons (Fig. 3, Extended Data Fig. 8c, d). As expected, almost all labelled P-EN neurons are labelled by both P-EN2 drivers. P-ENs often showed fluorescent signals whose strength varied across glomeruli, which could reflect varying innervation densities across the bridge. They could also reflect incomplete targeting of P-ENs by our driver lines.
Extended Data Figure 8 Simultaneous imaging of the protocerebral bridge and ellipsoid body for each cell type separately and dual-colour imaging of GCaMP6f and jRGECO1a in E-PGs in the ellipsoid body.
a, We imaged the bridge and ellipsoid body in the same fly, at the same time, using a tall z-stack that encompassed both structures, to determine the relationship between the signals measured in each structure. b, Phase-nulled P-EN1 signals measured in the bridge (orange) and ellipsoid body (grey). The signals measured in the bridge were replotted onto the ellipsoid body using the P-EN projection pattern. c, d, Same as b but for P-EN2 signals from VT020739-Gal4 (c) and 12D09-Gal4 (d). e, As in a but for imaging E-PGs, with the bridge in blue. f, Same as b for E-PGs, with the left and right bridge in blue. In b–d and f, the mean and s.e.m. across flies are shown (in f, the s.e.m. for the bridge curves (blue) are omitted for clarity). Both the bridge and ellipsoid body signals were nulled using the ellipsoid body phase. Note that the positions of the left- and right-bridge peaks are inverted between P-EN1 and P-EN2. These results are consistent with the dual-imaging experiments in Fig. 3, and support the idea that the results in Fig. 3 were not due to crosstalk between the red and green channels. g, Schematic illustrating imaging from the ellipsoid body. h–j, Phase-nulled ellipsoid body signals of GCaMP6f and jRGECO1a co-expressed in E-PGs, computed for when the fly turned left (h, −300°/s), walked straight (i, 0°/s) or turned right (j, +300°/s), 300 ms before the calcium signal, as in Fig. 3k–p. The mean and s.e.m. across flies are shown. We observed no consistent, strong asymmetries in the jRGECO1a and GCaMP6f signals during left or right turns when both indicators were expressed in E-PGs. These data argue that the asymmetries we observed in dual imaging of P-ENs and E-PGs in the ellipsoid body (Fig. 3m–p) were not an artefact of indicator kinetics. Data are averaged over bar and dark conditions.
Extended Data Figure 9 Analysis of ellipsoid body asymmetry in P-EN1s and P-EN2s relative to E-PGs in the ellipsoid body.
a, Mean E-PG and P-EN1 activity in the ellipsoid body triggered on when the fly was turning to the left (−300°/s, upper panel) or right (+300°/s, lower panel), as in Fig. 3m–p, but over time. The P-EN1 and E-PG signals were phase-nulled using the E-PG phase. b, Same as a but for P-EN2 activity. c, d, When analysing the two-colour imaging experiments in Fig. 3i–p, we calculated the cross correlation between the ellipsoid body asymmetry in P-EN1 (c) or P-EN2 (d) and the E-PG phase velocity in the ellipsoid body. A positive correlation indicates an increased P-EN signal in the direction in which the E-PG peak is moving. A positive lag indicates that the P-EN asymmetry comes after the change in the E-PG phase. Thus, the P-EN1 peak tends to lead the E-PG peak whereas the P-EN2 peak tends to lag behind the E-PG peak. Note that we also observed a smaller, negative, late peak in the signal driven by the P-EN1 Gal4 and a smaller positive, early peak in the signal driven by the P-EN2 Gal4, suggesting that each Gal4 line contains some number of both P-EN1 and P-EN2 cells, but with more of one than the other. e, f, Same as c, d, except that the P-EN ellipsoid body asymmetry is correlated with the fly’s turning velocity. A positive lag indicates that the P-EN asymmetry comes after the fly turns. Arrows indicate the lag where the mean correlation was greatest. In c–f, thin lines represent single flies and thick lines represent the mean across flies. Data are averaged over bar and dark conditions.
Extended Data Figure 10 The effects of blocking P-EN synaptic transmission on the ability of E-PGs to track a landmark, and controls for the P2X2 experiments.
a, b, E-PG activity in the bridge from the same fly as in Fig. 4e, f (P-EN1>shibirets), except with a closed-loop bar, at 22 °C (a) and 32 °C (b). c, Correlations between phase and bar velocity, for three P-EN-Gal4 lines driving shibirets, with parental controls. Each circle represents one fly. The mean and s.e.m. across flies are shown. d, Same as c, but plotting circular correlations between phase and bar position. Only data in which the bar was visible in the front 270° of the arena were used for calculating correlations. Trials during which the bar was visible for less than 10 s were excluded, with some flies having no trials passing this criterion. The total number of flies (without excluding flies that did not pass the above criterion) for each genotype is shown. The mean and s.e.m. across included flies are shown. Only VT020739–Gal4 seems to affect the ability of the E-PG signal to track a visual landmark, suggesting that perhaps this effect is due to non-P-EN neurons targeted by this line, for example visual lobe neurons, or that this effect requires stronger Gal4 expression in P-ENs in this line. e, The change in the phase-nulled E-PG>GCaMP6f and ATP (Alexa594) signals during an ATP pulse, with P-EN1s expressing P2X2. We subtracted the average E-PG signal at −0.3 to 0.0 s from the average at 0.7 to 1.0 s with respect to the time of the pressure pulse, highlighting the effect of the stimulation. We subtracted the average Alexa594 (ATP) signal at −0.3 to 0.0 s from the average immediately (1 frame) after stimulation. The irregular dips in the E-PG signal are due to the fact that the E-PG phase was not uniformly distributed immediately before stimulation. Both signals were phase-nulled using the position of the pipette. f, Same as e, but without ATP in the same flies. g, h, Same as e, f, but with P-EN2s expressing P2X2. i, j, Same as e, f but with no Gal4 as a control for the specificity of P2X2 expression. In e–j, thin lines represent single flies, and thick lines represent the means across flies.
Supplementary information
Supplementary Information
This file contains the Supplementary Discussion and Supplementary Table 1. (PDF 369 kb)
Sample raw GCaMP6f signals from E-PG neurons in the protocerebral bridge shown as the fly performs tethered walking behaviour (E-PG>GCaMP6f raw 1.00x fixation Sample)
Sample raw GCaMP6f signals from E-PG neurons in the protocerebral bridge are shown as the fly performs tethered walking behaviour. The fly is controlling the azimuthal position of a bright blue bar in closed loop during this experiment, and tends to maintain its heading within this virtual environment such that the bar remains in front of the fly. Note that the 2-3 peaks of GCaMP signal in the protocerebral bridge accurately track the position of the bar over time. Shown is the maximum z-projection across 3 z-slices. The resulting z-projection was smoothed with a 0.65-pixel (~0.5 µm) gaussian. Since the fly is displayed from the front, the right bridge is displayed on the left, and the left bridge on the right, contrary to all Figures and Extended Data Figures. The video is played at real time-speed. (MP4 1101 kb)
Sample raw GCaMP6m signals from E-PG neurons in the protocerebral bridge shown as the fly performs tethered walking behaviour (E-PG>GCaMP6m_raw_1.00x Sample)
Sample raw GCaMP6m signals from E-PG neurons in the protocerebral bridge are shown as the fly performs tethered walking behaviour. The fly is controlling the azimuthal position of a bright blue bar in closed loop during this experiment. Note that the 2-3 peaks of GCaMP signal in the protocerebral bridge accurately track the position of the bar over time. Shown is the maximum z-projection across 3 z-slices. The resulting z-projection was smoothed with a 0.65-pixel (~0.5 µm) gaussian. Since the fly is displayed from the front, the right bridge is displayed on the left, and the left bridge on the right, contrary to all Figures and Extended Data Figures. The video is played at real time-speed. (MP4 1575 kb)
Sample raw GCaMP6m signals from E-PG neurons in the protocerebral bridge shown as the fly performs tethered walking behaviour (E-PG>GCaMP6m_raw_0.50x Sample)
Sample raw GCaMP6m signals from E-PG neurons in the protocerebral bridge are shown as the fly performs tethered walking behaviour. The fly is controlling the azimuthal position of a bright blue bar in closed loop during this experiment. Note that the 2-3 peaks of GCaMP signal in the protocerebral bridge accurately track the position of the bar over time. Shown is the maximum z-projection across 3 z-slices. The resulting z-projection was smoothed with a 0.65-pixel (~0.5 µm) gaussian. Since the fly is displayed from the front, the right bridge is displayed on the left, and the left bridge on the right, contrary to all Figures and Extended Data Figures. The video is played at half-speed. (MP4 1970 kb)
Sample raw GCaMP6m signals from P-EN1 neurons in the protocerebral bridge shown as the fly performs tethered walking behaviour (P-EN1>GCaMP6m_raw_1.00x Sample)
Sample raw GCaMP6m signals from P-EN1 neurons in the protocerebral bridge are shown as the fly performs tethered walking behaviour. The fly is controlling the azimuthal position of a bright blue bar in closed loop during this experiment. Note that the 2-3 peaks of GCaMP signal in the protocerebral bridge are only strongly active when the fly turns, and that these peaks are asymmetrically active during these turns. In this video, the fly expresses only one copy of VT032906-Gal4 and UAS-GCaMPm, compared to the two copies of each used in generating Figs. 1-2. The P-EN1 signal in the single copy flies highlights the transient nature of P-EN1 activity during turns. Shown is the maximum z-projection across 3 z-slices. The resulting z-projection was smoothed with a 0.65-pixel (~0.5 µm) gaussian. Since the fly is displayed from the front, the right bridge is displayed on the left, and the left bridge on the right, contrary to all Figures and Extended Data Figures. The video is played at real time-speed. (MP4 2011 kb)
Sample raw GCaMP6m signals from P-EN1 neurons in the protocerebral bridge shown as the fly performs tethered walking behaviour (P-EN1>GCaMP6m_raw_0.50x Sample)
Sample raw GCaMP6m signals from P-EN1 neurons in the protocerebral bridge are shown as the fly performs tethered walking behaviour. The fly is controlling the azimuthal position of a bright blue bar in closed loop during this experiment. Note that the 2-3 peaks of GCaMP signal in the protocerebral bridge are only strongly active when the fly turns, and that these peaks are asymmetrically active during these turns. In this video, the fly expresses only one copy of VT032906-Gal4 and UAS-GCaMPm, compared to the two copies of each used in generating Figs. 1-2. The P-EN1 signal in the single copy flies highlights the transient nature of P-EN1 activity during turns. Shown is the maximum z-projection across 3 z-slices. The resulting z-projection was smoothed with a 0.65-pixel (~0.5 µm) gaussian. Since the fly is displayed from the front, the right bridge is displayed on the left, and the left bridge on the right, contrary to all Figures and Extended Data Figures. The video is played at half-speed. (MP4 2408 kb)
Sample raw GCaMP6m signals from P-EN2 neurons in the protocerebral bridge shown as the fly performs tethered walking behaviour (P-EN2>GCaMP6m_raw_1.00x Sample)
Sample raw GCaMP6m signals from P-EN2 neurons in the protocerebral bridge are shown as the fly performs tethered walking behaviour. The fly is controlling the azimuthal position of a bright blue bar in closed loop during this experiment. Note that the 2-3 peaks of GCaMP signal in the protocerebral bridge accurately track the position of the bar over time. The turn-related asymmetries are not as evident in the raw P-EN2 videos as they are in the P-EN1 videos - they are, however, evident from a quantitative analysis of the P-EN2 signals (Fig. 2-3). Shown is the maximum z-projection across 3 z-slices. The resulting z-projection was smoothed with a 0.65-pixel (~0.5 µm) gaussian. Since the fly is displayed from the front, the right bridge is displayed on the left, and the left bridge on the right, contrary to all Figures and Extended Data Figures. The video is played at real time-speed. (MP4 1918 kb)
Sample raw GCaMP6m signals from P-EN2 neurons in the protocerebral bridge shown as the fly performs tethered walking behaviour (P-EN2>GCaMP6m_raw_0.50x Sample)
Sample raw GCaMP6m signals from P-EN2 neurons in the protocerebral bridge are shown as the fly performs tethered walking behaviour. The fly is controlling the azimuthal position of a bright blue bar in closed loop during this experiment. Note that the 2-3 peaks of GCaMP signal in the protocerebral bridge accurately track the position of the bar over time. The turn-related asymmetries are not as evident in the raw P-EN2 videos as they are in the P-EN1 videos - they are, however, evident from a quantitative analysis of the P-EN2 signals (Fig. 2-3). Shown is the maximum z-projection across 3 z-slices. The resulting z-projection was smoothed with a 0.65-pixel (~0.5 µm) gaussian. Since the fly is displayed from the front, the right bridge is displayed on the left, and the left bridge on the right, contrary to all Figures and Extended Data Figures. The video is played at half-speed. (MP4 2384 kb)
Rights and permissions
About this article

Cite this article
Green, J., Adachi, A., Shah, K. et al. A neural circuit architecture for angular integration in Drosophila. Nature 546, 101–106 (2017). https://doi.org/10.1038/nature22343
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature22343
- Springer Nature Limited
This article is cited by
-
Converting an allocentric goal into an egocentric steering signal
Nature (2024)
-
Transforming a head direction signal into a goal-oriented steering command
Nature (2024)
-
A unifying perspective on neural manifolds and circuits for cognition
Nature Reviews Neuroscience (2023)
-
A rise-to-threshold process for a relative-value decision
Nature (2023)
-
Emergent behaviour and neural dynamics in artificial agents tracking odour plumes
Nature Machine Intelligence (2023)