

Abstract
The retina extracts visual features for transmission to the brain. Different types of bipolar cell split the photoreceptor input into parallel channels and provide the excitatory drive for downstream visual circuits. Mouse bipolar cell types have been described at great anatomical and genetic detail, but a similarly deep understanding of their functional diversity is lacking. Here, by imaging light-driven glutamate release from more than 13,000 bipolar cell axon terminals in the intact retina, we show that bipolar cell functional diversity is generated by the interplay of dendritic excitatory inputs and axonal inhibitory inputs. The resulting centre and surround components of bipolar cell receptive fields interact to decorrelate bipolar cell output in the spatial and temporal domains. Our findings highlight the importance of inhibitory circuits in generating functionally diverse excitatory pathways and suggest that decorrelation of parallel visual pathways begins as early as the second synapse of the mouse visual system.
Similar content being viewed by others


Main
The retina extracts visual features such as motion or edges1 and relays these to the brain through a diverse set of retinal ganglion cells (RGCs)2,3. This functional diversity begins to emerge as early as the first retinal synapse, where, in mice, the visual signal is distributed from the photoreceptors onto 14 types of bipolar cell (BC)4. The axon terminals of the BCs stratify at different depths of the inner plexiform layer (IPL) and provide the excitatory drive for the feature-extracting circuits of the inner retina.
The different types of mouse BC have been anatomically and genetically well characterized5,6,7,8,9,10. Functionally, BCs have been classified mostly into broad categories such as On and Off, transient and sustained or chromatic and achromatic11,12,13,14; however, a deeper understanding of the functional diversity of BCs and its origin is lacking.
Some of the observed functional differences among types of BC are established in the outer retina as early as the level of the BC dendrites7,15,16,17,18. In the inner retina, roughly 42 types of mostly inhibitory amacrine cell modulate BC output at the level of the synaptic terminals of BCs5,19,20. Although some amacrine cell circuits have been studied in depth (see, for example, refs 21, 22), we still understand little about the general principles by which amacrine cell circuits help to decompose the visual scene into the parallel channels carried by the BCs.
Glutamate release units of the IPL
To address this important issue, we used the glutamate-sensing fluorescent reporter iGluSnFR23,24. In contrast to presynaptic calcium changes, which have been used to assess BC function in mouse and zebrafish25,26,27,28, glutamate release represents the output ‘currency’ of BCs, accounting not only for presynaptic inhibition but also for any release dynamics of BC ribbon synapses29,30. Using two-photon imaging, we systematically characterized the glutamatergic output of mouse BCs at the level of individual axon terminals in whole-mounted retinas, in which long-range connections are preserved.
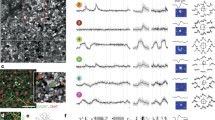
Intravitreal injection of AAV9–iGluSnFR yielded homogenous expression across the IPL (Fig. 1a), allowing to sample glutamate release at all IPL depths (Extended Data Fig. 1i). For each scan field, we registered the recording depth as its relative distance from the two plexi of SR101-stained blood vessels (Fig. 1b, Methods). To objectively define individual glutamate ‘release units’, we placed regions of interest (ROIs) in a single scan field (typically 48 × 12 μm at 31.25 Hz) using local image correlations (Fig. 1c, Extended Data Fig. 1, Supplementary Video 1, Methods and Supplementary Discussion). We verified the ROI placement using calcium imaging of BCs with the calcium biosensor GCaMP6f31, which allowed us to resolve individual terminal systems and single axon terminals (Extended Data Fig. 2).

a, Vertical projection of stack showing iGluSnFR expression (green) across the IPL and blood vessels in red. Grey plane, scan field orientation. GCL, ganglion cell layer; INL, inner nuclear layer; OV, outer vessels; IV, inner vessels. b, Choline acetyltransferase (ChAT) bands (white) relative to blood vessels (red) and average depth profiles (±s.d. shading); n = 9 stacks, n = 3 mice. c, Example scan field (64 × 16 pixels) with ROI mask. Numbers correspond to different ROIs shown in d–g. d, Glutamate response of ROI number 3 from c to local and full-field chirp stimulus and full-field flashes. Black, mean; grey, individual trials. Glutamate traces represent relative glutamate release (d[Glut]). e, Temporal and spatial receptive field of ROI from d. Dashed line, time of response. f, Superimposed mean glutamate traces in response to local (top) and full-field chirp (bottom) of red (1–3) and green (4–6) ROIs from c. g, Scan field and ROI mask from c with spatial receptive fields of red and green ROIs (2 s.d. outlines, n = 20 ROIs).
We used a standardized set of four light stimuli (Fig. 1d, see also ref. 2) to characterize BC output: (i) local (100 μm diameter) and (ii) full-field (600 × 800 μm) ‘chirp’ stimuli to probe response polarity and the contrast and frequency preferences of the BC centre and centre-surround, respectively, (iii) 1-Hz full-field steps to study response kinetics and (iv) binary dense noise to estimate receptive fields (Methods). The light conditions on the retina corresponded to the low-photopic range (Methods).
ROIs in a single scan field typically showed two or more distinct response profiles (Fig. 1f), suggesting that multiple types of BC could be recorded at a single IPL depth, as expected from their stratification overlap5,6,10 (compare with Fig. 2b). ROIs that shared a common response profile had receptive fields that either almost completely overlapped or were spatially offset (Fig. 1g), consistent with the reported tiling of BCs of the same type8. For example, the highly overlapping spatial receptive fields of the green ROIs suggest that they correspond to terminals not only of the same type of BC, but the same cell (Fig. 1g, Extended Data Fig. 2a–e). By contrast, the red ROIs are likely to correspond to terminals of two neighbouring cells of a second type (Fig. 1g). Therefore our ROIs are likely to reflect a reliable measure of BC output at the level of individual axon terminals.

a, Electron microscopy-reconstructed example BCs from ref. 5. b, Mean BC stratification profiles of all known BCs (see text). Colours indicate cluster allocation in all subsequent figures. c, Prior probabilities of scan fields recorded at two IPL depths (A, 1.7; B, 0 in arbitrary units (AU)). d, Mean glutamate responses (n = 8,452 ROIs) of every cluster to local and full-field chirps, full-field flashes and temporal receptive field kernels. e, Polarity index (Methods) as function of IPL depth (n = 8,452). Shading corresponds to median ± s.d. for every IPL bin (n = 13 bins). Cluster means (±s.d.) are overlaid. f–h, As in e for plateau index (f), response delay (g) and receptive field (RF) diameter (h) (Methods). Data in e–g estimated from local chirp step responses.
Anatomy-guided functional clustering of mouse BCs
In total, we recorded light-evoked BC glutamate release from 13,311 ROIs (n = 179 scan fields, n = 37 mice) across the IPL. We assumed that BCs are the main source of glutamate in the inner retina (Supplementary Discussion, Extended Data Fig. 3) and that the catalogue of BCs in the mouse retina is complete with 14 types (five Off-cone BCs (CBCs), eight On-CBCs and the rod BC (RBC))5,6,9,10, each tiling the retina8. Accordingly, the glutamate signals must map onto these 14 types of BC, including RBCs (Extended Data Figs 2g–j, 5k–n). We took advantage of available electron microscopy-based BC axonal stratification profiles6,10 to guide a functional clustering algorithm (Fig. 2a, b). For each scan taken at a specific IPL stratum, a prior probability for cluster allocation was computed from the relative axon terminal volume of all BC types in the respective IPL stratum (Fig. 2c, Extended Data Fig. 4a). We then extracted functional features from the glutamate responses that passed our quality criterion (76.9% of ROIs) using sparse principal component analysis (Extended Data Fig. 4b) and clustered the ROIs using a modified Mixture of Gaussian model (Methods).
This process yielded a functional fingerprint for every type of mouse BC (Fig. 2d, Extended Data Fig. 4c). Most functional clusters were well separated in feature space (Extended Data Fig. 4d–f); only a few cluster pairs showed some overlap (for example, for C3a and C3b or C5t and C5o the sensitivity index d′ ≈ 2). Because some types of BC have highly overlapping stratification profiles, clusters C1,2, C3a,b,4, C5t,o,i;X and C8,9,R (Extended Data Fig. 4c) might need to be permuted. For simplicity, we refer to functional clusters by the anatomical BC profiles from which they originate.
To validate our clustering approach, we visualized individual BC axon terminal systems by single-cell injection before glutamate imaging (Extended Data Fig. 5, Supplementary Discussion). At least 90% of all ROIs allocated to one terminal system were assigned to the same cluster or to functionally very similar clusters (Extended Data Fig. 5o). This suggests that each of our functional clusters predominantly contains ROIs from a single type of BC and therefore represents a valid approximation of its functional profile.
Organizational principles of the IPL
A fundamental principle of vertebrate inner retinal organization is the subdivision into Off- and On-cells13,32. Consistent with this, C1–4 increased activity at light offset (Off-BCs), whereas C5–R responded to light onset (On-BCs, Fig. 2e). However, the segregation into On and Off was not as clear-cut as expected: Off-BC clusters frequently responded with delayed spike-like events during the On phase of the light step (Extended Data Fig. 6a, g, Supplementary Discussion). These On events in Off-clusters did not correspond to spontaneous activity (Extended Data Fig. 6b). By contrast, On-BCs only rarely exhibited analogous Off responses (Extended Data Fig. 6b, g).
A second fundamental principle of inner retinal organization is the segregation into temporal ‘transient’ and ‘sustained’ channels, which map onto the IPL centre and borders, respectively11,24,28,33. While our results are broadly in line with this principle, the full picture is more complex. For example, although spike-like events were observed most frequently towards the IPL centre28 (Extended Data Fig. 6d–g), they could be found at all IPL depths. In addition, all On-clusters but none of the Off-clusters showed a sustained plateau following an initial fast peak (Fig. 2f), implying that On- and Off-BCs exhibit different release dynamics. Interestingly, temporal BC properties such as response delay (‘fast’ versus ‘slow’ onset), transience (speed of response decay) and the presence of spike-like events were not correlated (Extended Data Fig. 6i, j), suggesting that these properties can vary independently among BC types.
Finally, we found a spatial map across the IPL: receptive field size varied systematically among BC clusters and decreased significantly with increasing stratification depth (Fig. 2h, Extended Data Fig. 6h; P < 0.001, r = 0.4, n = 8,452 ROIs, linear correlation; mean: 66.3 and 55.9 μm for Off- and On-BC clusters, respectively). Despite this overall trend, receptive field sizes within one IPL depth differed substantially. For example, the receptive field diameters of C3b and C5i were nearly 10 μm larger than those of C4 and C7, respectively.
In summary, our results highlight important exceptions from fundamental principles of inner retinal organization and identify a new spatial organizing principle. They indicate that functionally opposite signals such as short and long delays or even On versus Off response polarities co-exist at a single depth.
BC surround activation increases functional diversity
The abovementioned organizing principles were extracted from the responses to the local chirp, but responses to the full-field chirp were substantially more heterogeneous (Figs 2d, 3). Additional surround stimulation significantly decorrelated chirp responses across clusters of the same polarity, and further anticorrelated responses of opposite polarity (Fig. 3a, b). This effect could be quite strong (Fig. 3c, Extended Data Fig. 6k): for example, the On-clusters C6 and C9 responded nearly identical to the local chirp, whereas their responses to the full-field chirp were decorrelated. This effect markedly broadened the response space covered by BC types (Extended Data Fig. 6l, m). For example, some On-BCs (for example, C5i or C9) lost their sustained plateau phase during full-field stimulation and became much more transient (compare Fig. 2f with Extended Data Fig. 6m). Accordingly, the major type-specific differences in the final outputs of BCs appear to be determined by concomitant centre and surround activation, rather than by centre activation alone.

a, Correlation between cluster means of local (top) and full-field (bottom) chirp responses. b, Mean correlation between local and full-field chirp responses for each cluster with all other clusters of the same (top) and opposite (bottom) response polarity (mean: ρlocal = 0.9 versus ρfull-field = 0.7 and ρlocal = −0.3 versus ρfull-field = −0.5, P < 0.001, n = 14, non-parametric paired Wilcoxon signed-rank test). Mean ± s.d. in black. c, Mean chirp responses of C6 and C9, with linear correlation coefficient (ρ) of whole trace or contrast ramp.
Different amacrine cells mediate and gate BC surround
We next dissected the cellular components underlying the observed surround effects using pharmacology. Two major groups of amacrine cells modulate BC output at the level of the BC axon terminal34,35,36: glycinergic small- and GABAergic wide-field amacrine cells37. In addition, there is extensive cross-talk among amacrine cells38. To test how these interactions modulate the BC surround, we pharmacologically blocked either GABA or glycine receptors while monitoring light-evoked glutamate release.
Pharmacological manipulation had little effect on overall response shape for local stimuli, but caused strong effects for full-field stimulation. Combined blocking of GABAA and GABAC receptors increased the response amplitude in both On- and Off-BCs (Extended Data Fig. 7a–c) and nearly eliminated the difference between local and full-field chirp responses (Fig. 4a–c, Extended Data Fig. 7d), consistent with attenuated surround inhibition39,40,41. This suggests that the BC surround is largely generated by presynaptic inhibition from GABAergic amacrine cells (Supplementary Discussion).

a, Local (grey) and full-field (black) chirp responses for control condition and GABA receptor block, with linear correlation coefficient (ρ) between each pair. b, Schematic illustrating the effects of GABA receptor block (1,2,5,6-tetrahydropyridin-4-yl)methylphosphinic acid and gabazine (TPMPA/Gbz), 75 μM and 10 μM, respectively). c, Linear correlation coefficients of local and full-field chirp responses across different clusters for GABA receptor block (P < 0.05, n = 10 from five scan fields and four mice, non-parametric paired Wilcoxon signed-rank test). d–f, As in a–c but for glycine receptor block (Strychnine, 0.5 μM). P < 0.001, n = 8 from four scan fields and three mice.
In contrast, blocking glycine receptors reduced the response to full-field flashes (Extended Data Fig. 7a–c), consistent with an increase in surround strength due to disinhibition of the GABAergic network. This effect reliably induced a polarity switch in BC responses to full-field stimulation, anti-correlating local and full-field chirp responses (Fig. 4d–f, Extended Data Fig. 7e). Compared to this strong network effect of glycinergic amacrine cells, direct glycinergic inputs to BCs via crossover inhibition acted more subtly on BC output, for example by decreasing tonic release of Off-BCs42,43 (Extended Data Fig. 8, Supplementary Discussion). This suggests that in the whole-mounted retina, glycinergic amacrine cells primarily modulate BC output in an indirect way by inhibiting GABAergic amacrine cells, leading to decreased surround inhibition.
In summary, the two major groups of mouse amacrine cells appear to act in tandem to set the ratio of excitation and inhibition in a group specific manner, thereby increasing functional diversity among BC types.
Centre–surround interactions underlie BC diversity
An increase in functional diversity upon surround stimulation is only possible if the surround networks of different BC types process visual stimuli differently. To obtain precise estimates of centre and surround spatio-temporal receptive fields, we calculated a series of linear time kernels at different distances from the BC receptive field centre by presenting a ‘ring noise’ stimulus (Fig. 5a, Extended Data Fig. 9a).

a, Schematic of ring noise stimulus (ring width, 25 μm). b, Centre–surround time kernels of an example cluster (CX) for eight rings (Methods, compare with Extended Data Fig. 9a). Dashed line at t = 0. Numbers on the left indicate distance in micrometres from stimulus centre. c, Normalized time (top) and space (bottom) kernels of CX. Space kernels are Gaussian fits of centre and surround activation, circles indicate original data points (Methods). d, Predicted centre surround ratios (CSRs) to local (top) and full-field (bottom) spot stimuli from activation of centre (light grey) and surround (dark grey). e, Normalized effective time kernels of CX for different stimulus diameters (left) and for 100 and 500 μm (right). f, Normalized spectra of predicted time kernels shown in e, right. g, Normalized spectra predicted for Off- (top) and On-clusters (middle) during local (left) and full-field (right) stimulation with mean ± s.d. shown in black and grey (bottom). h, Measured time kernels (left) and frequency spectra (right) for local and full-field spot noise stimuli (C1 and C6). i, Measured and predicted correlations of time kernels for local, full-field and surround-only stimulation (data: P < 0.001, n = 13, non-parametric paired Wilcoxon signed-rank test; model: both P < 0.001, n = 14). Black, mean ± s.d. j, Average correlation ± s.e.m. across predicted cluster time kernels for different stimulus diameters.
The temporal properties of centre and surround time kernels were consistent across ROIs assigned to the same cluster, with pronounced differences among clusters (Extended Data Figs 9b–d, 10a, d–g). In addition, the centre and surround time kernels of the same clusters were correlated with respect to width and time to peak (Extended Data Fig. 10d, e). Moreover, the spatial extent of surround kernels varied between types (Extended Data Fig. 10f; range, 255–410 μm), indicating that different amacrine cell networks are involved in different BC circuits.
We investigated how functional diversity could emerge from the differences in centre–surround properties. Using the spatial receptive fields, we first estimated the centre–surround activation ratio (CSR) for circular stimuli of different sizes (Fig. 5d, Extended Data Fig. 10h, Methods). For an example cluster (CX), a small stimulus (diameter, 100 μm) resulted in twofold stronger activation of the centre than of the surround (CSR = 2.3, ‘centre-dominant’). By contrast, the surround was stronger than the centre for a full-field spot (diameter, 500 μm; CSR = 0.8, ‘surround-dominant’). All clusters gradually switched from a centre-dominant to a surround-dominant mode for spot diameters between 200 and 600 μm (Extended Data Fig. 10h).
We used a simple model to predict how the temporal properties of BCs would change with stimulus diameter (Fig. 5e). For example, the predicted kernel of cluster CX doubled its centre frequency and temporal bandwidth with increasing stimulus size (Fig. 5f). The model predicted cluster-specific changes in the temporal coding properties with increasing stimulus size, leading to an increase in the overall diversity across clusters (Fig. 5g), with kernels for larger stimuli encompassing a broader range of temporal frequencies. To test this prediction experimentally, we recorded BC kernels for two different stimulus diameters (100 and 500 μm). In agreement with the model, time kernels differed consistently between stimulus sizes (Fig. 5h), leading to lower time kernel correlations across clusters for centre–surround compared to centre-only or surround-only stimulation (Fig. 5i; compare with Fig. 3a, Extended Data Fig. 10i, j). In addition, the model predicted that this effect would be strongest between around 200 and 500 μm (Fig. 5j), matching the distribution of receptive field centre sizes of mouse RGCs2.
Conclusions
Here, we systematically surveyed the functional diversity of mouse BCs by imaging their glutamatergic output. We have shown how the temporal diversity is created by the interplay of the excitatory drive received at the dendrites and local axonal inputs from amacrine cells. These two input streams combine at the BC axon terminal, the central computational unit of the IPL4. We have shown that the streams are of comparable strengths but act at different spatial scales. Hence, the spatial structure of the visual input sets the ratio of excitation and inhibition and thus, the temporal encoding in BCs and consequently, the visual system. It is possible that there is an even finer level of computational granularity, as individual axon terminals of a single BC could signal independently, if they received differential inputs from amacrine cells (Supplementary Discussion).
Why does the mouse retina split the visual signal into 14 parallel channels at the level of BCs? The finding that small stimuli in the range of a BC’s dendritic field evoke highly correlated responses among types with the same response polarity implies that the set of BC types is not optimized to decompose the visual signal at this scale. Instead, it is only upon spatially extended stimulation that the rich functional diversity among BC types is revealed.
Our data suggests that this diversity is generated by the interactions of correlated but not-identical pairs of temporal centre and surround receptive fields of BCs, with increasing stimulus size shifting the balance between centre and surround activation towards a stronger surround contribution. Here, different groups of amacrine cells play distinct roles in setting up and gating the antagonistic surround (Supplementary Discussion).
The notion that an antagonistic centre–surround receptive field organization can decorrelate neural response properties is a fundamental principle in neuroscience (for example, see refs 44, 45). For instance, surround-mediated decorrelation occurs in RGCs46 as well as in the visual cortex47 and other sensory systems48. However, these previous studies have focused on decorrelation between neurons independent of cell type. By contrast, less is known about the role of centre–surround receptive field organization in decreasing the redundancy of the encoding between different neuronal cell types of the same class, as demonstrated here for the parallel signal channels formed by mouse BC types. Such decorrelation is typically linked to efficient coding49 and our data place this computation as early as the second synapse of the mouse visual system.
Methods
Animals and tissue preparation
All animal procedures adhered to the laws governing animal experimentation issued by the German Government. For all experiments, we used 3- to 12-week-old C57Bl/6 (n = 3), Chattm2(cre)Lowl (n = 34; ChAT:Cre, JAX 006410, The Jackson Laboratory), and Tg(Pcp2-cre)1Amc (n = 5; Pcp2, JAX 006207) mice of either sex. The transgenic lines were cross-bred with the Cre-dependent red fluorescence reporter line Gt(ROSA)26Sortm9(CAG-tdTomato)Hze (Ai9tdTomato, JAX 007905) for a subset of experiments. Owing to the explanatory nature of our study, we did not use randomization and blinding. No statistical methods were used to predetermine sample size.
Animals were housed under a standard 12-h day–night rhythm. For recordings, animals were dark-adapted for ≥ 1 h, then anaesthetized with isoflurane (Baxter) and killed by cervical dislocation. The eyes were removed and hemisected in carboxygenated (95% O2, 5% CO2) artificial cerebral spinal fluid (ACSF) solution containing (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 20 glucose, and 0.5 l-glutamine (pH 7.4). Then, the tissue was moved to the recording chamber of the microscope, where it was continuously perfused with carboxygenated ACSF at ~37 °C. The ACSF contained ~0.1 μM sulforhodamine-101 (SR101, Invitrogen) to reveal blood vessels and any damaged cells in the red fluorescence channel. All procedures were carried out under very dim red (>650 nm) light.
Virus injection
A volume of 1 μl of the viral construct (AAV9.hSyn.iGluSnFR.WPRE.SV40 or AAV9.CAG.Flex.iGluSnFR.WPRE.SV40 (AAV9.iGluSnFR) or AAV9.Syn.Flex.GCaMP6f.WPRE.SV40, Penn Vector Core) was injected into the vitreous humour of 3- to 8-week-old mice anaesthetized with 10% ketamine (Bela-Pharm GmbH & Co. KG) and 2% xylazine (Rompun, Bayer Vital GmbH) in 0.9% NaCl (Fresenius). For the injections, we used a micromanipulator (World Precision Instruments) and a Hamilton injection system (syringe: 7634-01, needles: 207434, point style 3, length 51 mm, Hamilton Messtechnik GmbH). Owing to the fixed angle of the injection needle (15°), the virus was applied to the ventronasal retina. Imaging experiments were performed 3–4 weeks after injection.
Single cell injection
Sharp electrodes were pulled on a P-1000 micropipette puller (Sutter Instruments) with resistances >100 MΩ. Single cells in the inner nuclear layer were dye-filled with 10 mM Alexa Fluor 555 (Life Technologies) in a 200 mM potassium gluconate (Sigma-Aldrich) solution using the buzz function (50-ms pulse) of the MultiClamp 700B software (Molecular Devices). Pipettes were carefully retracted as soon as the cell began to fill. Approximately 20 min were allowed for the dye to diffuse throughout the cell before imaging started. After recording, an image stack was acquired to document the cell’s morphology, which was then traced semi-automatically using the Simple Neurite Tracer plugin implemented in Fiji (https://imagej.net/Simple_Neurite_Tracer).
Pharmacology
All drugs were bath applied for at least 10 min before recordings. The following drug concentrations were used (in μM): 10 gabazine (Tocris Bioscience)50, 75 TPMPA (Tocris Bioscience)50, 50 l-AP4 (l-(+)-2-amino-4-phosphonobutyric acid, Tocris Bioscience) and 0.5 strychnine (Sigma-Aldrich)51. Drug solutions were carboxygenated and warmed to ~37 °C before application. Pharmacological experiments were exclusively performed in the On and Off ChAT-immunoreactive bands, which are labelled in red fluorescence in ChAT:Cre × Ai9tdTomato crossbred animals.
Two-photon imaging and light stimulation
We used a MOM-type two-photon microscope (designed by W. Denk, MPI, Heidelberg; purchased from Sutter Instruments/Science Products). The design and procedures have been described previously52. In brief, the system was equipped with a mode-locked Ti:Sapphire laser (MaiTai-HP DeepSee, Newport Spectra-Physics), two fluorescence detection channels for iGluSnFR or GCaMP6f (HQ 510/84, AHF/Chroma) and SR101/tdTomato (HQ 630/60, AHF), and a water immersion objective (W Plan-Apochromat 20×/1.0 DIC M27, Zeiss). The laser was tuned to 927 nm for imaging iGluSnFR, GCaMP6f or SR101, and to 1,000 nm for imaging tdTomato. For image acquisition, we used custom-made software (ScanM by M. Müller and T.E.) running under IGOR Pro 6.3 for Windows (Wavemetrics), taking time-lapsed 64 × 16 pixel image scans (at 31.25 Hz) for glutamate and 32 × 32 pixel image scans (at 15.625 Hz) for calcium imaging. For visualizing morphology, 512 × 512 pixel images were acquired.
For light stimulation, we focused a DLP projector (K11, Acer) through the objective, fitted with band-pass-filtered light-emitting diodes (LEDs) (green, 578 BP 10; and blue, HC 405 BP 10, AHF/Croma) to match the spectral sensitivity of mouse M- and S-opsins. LEDs were synchronized with the microscope’s scan retrace. Stimulator intensity (as photoisomerization rate, 103 P* per s per cone) was calibrated as described previously52 to range from 0.6 and 0.7 (black image) to 18.8 and 20.3 for M- and S-opsins, respectively. Owing to technical limitations, intensity modulations were weakly rectified below 20% brightness. An additional, steady illumination component of ~104 P* per s per cone was present during the recordings because of two-photon excitation of photopigments (for detailed discussion, see refs 52 and 53). The light stimulus was centred before every experiment, such that its centre corresponded to the centre of the recording field. For all experiments, the tissue was kept at a constant mean stimulator intensity level for at least 15 s after the laser scanning started and before light stimuli were presented. Because the stimulus was projected though the objective lens, the stimulus projection plane shifted when focusing at different IPL levels. We therefore quantified the resulting blur of the stimulus at the level of photoreceptor outer segments. We found that a vertical shift of the imaging plane by 50 μm blurred the image only slightly (2% change in pixel width), indicating that different IPL levels (total IPL thickness = 41.6 ± 4.8 μm, mean ± s.d., n = 20 scans) can be imaged without substantial change in stimulus quality.
Four types of light stimuli were used (Fig. 1): (i) full-field (600 × 800 μm) and (ii) local (100 μm in diameter) chirp stimuli consisting of a bright step and two sinusoidal intensity modulations, one with increasing frequency (0.5–8 Hz) and one with increasing contrast; (iii) 1-Hz light flashes (500 μm in diameter, 50% duty cycle); and (iv) binary dense noise (20 × 15 matrix of 20 × 20 μm pixels; each pixel displayed an independent, balanced random sequence at 5 Hz for 5 min) for space–time receptive field mapping. In a subset of experiments, we used three additional stimuli: (v) a ring noise stimulus (10 annuli with increasing diameter, each annulus 25 μm wide), with each ring’s intensity determined independently by a balanced 68-s random sequence at 60 Hz repeated four times; (vi) a surround chirp stimulus (annulus; full-field chirp sparing the central 100 μm corresponding to the local chirp); and (vii) a spot noise stimulus (100 or 500 μm in diameter; intensity modulation like ring noise) flickering at 60 Hz. For all drug experiments, we showed in addition: (viii) a stimulus consisting of alternating 2-s full-field and local light flashes (500 and 100 μm in diameter, respectively). All stimuli were achromatic, with matched photo-isomerization rates for mouse M- and S-opsins.
Estimating recording depth in the IPL
For each scan field, we used the relative positions of the inner (ganglion cell layer) and outer (inner nuclear layer) blood vessel plexus to estimate IPL depth. To relate these blood vessel plexi to the ChAT bands, we performed separate experiments in ChAT:Cre × Ai9tdTomato mice. High-resolution stacks throughout the inner retina were recorded in the ventronasal retina. The stacks were then first corrected for warping of the IPL using custom-written scripts in IGOR Pro. In brief, a raster of markers (7 × 7) was projected in the x–y plane of the stack and for each marker the z positions of the On ChAT band were manually determined. The point raster was used to calculate a smoothed surface, which provided a z offset correction for each pixel beam in the stack. For each corrected stack, the z profiles of tdTomato and SR101 labelling were extracted by manually drawing ROIs in regions where only blood vessel plexi or the ChAT bands were visible. The two profiles were then matched such that 0 corresponded to the inner vessel peak and 1 corresponded to the outer vessel peak. We averaged the profiles of n = 9 stacks from three mice and determined the IPL depth of the On and Off ChAT bands to be 0.48 ± 0.011 and 0.77 ± 0.014 AU (mean ± s.d.), respectively. The s.d. corresponds to an error of 0.45 and 0.63 μm for the On and Off ChAT bands, respectively. In the following, recording depths relative to blood vessel plexi were transformed into IPL depths relative to ChAT bands for all scan fields (Fig. 1b), with 0 corresponding to the On ChAT band and 1 corresponding to the Off ChAT band.
Data analysis
Data analysis was performed using Matlab 2014b/2015a (Mathworks Inc.) and IGOR Pro. Data were organized in a custom written schema using the DataJoint for Matlab framework (github.com/datajoint/datajoint-matlab)54.
Pre-processing
Regions-of-interest (ROIs) were defined automatically by a custom correlation-based algorithm in IGOR Pro. First, the activity stack in response to the dense noise stimulus (64 × 16 × 10,000 pixels) was de-trended by high-pass filtering the trace of each individual pixel above ~0.1 Hz. For the 100 best-responding pixels in each recording field (highest s.d. over time), the trace of each pixel was correlated with the trace of every other pixel in the field. Then, the correlation coefficient (ρ) was plotted against the distance between the two pixels and the average across ROIs was computed (Extended Data Fig. 1a). A scan field-specific correlation threshold (ρThreshold) was determined by fitting an exponential between the smallest distance and 5 μm (Extended Data Fig. 1b). ρThreshold was defined as the correlation coefficient at λ, where λ is the exponential decay constant (space constant; Extended Data Fig. 1b). Next, we grouped neighbouring pixels with ρ > ρThreshold into one ROI (Extended Data Fig. 1c–e). To match ROI sizes with the sizes of BC axon terminals, we restricted ROI diameters (estimated as effective diameter of area-equivalent circle) to range between 0.75 and 4 μm (Extended Data Fig. 1b, g). For validation, the number of ROIs covering single axon terminals was quantified manually for n = 31 terminals from n = 5 GCaMP6-expressing BCs (Extended Data Figs 1g, 2a–c).
The glutamate (or calcium) traces for each ROI were extracted (as ΔF/F) using the image analysis toolbox SARFIA for IGOR Pro55 and resampled at 500 Hz. A stimulus time marker embedded in the recorded data served to align the traces relative to the visual stimulus with 2 ms precision. For this, the timing for each ROI was corrected for sub-frame time-offsets related to the scanning. Stimulus-aligned traces for each ROI were imported into Matlab for further analysis.
For the chirp and step stimuli, we down-sampled to 64 Hz for further processing, subtracted the baseline (median of first 20–64 samples), computed the median activity r(t) across stimulus repetitions (5 repetitions for chirp, >30 repetitions for step) and normalized it such that  .
.
For dye-injected BCs, axon terminals were labelled manually using the image analysis toolbox SARFIA for IGOR Pro. Then, ROIs were estimated as described above and assigned to the reconstructed cell, if at least two pixels overlapped with the cell´s axon terminals.
Receptive fields/ring response kernel
We mapped the receptive field from the dense noise stimulus and the response kernel to the ring noise stimulus by computing the glutamate/calcium transient-triggered average. To this end, we used Matlab’s findpeaks function to detect the times ti at which transients occurred. We set the minimum peak height to 1 s.d., where the s.d. was robustly estimated using:

We then computed the glutamate/calcium transient-triggered average stimulus, weighting each sample by the steepness of the transient:

Here,  is the stimulus, τ is the time lag and M is the number of glutamate/calcium events.
is the stimulus, τ is the time lag and M is the number of glutamate/calcium events.
For the receptive field from the dense noise stimulus, we smoothed this raw receptive field estimate using a 3 × 3-pixel Gaussian window for each time lag separately and used singular value decomposition (SVD) to extract temporal and spatial receptive field kernels. To extract the receptive field’s position and scale, we fitted it with a 2D Gaussian function using Matlab’s lsqcurvefit. Receptive field quality (QiRF) was measured as one minus the fraction of residual variance not explained by the Gaussian fit  ,
,

Other response measures
Response quality index. To measure how well a cell responded to a stimulus (local and full-field chirp, flashes), we computed the signal-to-noise ratio

where C is the T by R response matrix (time samples by stimulus repetitions), while  and
and  denote the mean and variance across the indicated dimension, respectively2.
denote the mean and variance across the indicated dimension, respectively2.
For further analysis, we used only cells that responded well to the local chirp stimulus (QiLchirp > 0.3) and resulted in good receptive fields (QiRF > 0.2).
Polarity index. To distinguish between On and Off BCs, we calculated the polarity index (POi) from the step response to local and full-field chirp, respectively, as

where b = 2 s (62 samples). For cells responding solely during the On-phase of a step of light POi = 1, while for cells only responding during the step’s Off-phase POi = −1.
Opposite polarity index. The number of opposite polarity events (OPi) was estimated from individual trials of local and full-field chirp step responses (first 6 s) using IGOR Pro’s FindPeak function. Specifically, we counted the number of events that occurred during the first 2 s after the step onset and offset for Off and On BCs, respectively. For each trial the total number of events was divided by the number of stimulus trials. If OPi = 1, there was on average one opposite polarity event per trial.
High frequency index. The high frequency index (HFi) was used to quantify spiking (compare with ref. 28) and was calculated from responses to individual trials of the local and full-field chirps. For the first 6 s of each trial, the frequency spectrum was calculated by fast Fourier transform (FFT) and spectra were averaged across trials for individual ROIs. Then, HFi = log(F1/F2), where F1 and F2 are the mean power between 0.5–1 Hz and 2–16 Hz, respectively.
Response transience index. The step response (first 6 s) of local and full-field chirps was used to calculate the response transience (RTi). Traces were up-sampled to 500 Hz and the response transience was calculated as

where α = 400 ms is the read-out time following the peak response tmax. For a transient cell with complete decay back to baseline RTi = 1, whereas for a sustained cell with no decay RTi = 0.
Response plateau index. Local and full-field chirp responses were up-sampled to 500 Hz and the plateau index (RPi) was determined as:

with the read-out time α = 2 s. A cell showing a sustained plateau has an RPi = 1, while for a transient cell RPi = 0.
Tonic release index. Local chirp frequency and contrast responses were up-sampled to 500 Hz and the baseline (response to 50% contrast step) was subtracted. Then, the glutamate traces were separated into responses above (r+) and below (r−) baseline and the tonic release index (TRi) was determined as:

For a cell with no tonic release TRi = 0, whereas for a cell with maximal tonic release TRi = 1.
Response delay. The response delay (tdelay) was defined as the time from stimulus onset/offset until response onset and was calculated from the up-sampled local chirp step response. Response onset (tonset) and delay (tdelay) were defined as  and
and  , respectively.
, respectively.
Feature extraction
We used sparse principal component analysis, as implemented in the SpaSM toolbox by K. Sjöstrang et al. (http://www2.imm.dtu.dk/projects/spasm/), to extract sparse response features from the mean responses across trials to the full-field (12 features) and local chirp (6 features), and the step stimulus (6 features) (as described in ref. 2; see Extended Data Fig. 4b). Before clustering, we standardized each feature separately across the population of cells.
Anatomy-guided clustering
BC-terminal volume profiles were obtained from electron microscopic reconstructions of the inner retina6,10. To isolate synaptic terminals, we removed those parts of the volume profiles that probably corresponded to axons. We estimated the median axon density for each type from the upper 0.06 units of the IPL and subtracted twice that estimate from the profiles, clipping at zero. Profiles were smoothed with a Gaussian kernel (s.d. = 0.14 units IPL depth) to account for jitter in depth measurements of two-photon data. For the GluMI cell, we assumed the average profile of CBC types 1 and 2.
We used a modified mixture of Gaussian model56 to incorporate the prior knowledge from the anatomical BC profiles. For each ROI i with IPL depth  , we define a prior over anatomical types c as
, we define a prior over anatomical types c as

Where IPL(d,c) is the IPL terminal density profile as a function of depth and anatomical cell type. For example, all ROIs of a scan field taken at an IPL depth of 1.7 were likely to be sorted into clusters for CBC types 1 and 2, while a scan field taken at a depth of 0 received a bias for CBC types 5–7 (Extended Data Fig. 4a).
The parameters of the mixture of Gaussian model are estimated as usual, with the exception of estimating the posterior over clusters. Here, the mixing coefficients are replaced by the prior over anatomical types, resulting in a modified update formula for the posterior:

All other updates remain the same as for the standard mixture of Gaussians algorithm57. We constrained the covariance matrix for each component to be diagonal, resulting in 48 parameters per component (24 for the mean, 24 for the variances). We further regularized the covariance matrix by adding a constant (10−5) to the diagonal.
The clustering was based on a subset (~83%) of the data (the first 11,101 recorded cells). The remaining ROIs were then automatically allocated to the established clustering (n = 2,210 ROIs).
For each pair of clusters, we computed the direction in feature space that optimally separated the clusters  , where
, where  are the cluster means in feature space and
are the cluster means in feature space and  is the pooled covariance matrix. We projected all data on this axis and standardized the projected data according to cluster 1 (that is, subtracted the projected mean of cluster 1 and divided by its s.d.). We computed d′ as a measure of the separation between the clusters:
is the pooled covariance matrix. We projected all data on this axis and standardized the projected data according to cluster 1 (that is, subtracted the projected mean of cluster 1 and divided by its s.d.). We computed d′ as a measure of the separation between the clusters:  , where
, where  are the means of the two clusters in the projected, normalized space.
are the means of the two clusters in the projected, normalized space.
We also performed a more constrained clustering in which we divided the IPL into five portions without overlap based on stratification profiles. We then clustered each zone independently using a standard mixture of Gaussian approach and a cluster number determined by the number of BC types expected in each portion. The correlation between the cluster means of our clustering and the more constrained clustering was 0.97 for the full-field chirp traces, indicating high agreement.
Further statistical analysis
Field entropy. Field entropy (SField) was used as a measure of cluster heterogeneity within single recording fields and was defined as  , where i is the number of clusters in one recording field and pi corresponds to the number of ROIs assigned to the ith cluster. SField = 0 if all ROIs of one recording field are assigned to one cluster and SField increases if ROIs are equally distributed across multiple clusters. In general, high field-entropy indicates high cluster heterogeneity within a single field.
, where i is the number of clusters in one recording field and pi corresponds to the number of ROIs assigned to the ith cluster. SField = 0 if all ROIs of one recording field are assigned to one cluster and SField increases if ROIs are equally distributed across multiple clusters. In general, high field-entropy indicates high cluster heterogeneity within a single field.
Analysis of response diversity. To investigate the similarity of local and full-field chirp responses across clusters (Fig. 3), we determined the linear correlation coefficient between any two cluster pairs. The analysis was performed on cluster means. For every cluster, correlation coefficients were averaged across clusters with the same and opposite response polarity, respectively. We used principal component analysis (using Matlab’s pca function) to obtain a 2D embedding of the mean cluster responses. The principal component analysis was computed on all 14 local and 14 full-field cluster means. If not stated otherwise, the non-parametric Wilcoxon signed-rank test was used for statistical testing.
Pharmacology. To analyse drug-induced effects on BC clusters (Fig. 4, Extended Data Figs 7, 8), response traces and receptive fields of ROIs in one recording field belonging to the same cluster were averaged if there were at least 5 ROIs assigned to this cluster. Spatial receptive fields were aligned relative to the pixel with the highest s.d. before averaging.
Centre-surround properties. To estimate the signal-to-noise ratio of ring maps of single ROIs, we extracted temporal centre and surround kernels and normalized the respective kernel to the s.d. of its baseline (first 50 samples). For further analysis, we included only ROIs with |PeakCentre| > 12 s.d. and |PeakSurround| > 7 s.d. Ring maps of individual ROIs were then aligned relative to its peak centre activation and averaged across ROIs assigned to one cluster. To isolate the BC surround, the centre rings (first two rings) were cut and the surround time and space components were extracted by singular value decomposition (SVD). The surround space component was then extrapolated across the centre by fitting a Gaussian and an extrapolated surround map was generated. To isolate the BC centre, the estimated surround map was subtracted from the average map and centre time and space components were extracted by SVD. The estimated centre and surround maps were summed to obtain a complete description of the centre–surround structure of BC receptive fields. Across clusters, the estimated centre–surround maps captured 92.5 ± 1.9% of the variance of the original map. Owing to the low signal-to-noise ratio, the temporal centre–surround properties of individual ROIs were extracted as described above using the centre and surround space kernels obtained from the respective cluster average.
The 1D Gaussian fits of centre and surround space activation were used to calculate centre and surround ratios (CSRs) for various stimulus sizes. Specifically, the CSR was defined as

where Sr corresponds to the stimulus radius and ranged from 10 to 500 μm, with a step size dx of 1 μm. Time kernels for different stimulus sizes were generated by linearly mixing centre and surround time kernels, weighted by the respective CSR.
BC spectra. The temporal spectra of BC clusters were calculated by Fourier transform of the time kernels estimated for a local (100 μm in diameter) and full-field (500 μm in diameter) light stimulus (see centre–surround properties). Owing to the lower SNR of time kernels estimated for the full-field stimulus, kernels were cut 100 ms before and at the time point of response, still capturing 86.7 ± 14.7% of the variance of the original kernel. The centre of mass (Centroid) was used to characterize spectra of different stimulus sizes and was determined as

where x(n) corresponds to the magnitude and f(n) represents the centre frequency of the nth bin.
Surround chirp and spot noise data. To investigate the effects of surround-only activation and stimulus size on temporal encoding properties across BC clusters, response traces and estimated kernels of ROIs in one recording field belonging to the same cluster were averaged if there were at least five ROIs assigned to this cluster. The spectra for kernels estimated from local and full-field spot noise stimuli were calculated as described above.
Time kernel correlation. To analyse the similarity of temporal kernels estimated for a specific stimulus size (Fig. 5i, j), we computed the linear correlation coefficient of each kernel pair from clusters with the same response polarity. We then calculated the average correlation coefficient for every cluster (Fig. 5i) and across all cluster averages (Fig. 5j).
Code and data availability
Data (original data and clustering results) as well as Matlab code are available from http://www.retinal-functomics.org.
References
Masland, R. H. The neuronal organization of the retina. Neuron 76, 266–280 (2012)
Baden, T. et al. The functional diversity of retinal ganglion cells in the mouse. Nature 529, 345–350 (2016)
Sanes, J. R. & Masland, R. H. The types of retinal ganglion cells: current status and implications for neuronal classification. Annu. Rev. Neurosci. 38, 221–246 (2015)
Euler, T., Haverkamp, S., Schubert, T. & Baden, T. Retinal bipolar cells: elementary building blocks of vision. Nat. Rev. Neurosci. 15, 507–519 (2014)
Helmstaedter, M. et al. Connectomic reconstruction of the inner plexiform layer in the mouse retina. Nature 500, 168–174 (2013)
Kim, J. S. et al. Space–time wiring specificity supports direction selectivity in the retina. Nature 509, 331–336 (2014)
Behrens, C., Schubert, T., Haverkamp, S., Euler, T. & Berens, P. Connectivity map of bipolar cells and photoreceptors in the mouse retina. eLife 5, e20041 (2016)
Wässle, H., Puller, C., Müller, F. & Haverkamp, S. Cone contacts, mosaics, and territories of bipolar cells in the mouse retina. J. Neurosci. 29, 106–117 (2009)
Shekhar, K. et al. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell 166, 1308–1323 (2016)
Greene, M. J., Kim, J. S. & Seung, H. S. Analogous convergence of sustained and transient inputs in parallel on and off pathways for retinal motion computation. Cell Reports 14, 1892–1900 (2016)
Awatramani, G. B. & Slaughter, M. M. Origin of transient and sustained responses in ganglion cells of the retina. J. Neurosci. 20, 7087–7095 (2000)
Breuninger, T., Puller, C., Haverkamp, S. & Euler, T. Chromatic bipolar cell pathways in the mouse retina. J. Neurosci. 31, 6504–6517 (2011)
Euler, T., Schneider, H. & Wässle, H. Glutamate responses of bipolar cells in a slice preparation of the rat retina. J. Neurosci. 16, 2934–2944 (1996)
DeVries, S. H. Bipolar cells use kainate and AMPA receptors to filter visual information into separate channels. Neuron 28, 847–856 (2000)
DeVries, S. H., Li, W. & Saszik, S. Parallel processing in two transmitter microenvironments at the cone photoreceptor synapse. Neuron 50, 735–748 (2006)
Lindstrom, S. H., Ryan, D. G., Shi, J. & DeVries, S. H. Kainate receptor subunit diversity underlying response diversity in retinal off bipolar cells. J. Physiol. (Lond.) 592, 1457–1477 (2014)
Puller, C., Ivanova, E., Euler, T., Haverkamp, S. & Schubert, T. OFF bipolar cells express distinct types of dendritic glutamate receptors in the mouse retina. Neuroscience 243, 136–148 (2013)
Puthussery, T. et al. Kainate receptors mediate synaptic input to transient and sustained OFF visual pathways in primate retina. J. Neurosci. 34, 7611–7621 (2014)
Masland, R. H. The tasks of amacrine cells. Vis. Neurosci. 29, 3–9 (2012)
Eggers, E. D. & Lukasiewicz, P. D. Multiple pathways of inhibition shape bipolar cell responses in the retina. Vis. Neurosci. 28, 95–108 (2011)
Grimes, W. N., Li, W., Chávez, A. E. & Diamond, J. S. BK channels modulate pre- and postsynaptic signaling at reciprocal synapses in retina. Nat. Neurosci. 12, 585–592 (2009)
Demb, J. B. & Singer, J. H. Intrinsic properties and functional circuitry of the AII amacrine cell. Vis. Neurosci. 29, 51–60 (2012)
Marvin, J. S. et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 10, 162–170 (2013)
Borghuis, B. G., Marvin, J. S., Looger, L. L. & Demb, J. B. Two-photon imaging of nonlinear glutamate release dynamics at bipolar cell synapses in the mouse retina. J. Neurosci. 33, 10972–10985 (2013)
Chen, M., Lee, S., Park, S. J. H., Looger, L. L. & Zhou, Z. J. Receptive field properties of bipolar cell axon terminals in direction-selective sublaminas of the mouse retina. J. Neurophysiol. 112, 1950–1962 (2014)
Rosa, J. M., Ruehle, S., Ding, H. & Lagnado, L. Crossover inhibition generates sustained visual responses in the inner retina. Neuron 90, 308–319 (2016)
Dreosti, E., Esposti, F., Baden, T. & Lagnado, L. In vivo evidence that retinal bipolar cells generate spikes modulated by light. Nat. Neurosci. 14, 951–952 (2011)
Baden, T., Berens, P., Bethge, M. & Euler, T. Spikes in mammalian bipolar cells support temporal layering of the inner retina. Curr. Biol. 23, 48–52 (2013)
Burrone, J. & Lagnado, L. Synaptic depression and the kinetics of exocytosis in retinal bipolar cells. J. Neurosci. 20, 568–578 (2000)
Nikolaev, A., Leung, K.-M., Odermatt, B. & Lagnado, L. Synaptic mechanisms of adaptation and sensitization in the retina. Nat. Neurosci. 16, 934–941 (2013)
Chen, T.-W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013)
Werblin, F. S. & Dowling, J. E. Organization of the retina of the mudpuppy, Necturus maculosus. II. Intracellular recording. J. Neurophysiol. 32, 339–355 (1969)
Roska, B. & Werblin, F. Vertical interactions across ten parallel, stacked representations in the mammalian retina. Nature 410, 583–587 (2001)
Lukasiewicz, P. D. & Werblin, F. S. A novel GABA receptor modulates synaptic transmission from bipolar to ganglion and amacrine cells in the tiger salamander retina. J. Neurosci. 14, 1213–1223 (1994)
Euler, T. & Masland, R. H. Light-evoked responses of bipolar cells in a mammalian retina. J. Neurophysiol. 83, 1817–1829 (2000)
Euler, T. & Wässle, H. Different contributions of GABAA and GABAC receptors to rod and cone bipolar cells in a rat retinal slice preparation. J. Neurophysiol. 79, 1384–1395 (1998)
Vaney, D. I. The mosaic of amacrine cells in the mammalian retina. Prog. Retinal Res. 9, 49–100 (1990)
Eggers, E. D. & Lukasiewicz, P. D. Interneuron circuits tune inhibition in retinal bipolar cells. J. Neurophysiol. 103, 25–37 (2010)
Roska, B., Nemeth, E., Orzo, L. & Werblin, F. S. Three levels of lateral inhibition: A space–time study of the retina of the tiger salamander. J. Neurosci. 20, 1941–1951 (2000)
Ichinose, T. & Lukasiewicz, P. D. Inner and outer retinal pathways both contribute to surround inhibition of salamander ganglion cells. J. Physiol. (Lond.) 565, 517–535 (2005)
Buldyrev, I. & Taylor, W. R. Inhibitory mechanisms that generate centre and surround properties in ON and OFF brisk-sustained ganglion cells in the rabbit retina. J. Physiol. (Lond.) 591, 303–325 (2013)
Liang, Z. & Freed, M. A. The ON pathway rectifies the OFF pathway of the mammalian retina. J. Neurosci. 30, 5533–5543 (2010)
Zaghloul, K. A., Boahen, K. & Demb, J. B. Different circuits for ON and OFF retinal ganglion cells cause different contrast sensitivities. J. Neurosci. 23, 2645–2654 (2003)
Atick, J. J. & Redlich, A. N. Towards a theory of early visual processing. Neural Comput. 320, 1–13 (1990)
Barlow, H. Possible principles underlying the transformations of sensory messages. Sensory Comm. 6, 57–58 (1961)
Pitkow, X. & Meister, M. Decorrelation and efficient coding by retinal ganglion cells. Nat. Neurosci. 15, 628–635 (2012)
Vinje, W. E. & Gallant, J. L. Sparse coding and decorrelation in primary visual cortex during natural vision. Science 1273, 1273–1276 (2007)
Giridhar, S., Doiron, B. & Urban, N. N. Timescale-dependent shaping of the correlation by olfactory bulb lateral inhibition. Proc. Natl Acad. Sci. USA 108, 5843–5848 (2011)
Denève, S. & Machens, C. K. Efficient codes and balanced networks. Nat. Neurosci. 19, 375–382 (2016)
Kemmler, R., Schultz, K., Dedek, K., Euler, T. & Schubert, T. Differential regulation of cone calcium signals by different horizontal cell feedback mechanisms in the mouse retina. J. Neurosci. 34, 11826–11843 (2014)
Schubert, T. et al. Development of presynaptic inhibition onto retinal bipolar cell axon terminals is subclass-specific. J. Neurophysiol. 100, 304–316 (2008)
Euler, T. et al. Eyecup scope—optical recordings of light stimulus-evoked fluorescence signals in the retina. Pflugers Arch. 457, 1393–1414 (2009)
Baden, T. et al. A tale of two retinal domains: near-optimal sampling of achromatic contrasts in natural scenes through asymmetric photoreceptor distribution. Neuron 80, 1206–1217 (2013)
Yatsenko, D. et al. DataJoint: managing big scientific data using MATLAB or Python. Preprint at http://biorxiv.org/content/early/2015/11/14/031658 (2015)
Dorostkar, M. M., Dreosti, E., Odermatt, B. & Lagnado, L. Computational processing of optical measurements of neuronal and synaptic activity in networks. J. Neurosci. Methods 188, 141–150 (2010)
Szczurek, E., Biecek, P., Tiuryn, J. & Vingron, M. Introducing knowledge into differential expression analysis. J. Comput. Biol. 17, 953–967 (2010)
Bishop, C. M. Pattern Recognition and Machine Learning (Springer, 2006)
Acknowledgements
We thank G. Eske for technical support, C. Behrens for help with EM data, J. Jüttner for help with virus injection; X. Pitkow and R. Taylor for discussions; and L. L. Looger, the Janelia Research Campus of the Howard Hughes Medical Institute and the Genetically-Encoded Neuronal Indicator and Effector (GENIE) Project for making the viral constructs (AAV9.hSyn.iGluSnFR.WPRE.SV40, AAV9.CAG.Flex.iGluSnFR.WPRE.SV40 and AAV9.Syn.Flex.GCaMP6f.WPRE.SV40) publically available. This work was supported by the Deutsche Forschungsgemeinschaft (DFG; EXC307 to T.E.; BA 5283/1-1 to T.B.; BE 5601/1-1 to P.B.), the German Federal Ministry of Education and Research (BMBF; FKZ 01GQ1002 to M.B. and T.E.; FKZ 01GQ1601 to P.B.), the BW-Stiftung (AZ 1.16101.09 to T.B.), the intramural fortüne program of the University of Tübingen (2125-0-0 to T.B.), the European Commission (H2020 ERC-StG 677687 ‘NeuroVisEco’ to T.B.), the National Institute of Neurological Disorders and Stroke (U01NS090562 to T.E.) and the National Eye Institute (1R01EY023766 to T.E.) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Author information
Authors and Affiliations
Contributions
K.F., P.B., T.E. and T.B. designed the study; K.F. performed experiments and pre-processing; K.F. performed viral injections with help from T.S.; P.B. developed clustering with input from M.B.; K.F., P.B. and T.B. analysed the data with input from T.E.; K.F., P.B., T.E. and T.B. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks J. Diamond and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Figure 1 ROI detection.
a, Mean correlation (±s.d. shading, n = 100 pixels) between noise-response traces of two individual pixels from scan field shown in Fig. 1c plotted against the distance of each pixel pair. Dotted line shows linear fit to the data above x = 10 μm and its extrapolation towards x = 0 μm. Exponential fit for distances <5 μm in orange. b, Zoomed-in version of a. The space constant obtained from the exponential fit (orange) was used to determine a scan field’s specific correlation minimum for ROI detection. Blue shading indicates the range of allowed ROI radii (0.375–2 μm). c, Scan field from Fig. 1c, with each pixel colour-coded by its correlation with the noise trace of pixel indicated in white. d, As c, with the red shading corresponding to pixels with a correlation coefficient greater than the correlation minimum from b, resulting in the red ROI in e. f, As a, averaged for n = 71 scan fields recorded at 48 × 12 μm. g, Histogram of ROI (black) and BC axon terminal (red) areas. The terminal area was determined from BC axonal arborizations labelled in GCaMP6-injected Pcp2 mice where individual axon terminals can be distinguished (see Extended Data Fig. 2a). Inset shows a histogram of the number of ROIs per BC axon terminal. h, Distribution of ROI numbers per scan field. i, Histogram illustrating sampling of ROIs against IPL depth. Data collected specifically for drug experiments (Methods) contributed to the ‘oversampling’ of the two ChAT bands.
Extended Data Figure 2 GCaMP6 signals in mouse BC axon terminals.
a–c, High-resolution scan of GCaMP6f-expressing BC axon terminal systems in the IPL of a Pcp2 mouse (a) and corresponding scan field (b) with automatically generated ROI mask overlaid (c, see Extended Data Fig. 1). Our ROI algorithm reliably detected individual axon terminals and preferably assigned two ROIs to a single terminal before merging two terminals into one ROI (compare with Extended Data Fig. 1g). d, Exemplary mean local-chirp responses of individual ROIs shown in c of the two different BCs shown in a. e, Scan field from c with ROI mask and spatial receptive fields (2 s.d. outlines of Gaussian fit) overlaid. f, Distribution of receptive field diameters estimated from On BC terminal calcium (GCaMP6) and glutamate release (iGluSnFR). Black dots correspond to mean receptive field diameters. Receptive field sizes estimated from calcium signals of single terminals closely fit those estimated from single iGluSnFR ROIs (56.1 ± 10 μm for GCaMP6 and 57.5 ± 10.6 μm for iGluSnFr; P > 0.05, n = 261 (GCaMP6) and n = 3,540, non-parametric non-paired Wilcoxon signed-rank test) and matched the anatomical dimensions of BC dendritic fields7,8. The findings shown here suggest that each ROI probably captured the light-driven glutamate signal of at most one individual BC axon terminal. g, h, High-resolution scan of GCaMP6-expressing RBC axon terminals (g) and corresponding scan field (h), with two individual terminals indicated. i, j, Mean responses of RBC terminals shown in h to full-field flashes (i, n = 20 trials) and local chirp stimulus (j, n = 5 trials).
Extended Data Figure 3 Alternative clustering including the glutamatergic monopolar interneuron (GluMI).
a, b, Heat maps of local chirp responses of C1, C2 and CGluMI (a) and their respective cluster means (b). Superimposed cluster means (b, bottom) illustrate response similarity between clusters. c, d, As in a and b, respectively, but for full-field responses.
Extended Data Figure 4 Clustering.
a, Exemplary distributions of prior probabilities for cluster allocation (right) taken from mean stratification profiles (left) of scan fields recorded at two different IPL depths (A: 1.7; B: 0). b, Temporal features extracted from glutamate traces in response to local (n = 6 features, top) and full-field (n = 12 features, middle) chirp and full-field flashes (n = 6 features, bottom). c, Heat maps of all recorded glutamate responses (n = 14 clusters plus ROIs discarded based on signal-to-noise (S/N) ratio) to the four visual stimuli (compare with Fig. 1); n = 11,101 ROIs from 29 retinas. Each line corresponds to the responses of individual ROIs with activity colour-coded. Block height represents the number of included ROIs per cluster. Within one cluster, ROIs are sorted on the basis of the quality of their receptive fields and local chirp response (Methods). Overlaid stratification profiles (left) illustrate overlap for some BC types. d, e, Cluster separation was determined for every cluster pair using the sensitivity index d′. Dotted lines in d illustrate transition between On and Off clusters and dotted line in e at d′ = 2, which corresponds to about 15% false positive/false negative rates. f, Separation of exemplary cluster pairs with a low (d′ = 2.0, top) and an average (d′ = 4.5, bottom) sensitivity index. g, Distribution of field entropies (left, Methods). Two exemplary scan fields (right) with ROIs colour-coded by cluster allocation illustrate low (1) and high (2) field entropy, respectively.
Extended Data Figure 5 Anatomical verification of clustering approach.
a, High-resolution scan of the axon terminal system of a filled BC (middle), with the iGluSnFR staining overlaid in green (top) and the corresponding scan field (bottom). b, Colour-coded ROI mask (cyan), with ROIs assigned to the labelled axon terminal system in red (top; Methods). Cluster allocations of all ROIs, and ROIs assigned to the labelled cell that passed the quality criterion, are shown in the middle and bottom panel, respectively. c, Reconstructed BC and its smoothed stratification profile (black), with the profile of the BC underlying the assigned functional cluster (CBC2) overlaid (red). d, Receptive field outlines of ROIs with QiRF > 0.4 allocated to one reconstructed cell. e, Mean local (top) and full-field chirp (bottom) responses of all ROIs assigned to the labelled cell (black), with s.d. shading (grey) and cluster mean of the assigned functional cluster (C2, red). f–j, As in a–e; k–n, as in b–e. o, Box plots illustrate the fraction of ROIs from one cell that were assigned to the same cluster for all reconstructed BCs (n = 8 cells, n = 3 mice). For the plot in the bottom, functionally very similar clusters (C3a and C3b; C5o, C5i and C5t) were merged. Owing to lower signal-to-noise ratios and a slower sampling rate (15.625 Hz vs. 31.25 Hz) for these single-cell data compared to all other data, the fraction of ‘correctly’ assigned ROIs is likely to be underestimated.
Extended Data Figure 6 Functional organization of the IPL.
a, Glutamate responses to the local chirp step stimulus (first 8 s) of single ROIs assigned to C2 and C9. Shown are responses from three trials and a histogram of response amplitudes across each cluster’s 100 best-responding ROIs. On responses in Off BC cluster C2 are highlighted (arrows). b, Percentage of ROIs with at least one opposite polarity event in response to the local chirp step response for On and Off BC clusters. Dotted line illustrates mean incidence of spontaneous events (<1%). c, Time to peak of On events observed in the Off layer (grey) and On responses in On layer (red). Events were estimated from responses to single trials of the local chirp stimulus. Owing to the variability in timing, On events are not evident in traces averaged across the population of ROIs for each cluster (Fig. 2d). d, As in a, showing ‘spiking’ (C3b, CX) and non-spiking (C1, CR) responses in clusters of either polarity. e, f, Mean spectra (n = 5 trials) of local chirp step responses for two Off (C3b and C1) and two On (CX and CR) ROIs shown in d, with HFi estimated from the relative power of low (0.5–1 Hz) and high (2–16 Hz) frequencies (Methods). g, Response measures estimated from local chirp responses for all ROIs plotted against IPL depth (see Fig. 2e–h). h, Spatial receptive fields of individual ROIs assigned to C3a and C6. i, Mean response transience of BC clusters is not correlated with mean response delay (r = 0.01, P > 0.05, n = 14, linear correlation). j, Mean HFi and mean response delay are not correlated across BC clusters (r = 0.3, P > 0.05, n = 14, linear correlation). k, Mean chirp responses of two Off (C2 and C3b) clusters, with linear correlation coefficient (ρ) of whole trace or contrast ramp indicated. l, Cluster means of local (left) and full-field (right) chirp responses embedded in 2D feature space based on first and second principal components (PC). m, Response measures estimated from full-field chirp responses for all ROIs plotted against IPL depth (compare with Fig. 2e–h).
Extended Data Figure 7 GABA and glycinergic inhibition differentially shapes BC responses.
a, Mean responses (n = 5 trials) of individual ROIs to alternating local and full-field flashes under control conditions and with GABA (top) or glycine (bottom) receptor block. b, Change in mean peak amplitudes (±s.d. shading) of n = 15 ROIs originating from two scan fields during wash-in of GABA and glycine receptor blockers. c, Drug-induced changes in peak response amplitude across different BC clusters upon blocking GABA (top; P < 0.001, n = 9 clusters from 5 scan fields and 4 mice, non-parametric paired Wilcoxon signed-rank test) and glycine receptors (bottom; P < 0.001, n = 6 clusters from 5 scan fields and 4 mice). Mean ± s.d. in black. d, e, Local (grey) and full-field (black) chirp responses for control and drug conditions (d, GABA receptor block; e, glycine receptor block), with linear correlation coefficient (ρ) between each pair indicated. f, Local (grey) and surround (black) chirp responses for an exemplary On (C7, top) and Off (C3a, bottom) BC cluster. g, h, Spatial receptive fields with 2 s.d. outline of Gaussian fit shown in red (left) and quantification of changes in receptive field diameter across different BC clusters (right) upon blocking GABA (g; P < 0.05, n = 6 cluster from 3 scan fields and 2 mice, non-parametric paired Wilcoxon signed-rank test) and glycine receptors (h; P > 0.05, n = 5 cluster from 3 scan fields and 2 mice).
Extended Data Figure 8 Glycine-mediated crossover inhibition from the On pathway rectifies Off BCs.
a, Local chirp traces of an exemplary Off BC cluster (C3a) during control condition (left) and glycine receptor block (right). Magnified traces in the bottom illustrate an increase in tonic release upon drug application, with respective tonic release indices (TRi, Methods) indicated. Dotted line corresponds to baseline. Schematic (top right) illustrates effect of drug on inner retinal network. b, Quantification of drug-induced changes in tonic release across different Off BC clusters (P < 0.001, n = 5 clusters from 5 scan fields and 4 mice, non-parametric paired Wilcoxon signed-rank test). c, d, Local chirp responses of an exemplary On (c, C6) and Off (d, C2) BC cluster for control condition (left) and upon blocking the On BC pathway using l-AP4 (right). e, Tonic release indices of different Off BC clusters for control and l-AP4 conditions (P < 0.001, n = 5 clusters from 3 scan fields and 2 mice, non-parametric paired Wilcoxon signed-rank test). f, Average local chirp frequency and contrast responses of an exemplary Off (C1, top) and On (CX, bottom) BC cluster illustrate rectification of Off BC responses. g, Comparison of tonic release indices of On and Off BC clusters under control condition and Off BCs upon blocking crossover inhibition with l-AP4 or strychnine (*P < 0.05, **P < 0.01, non-parametric Kruskal–Wallis test).
Extended Data Figure 9 Extraction of BC centre–surround receptive fields.
a, Centre–surround maps obtained from the ring noise were averaged across ROIs assigned to one cluster (1). To isolate the surround component, the innermost two rings, which contained the majority of the centre component, were clipped (2) and the surround map estimated by SVD (3) was extrapolated across the centre by fitting a Gaussian (4). Next, the extrapolated surround map was subtracted from the average map and the centre component was extracted using SVD (6). The resultant centre–surround map was then subtracted from the average map to estimate the residual variance (7, Methods). Dotted lines at t = 0. b, Centre–surround maps of three ROIs assigned to CX from three independent experiments (top; bottom, left). Overlaid centre and surround time kernels (bottom, right) of ROIs (grey) and cluster mean (black) illustrate temporal precision and reproducibility. c, d, Heat maps of all centre (C) and surround (S) time kernels and cluster means ± s.d. for Off (c) and On (d) BC clusters.
Extended Data Figure 10 Centre–surround receptive fields of BC clusters.
a, Normalized time (top) and space (bottom) kernels of all On and Off BC clusters. Space kernels represent extrapolated Gaussian fits of centre and surround activation across rings shown in Fig. 5c, with circles corresponding to the original data points (Methods). b, c, Effect of GABA (b) and glycine (c) receptor block on centre–surround receptive fields of two exemplary BC clusters. Centre–surround maps correspond to averages of n > 5 ROIs of one scan field. d, Time to peak of centre time kernels were correlated with time to peak of surround time kernels (cluster mean ± s.e.m.; r = 0.91, P < 0.001, n = 14, linear correlation) and centre time kernel peaks preceded surround time kernel peaks, consistent with at least two additional synapses in the inhibitory pathway. Black line corresponds to linear fit and for the dashed line the slope is equal to 1. e, Half-maximal widths (cluster mean ± s.e.m.) of centre and surround time kernels were correlated (r = 0.83, P < 0.001, n = 14, linear correlation) and centre time kernel widths were consistently broader than surround time kernel widths. f, Half-maximal width of surround space kernels did not correlate with mean cluster IPL depth (r = 0.06, P > 0.05, n = 14, linear correlation). g, Time to peak of centre time kernels correlated with response delay estimated from local chirp step responses (mean ± s.e.m.; r = 0.82, P < 0.001, n = 14, linear correlation), indicating that the centre kernels adequately reflect BC responses to local stimuli. h, Predicted CSRs of BC clusters for different stimulus diameters (compare with Fig. 5d). i, j, Normalized temporal kernels predicted for local (100 μm diameter), full-field (500 μm diameter) and surround-only (500 and 100 μm outer and inner diameter, respectively) stimulation for Off (i) and On (j) BC clusters (compare with Fig. 5e).
Supplementary information
Supplementary Information
This file contains a Supplementary Discussion and additional references. (PDF 296 kb)
Glutamate responses in the IPL
Background-subtracted and colour-coded glutamate signals recorded in the On ChAT band (64x16 pixels @31.25 Hz), where yellow corresponds to higher activity. Responses to the noise stimulus (exemplary 30 s, single trial) and local chirp (mean across n=5 trials) are shown. Scan field corresponds to the one shown in (Fig. 1c). Video in real-time. (MP4 29084 kb)
Rights and permissions
About this article

Cite this article
Franke, K., Berens, P., Schubert, T. et al. Inhibition decorrelates visual feature representations in the inner retina. Nature 542, 439–444 (2017). https://doi.org/10.1038/nature21394
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature21394
- Springer Nature Limited
This article is cited by
-
Ancient origin of the rod bipolar cell pathway in the vertebrate retina
Nature Ecology & Evolution (2024)
-
A presynaptic source drives differing levels of surround suppression in two mouse retinal ganglion cell types
Nature Communications (2024)
-
Distributed feature representations of natural stimuli across parallel retinal pathways
Nature Communications (2024)
-
Ancestral photoreceptor diversity as the basis of visual behaviour
Nature Ecology & Evolution (2024)
-
Heterogeneity of synaptic connectivity in the fly visual system
Nature Communications (2024)