

Abstract
Mitochondrial complex I (also known as NADH:ubiquinone oxidoreductase) contributes to cellular energy production by transferring electrons from NADH to ubiquinone coupled to proton translocation across the membrane1,2. It is the largest protein assembly of the respiratory chain with a total mass of 970 kilodaltons3. Here we present a nearly complete atomic structure of ovine (Ovis aries) mitochondrial complex I at 3.9 Å resolution, solved by cryo-electron microscopy with cross-linking and mass-spectrometry mapping experiments. All 14 conserved core subunits and 31 mitochondria-specific supernumerary subunits are resolved within the L-shaped molecule. The hydrophilic matrix arm comprises flavin mononucleotide and 8 iron–sulfur clusters involved in electron transfer, and the membrane arm contains 78 transmembrane helices, mostly contributed by antiporter-like subunits involved in proton translocation. Supernumerary subunits form an interlinked, stabilizing shell around the conserved core. Tightly bound lipids (including cardiolipins) further stabilize interactions between the hydrophobic subunits. Subunits with possible regulatory roles contain additional cofactors, NADPH and two phosphopantetheine molecules, which are shown to be involved in inter-subunit interactions. We observe two different conformations of the complex, which may be related to the conformationally driven coupling mechanism and to the active–deactive transition of the enzyme. Our structure provides insight into the mechanism, assembly, maturation and dysfunction of mitochondrial complex I, and allows detailed molecular analysis of disease-causing mutations.
Similar content being viewed by others


Main
The electrochemical proton gradient across the inner mitochondrial membrane required by ATP synthase is maintained by the electron transport chain proton-pumping complexes I, III and IV (refs 1, 2). Complex I is crucial for the entire process, and even mild complex I deficiencies can cause severe pathologies4. Mammalian complex I is built of 45 (44 unique) subunits. Fourteen ‘core’ subunits, conserved from bacteria, comprise the ‘minimal’ form of the enzyme1,5, an L-shaped structure with seven subunits in the hydrophilic peripheral arm and another seven in the membrane arm. Mammalian complex I also contains 31 ‘supernumerary’ or ‘accessory’ subunits5, forming a shell around the core6. The role of these subunits is unclear. Complex I probably translocates four protons for every two electrons transferred from NADH to ubiquinone7,8.
Complex I is the least characterized enzyme of the electron transport chain. The crystal structure of bacterial (Thermus thermophilus) complex I is the only full atomic model of the enzyme9,10,11. In a later structure of the mitochondrial enzyme from aerobic yeast Yarrowia lipolytica, the atomic model comprises only about 25% of the protein12. Studies of bovine complex I resulted in poly-alanine models for the core and 22 supernumerary subunits6,13. Here we present the nearly complete atomic structure of mammalian complex I, containing all subunits and all known cofactors.
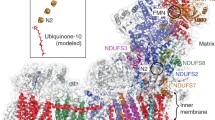
We used the ovine (O. aries) enzyme (Methods). Classification of cryo-electron microscopy (cryo-EM) images indicated that the relative orientation between the two arms of the complex is variable, producing classes with either an ‘open’ or ‘closed’ angle between them (Extended Data Fig. 1). Particles in the ‘open’ conformation produced a higher resolution map at ~3.9 Å (Extended Data Fig. 2). The resolution drops at the periphery of the molecule (Extended Data Fig. 3), owing to remaining differences in conformation. Therefore, we performed 3D refinements focusing on the peripheral arm and membrane arm separately, resulting in more uniformly resolved maps for the peripheral arm at 3.9 Å resolution and for the membrane arm at 4.1 Å (Extended Data Fig. 3). The best maps were combined for model building (Fig. 1a, Extended Data Figs 3d and 4).

a, Cryo-EM density coloured by subunit, with core subunits in grey (left–right view). b, Structure depicted as a cartoon, with core subunits coloured and labelled, and supernumerary subunits in grey and transparent. Approximate lipid bilayer boundaries are indicated. c, Structure depicted with core subunits in grey and supernumerary subunits coloured and labelled (left–right, IMS–matrix views). Amphipathic helices at the ‘heel’ of the complex, probably attached to the lipid bilayer, are indicated as AH.
Modelling of the core subunits was facilitated by the conservation of their fold from bacteria6,9. The assignment of the 31 supernumerary subunits (~0.5 MDa) to the remaining density is challenging. To provide experimental verification for previous assignments, to locate remaining subunits and to obtain restraints on the fold of individual subunits, we performed extensive cross-linking/mass-spectrometry mapping experiments (Extended Data Fig. 5, Supplementary Tables). The initial structure was improved by density-guided re-building in Rosetta14, resulting in final model of high quality (Extended Data Fig. 2c).
While this manuscript was under review, a 4.2 Å resolution cryo-EM model for the bovine complex I was published15. The assignments of all subunits agree with our structure, and two major conformations of the complex (somewhat different from ovine) are also observed. However, owing to lower resolution, the completeness of the atomic model is low for the supernumerary subunits (73% of residues are without side chains) and for the core 51-kDa, 24-kDa and 75-kDa subunits (extended data tables 1 and 2 in ref. 15).
In our ovine structure (Fig. 1), subunits were built almost entirely as atomic models with only some surface-exposed loops missing. Subunit B14.7 is disordered, so this area was modelled as poly-alanine according to its clear density in our ovine supercomplex map16. The model is at the atomic level for 88% of the protein (Extended Data Table 1), presenting, to our knowledge, the most complete atomic structure of mitochondrial complex I so far.
The fold of core subunits is generally conserved from bacteria (Supplementary Discussion). The Fe–S clusters are arranged in the redox chain with distances similar to bovine6 and T. thermophilus10 (Fig. 2a). The NADH-binding site is also conserved (Fig. 2b), preserving the entire path for electron transfer from NADH towards quinone. Key features in the membrane domain are also conserved, with four proton channels built around the central axis of polar residues propagating from the quinone-binding (Q) site into the three antiporter-like subunits.

a, Fe–S clusters are shown as spheres with centre-to-centre and edge-to-edge (in brackets) distances indicated in Å, overlaid with transparent grey depictions from T. thermophilus. Both traditional and structure-based (in brackets) nomenclature for clusters is shown. b, NADH-binding site (overlay with T. thermophilus structure in grey, containing NADH). Cryo-EM density for flavin mononucleotide (FMN) is shown in blue. Key residues involved in interactions with FMN and NADH are shown as sticks. c, Quinone-binding site with subunits coloured as Fig. 1. Key β1–β249-kDa loop deviates from bacterial structure (grey) and is more similar to Y. lipolytica (orange, PDB 4WZ7; ref. 12), clashing with the decyl-ubiquinone (DQ) head group position in T. thermophilus (grey; ref. 9). d, Environment surrounding the Q cavity (brown surface, entrance point indicated by an arrow), with some of the functionally important residues shown as sticks and labelled with non-ND1 subunit names in brackets. The quinone from the aligned T. thermophilus structure is shown in grey (DQ), demonstrating that the distal part of the cavity is blocked in the ovine enzyme.
Throughout the article, we use bovine nomenclature with numbering of residues according to mature17 ovine sequences; see Extended Data Table 1 for human nomenclature. The Q site lies at the interface of the hydrophilic 49-kDa and PSST subunits, and the membrane ND1 and ND3 subunits. The unique structure of the Q site, which forms an enclosed tunnel extending from the membrane towards cluster N2 about 25 Å away, is conserved with one difference: a loop connecting two strands of the N-terminal β-sheet from the 49-kDa subunit (β1–β249-kDa loop) extends further into the cavity, clashing with the position of the bound quinone from the bacterial structure9, where it interacts with conserved His5949-kDa and Tyr10849-kDa (Fig. 2c, d). A similar conformation was observed in the yeast enzyme, leading to the proposal that it represents the ‘deactive’ state12. In the absence of substrates, mitochondrial complex I exists in the deactive state (which may prevent oxygen radical production in vivo18), and converts into the ‘active’ state19 only upon turnover. Because the β1–β249-kDa loop in our structure will prevent quinone access closer than 20 Å to cluster N2 (blocking electron transfer), it probably also represents the deactive state. The ‘closed’ class conformation resembles one in supercomplex16, so may be more physiological. It remains to be established whether, as discussed previously12,15, different observed conformations are related to the catalytic cycle or indeed to active/deactive transitions, but the overall conformational flexibility of the complex is clear.
Supernumerary subunits form a shell around the core subunits6,12, especially around the membrane domain and its interface with the peripheral arm. With few exceptions, most supernumerary subunits are not globular, but form extended structures containing α-helices and coils (Extended Data Fig. 6), allowing for numerous interactions at interfaces with other subunits (Extended Data Table 2). They interweave extensively with each other and the core subunits (Extended Data Fig. 7), making the whole mitochondrial complex assembly much more interlinked and thus more stable, with a large total buried surface area (Extended Data Table 2). The intertwined nature of subunit structures suggests that they can be added to the complex only in a certain order, and, therefore, that the assembly of subunits must be tightly controlled20.
The fold of supernumerary subunits is described in the Supplementary Discussion. In summary, those associated with the membrane arm include 12 single transmembrane helix domain21 subunits scattered around the entire arm. Six of these surround the membrane arm tip and contribute their intertwined N-terminal domains to a large matrix ‘bulge’, the bulk of which is formed by an acyl-carrier protein (ACP)–LYR motif subunit pair (SDAP-β–B22). The large globular 42-kDa subunit from the nucleoside kinase family is attached to the matrix side of ND2 near the peripheral arm interface. On the intermembrane space (IMS) side, subunits SGDH and PDSW are ‘interlocked’ via their backbone and contain three long α-helices traversing nearly the entire membrane arm (Fig. 1c). PDSW and the subunits with CHCH domains (PGIV, 15 kDa and B18) contain disulfide bonds that further stabilize the fold in the oxidizing environment of the IMS. PGIV clamps the ‘heel’ of the complex to the middle of the membrane arm. The disulfide-rich, interlocked helices of the IMS subunits, with their rigid and stable structure, appear to replace the hairpin/helix motif (βH) found in bacterial complex I (refs 9, 11).
Subunits associated with the peripheral arm include the NADPH-containing 39-kDa subunit, the Zn-containing 13-kDa subunit and another ACP–LYR motif pair, SDAP-α–B14, with the latter pair and B13 jointly ‘embracing’ the 42-kDa subunit. The interface between the peripheral arm and membrane domain is stabilized by the exceptionally long membrane-traversing helix of subunit B16.6, as well as by B17.2 and B14.5a, both of which contain N-terminal amphipathic α-helices bound at the membrane interface, with the rest of their polypeptides wrapping around the hydrophilic arm. Subunits PSST, TYKY and B9 also contain such amphipathic helices, all located at the heel of the complex (Fig. 1c), probably assisting in proper peripheral arm position over the lipid bilayer.
Several cofactors present in supernumerary subunits are well resolved in the structure (Fig. 3). The 39-kDa subunit is wedged into the side of the peripheral arm near the membrane arm interface. It contains a tightly bound non-catalytic NADPH (Fig. 3b) that interacts with conserved Arg178PSST, providing a possible mitochondria redox state-sensitive conformational link to cluster N2. In SDAP-α, a phosphopantetheine that is covalently linked to Ser44 extends its attached acyl chain in the flipped-out22 conformation into the hydrophobic crevice between the helices of the LYR subunit B14 (Fig. 3d). A similar interaction is observed in the SDAP-β–B22 pair. These are the first structures of ACP–LYR complexes showing that their interaction depends on the extended acyl chain and revealing the role of LYR motif. Complex I is active only when fully assembled with the SDAP-α–B14 pair23. This interaction, which depends on the acyl chain attached to the ACP, may provide a regulatory link between fatty acid synthesis and oxidative phosphorylation in mitochondria. The 13-kDa subunit contains a Zn-binding motif, coordinating a Zn2+ ion in the vicinity of clusters N6a and N5. Zn-containing proteins are sensitive to oxidative stress24, and loss of the 13-kDa subunit leads to loss of cluster N6a (ref. 25) as it becomes exposed. In this way, complex I may be equipped with an oxidative-stress ‘sensor’, in addition to bound NADPH.

a, Overview of the model, coloured as in Fig. 1c, with cofactors shown as sticks. CDL, cardiolipin; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PPT, phosphopantetheine. b, NADPH in the 39-kDa subunit. Interacting residues are shown. c, Zn2+ ion in the 13-kDa subunit, with coordinating residues. d, Phosphopantetheine in SDAP-α. e, Phosphopantetheine in SDAP-β. f. Lipids phosphatidylethanolamine, phosphatidylcholine and cardiolipin. All cofactors are shown with cryo-EM density carved to within 5 Å.
Twelve bound lipids were identified in crevices between hydrophobic subunits. Several observed lipid molecules have four acyl chains and were therefore assigned as cardiolipins, known to be essential for activity26. Notably, a cardiolipin (CDL1; Fig. 3a) and three other lipid molecules fill the void left by the missing (in metazoans) three ND2 N-terminal helices6. This void is encircled by the two-transmembrane helix subunit B14.5b and the single transmembrane helix domain subunit KFYI, indicating that the ND2 helices may have been lost in evolution to accommodate a specific binding site for lipids. Two cardiolipins (CDL2 and CDL3) fill a large gap between the antiporter-like ND4 and ND5 subunits, preventing potential proton leaks and instability. Another cardiolipin (CDL4) stabilizes amphipathic helices at the heel of the complex. The structure thus shows the basis for the essential role of cardiolipin and other lipids.
The mechanism of coupling between electron transfer and proton translocation is still enigmatic. Conservation of key features from bacteria to mammals suggests that the basic mechanism is probably the same, with add-on ‘stabilizers’ and ‘regulators’. As we proposed previously9, the central axis of polar residues in the membrane probably has a key role (Fig. 4a). In each catalytic cycle, the negative charge stored either on Q or on nearby residues in the enclosed Q site may drive conformational changes in ND1 and the proton channel near the Q site, which would propagate via the central axis to channels in antiporter-like subunits ND2, ND4 and ND5, resulting in changes in pKa and accessibility of key residues. The net result would be the pumping of four protons per cycle, one per each channel. The observed conformation of loops in the Q site probably reflects the deactive state. This conformation might also occur during normal function when quinol is ejected from the site into the lipid bilayer, if active/deactive transitions are related to conformations encountered during the catalytic cycle12. Supernumerary subunits implicated in active/deactive transitions (39 kDa, B13 and SDAP-α–B14 pair) could also participate in catalytic conformational changes by interacting with the key TM1–TM2ND3 loop flanking the Q site, and possibly through interactions with the 42-kDa subunit (Fig. 4b). In the ‘closed’ class, B13 and SDAP-α move towards the 42-kDa subunit (Extended Data Fig. 8), hinting at such a possibility. Because the 42-kDa subunit is metazoan-specific27, its role may be to fine tune movements during turnover. The traverse helix HL from ND5 appears to mainly have a stabilizing ‘stator’ role28 rather than being a moving element29. Rigid disulfide-rich supernumerary subunits traversing the IMS side of the membrane domain may represent another stator element unique to the mitochondrial enzyme (Fig. 4b).

a, Structure of the core subunits of ovine complex I, coloured as in Fig. 1b, with polar residues in proton channels shown as sticks, with carbon in blue, orange and green for input, connecting and output parts, respectively. Key residues, Glu (TM5), Lys (TM7), Lys/His (TM8) and Lys/Glu (TM12) from the antiporters and the corresponding residues in the E-channel (near Q site), are shown as small spheres and labelled. These residues sit on flexible loops in discontinuous transmembrane helices shown as cylinders. Polar residues linking the E-channel to the Q cavity (brown) are shown in magenta. Tyr10849-kDa and His5949-kDa are shown in cyan near the position of bound Q in bacteria. Possible proton translocation pathways are indicated by blue arrows. b, Graphic of the coupling mechanism. Core and some putatively regulatory supernumerary subunits are shown. Conformational changes, indicated by red arrows, propagate from the Q site/E-channel to antiporter-like subunits via the central hydrophilic axis. Shifts of helices near the cluster N2 (ref. 31; blue arrows) may help initiate the process. ND5 helix HL and traverse helices from four supernumerary subunits on the IMS side may serve as stators. Dashed line indicates the shift of peripheral arm in the closed conformation (Extended Data Fig. 8). The NADPH-containing 39-kDa subunit and Zn-containing 13-kDa subunit are essential for activity and may serve as redox sensors. Both SDAP subunits interact with their LYR partners via flipped-out phosphopantetheine (black line). The net result of one conformational cycle, driven by NADH:ubiquinone oxidoreduction, is the translocation of four protons across the membrane (black lines indicate possible pathways).
Our structure clearly shows that supernumerary subunits stabilize the complex. Some of them, especially those containing additional cofactors (39 kDa, SDAPs, B14, B22 and 13 kDa) and phosphorylated residues (42 kDa, ESSS, MWFE, B14.5a, B14.5b and B16.6)30, may provide regulatory links to the redox status of the cell, lipid biosynthesis and mitochondrial homeostasis. Known human pathological mutations are present in all of the core and many of the supernumerary subunits4. Our structure provides the framework for understanding the molecular basis of mutations and mechanisms of complex I function and regulation.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Protein purification and electron microscopy
Protein was purified from O. aries heart mitochondria following the protocol adapted with some modifications from a previously published procedure for the bovine enzyme (ref. 32 and J.A.L. et al., manuscript submitted). We explored O. aries as a source of complex I that may be more suitable for high-resolution structural studies than the extensively studied bovine enzyme. We find that ovine enzyme appears more stable, as it is highly active after purification and retains the 42-kDa subunit, easily lost from bovine complex (J.A.L. et al., manuscript submitted). In terms of overall sequence similarity ovine is as good a model of the human enzyme as bovine (~84%), and all 44 different subunits of complex I were identified in the preparation by mass spectrometry (J.A.L. et al., manuscript submitted). In brief, fresh ovine hearts were purchased from the local abattoir and mitochondria prepared as described previously32. Mitochondrial membranes were solubilized in the branched chain detergent lauryl maltose neopentyl glycol (LMNG, 1%) and the sample applied to Q-sepharose HP anion exchange column (GE Healthcare) equilibrated with 20 mM Tris-HCl, pH 7.4, 10% (v/v) glycerol, 1 mM EDTA, 1 mM DTT and 0.1% LMNG. Protein was eluted with a NaCl gradient, peak fractions concentrated and applied to Superose 6 HiLoad 16/60 column equilibrated in 20 mM HEPES, pH 7.4, 2 mM EDTA, 1.5% (v/v) glycerol, 100 mM NaCl and 0.02% Brij-35. The peak fraction was concentrated to ~5 mg ml−1 protein and ~0.2% Brij-35. Then, 2.7 μl of sample was applied to glow discharged Quantifoil R 0.6/1 copper grids and blotted for 34 s at 90% humidity in the chamber of FEI Vitrobot III. Immediately after, the sample was snap-frozen in liquid ethane. Extensive trials with different detergents, including previously used Cymal-7 (ref. 6), revealed Brij-35 as the detergent giving the most homogeneous spread of particles. Imaging was performed with a 300 kV Titan Krios electron microscope equipped with direct electron detector FEI Falcon-II (ETH Zurich, ScopeM centre) in automated data collection mode at a calibrated magnification of 1.39 Å pixel−1 (×100,720) and dose of 26 e s−1 Å−2 with total 3-s exposure time. The data were collected as seven movie frames fractionated over the first second of exposure and an averaged image over 3 s.
Image processing
We collected a total of 2.6 k micrographs in two datasets, which were combined. All processing steps were done using RELION33 unless otherwise stated. We used averaged images from high dose 3 s exposure for initial CTF estimations using CTFFIND4 (ref. 34) and for automated particle picking in Relion, resulting in ~241 k particles. MOTIONCORR35 was used for whole-image drift correction of movie frames 1–7 (1 s) of each micrograph. Contrast transfer function (CTF) parameters of the corrected micrographs were estimated using Gctf and refined locally for each particle36. The particles were extracted using 2962 pixel box and sorted by reference-free 2D classification, resulting in ~171 k particles selected from good 2D classes. These were used for 3D classification with a regularization parameter T of 8 and a 30 Å low-pass filtered initial model from a previous low resolution model of the bovine enzyme6. That resulted in ~130 k particles of good quality; however, it was clear that the relative orientation between the two arms of the complex is slightly variable, producing 3D classes with either an open or closed angle between the arms (Extended Data Fig. 1). Particles in the open conformation (~82 k particles) produced higher resolution maps and were selected for a final reconstruction. For all high-resolution refinements, particles were re-extracted from the motion corrected micrographs with a 5122 pixel box to allow for high-resolution CTF correction37. After initial auto-refinement, particle-based beam-induced motion correction and radiation-damage weighing (particle polishing) was performed38. Refinement of polished particles gave a map resolved to 3.9 Å. All resolutions are based on the gold-standard (two halves of data refined independently) FSC = 0.143 criterion39. This 3D class selection probably still allows for small variations in the conformation, therefore the local resolution varies within the map, especially at the extremities of both arms (Extended Data Fig. 3). At the periphery of the molecule the resolution drops not only owing to the usual decrease in the precision of particle alignments in these areas, but also due to differences in the protein conformation, greatest at the edges of the molecule. To overcome this limitation we performed 3D refinement focused on the peripheral and membrane domains separately (with the subtraction of signal from the remaining parts of the complex40). This resulted in a 3.9 Å map of the peripheral arm, very well resolved in all areas, including the edges of the domain. The membrane domain refined to 4.1 Å, however, the map was more uniform and so better resolved for the distal part of the domain (near subunit ND5) as compared to the density from the refinement of the entire complex (Extended data Fig. 3). Higher quality refinement of the peripheral arm probably stems from the fact that high electron density of eight Fe–S clusters helps in particle alignment. The least ordered part of the complex is the 42-kDa subunit, loosely attached to the membrane domain. We have performed extended 3D classification of the open class to identify the most homogeneous population, especially with respect to 42-kDa subunit. This class (64 k particles) was refined to 4.0 Å, and the resulting density was used to model the 42-kDa subunit. To assist with overall model building and refinement, several maps were carved around specific parts of the complex and combined into one map in UCSF Chimera41: peripheral arm from peripheral-arm-focused refinement, the area around ND4/5 subunits (tip of the membrane domain) from membrane-arm-focused refinement, the 42-kDa subunit density as above, and the rest of the complex from the overall 3.9 Å map for the open class (Fig. 1a and Extended Data Fig. 3). The final model was refined against this combined map.
The final map is of high quality, with about three-quarters of the map at 3.9 Å resolution and the rest at 4.1 Å. Large- and medium-size side chains, as well as relatively small Val and Thr, are clearly seen in the density (Extended Data Fig. 4). Carboxylates (Asp, Glu) have much lower density than other residues owing to early radiation damage, as observed previously42. Disulfide bridges also are subject to early damage, as in X-ray crystallography43. Few features at the interfaces of maps used for the combined map may be better resolved in individual maps, since in overlapping regions both maps contribute. For example, the β1–β249-kDa loop is better resolved in the peripheral-arm-focused map, which is deposited along with other constituent maps. Overall map filtered to lower resolution is very similar to the previous 5 Å resolution map for the bovine enzyme6, suggesting that the mammalian complex I structure is very well conserved. One difference is that in ovine complex the accessory four-transmembrane subunit B14.7 is disordered in the detergent used for the microscopy samples (Brij-35). It is likely to be disordered rather than detached as B14.7 was identified by mass spectrometry in the sample used for electron microscopy (data not shown). Since Brij-35 gave us the best yield of particles, we kept its use for data collection, but took advantage of the availability of cryo-EM maps of ovine respiratory supercomplexes in our laboratory. In these maps all the subunits of complex I are well ordered, and so in our final complex I model we included the poly-ALA model of B14.7 based on 5.8 Å resolution map of the ‘tight’ respirasome16. Loss of B14.7 also results in the disorder of the nearby C-terminal half of transverse helix HL and TM16 from ND5, as well as TM4 from ND6, which were also modelled as poly-alanine (these stretches can be recognized by B-factor set to 200) based on the tight respirasome map. The register in poly-alanine stretches is approximate. The density for the 42-kDa subunit is rather weak but this subunit clearly preserves the nucleoside kinase family fold, which allowed us to model most of it using Rosetta and visible large side-chains as a guide.
Model building and refinement
For the 14 core subunits the initial homology models were generated manually based on the T. thermophilus structure9 with side-chains rebuilt to ovine sequence using SQWRL4 software44. Homology models were generated with Phyre2 (ref. 45) and Swiss-model46 servers for all supernumerary subunits, although they were mostly useful only for subunits with large globular domains, such as the 42-kDa and 39-kDa subunits, as well as for those with known structure of close homologues (SDAPs and B8). Secondary structure predictions for all subunits were generated with PredictProtein47, PsiPred48 and TMHMM49 servers, and were helpful during model building. Initial assignments of the location and the fold of supernumerary subunits were based on our cross-linking data and the secondary structure features and side-chain density observed in the cryo-EM map, with checks for consistency with the knowledge on subcomplexes and assembly intermediates in complex I. The initial models were adjusted to cryo-EM density (in cases when homology models were useful) or built manually in COOT50. Lipids were tentatively assigned on the basis of appearance in the density as cardiolipins, phosphatidylcholines and phosphatidylethanolamines, known to co-purify with the complex26,51.
Initial models were re-built and refined in Rosetta release version 2016.02.58402 using protocols optimized for cryo-EM maps14. For each subunit, 100 different models were produced in Rosetta with optimization of density fit using elec_dens_fast function (with -denswt = 40, chosen from several trials), selection of the best fitting structure and structure relaxation using -FastRelax flag. From the produced structures several best-scoring by density fit and geometry were selected and used in COOT to guide further model building/optimization. This procedure resulted mainly in improvements to backbone geometry, especially in coils, still allowing for the good fit of side-chains into density.
After several rounds of re-building the final model was refined with the Phenix suite52 phenix.real_space_refine program for 5 macro-cycles using the electron scattering table with default and secondary structure restrains. This resulted in a high quality model in terms of geometry (Molprobity score 2.5, that is, corresponding to average structure at 2.5 Å resolution) and fit to density (Extended Data Fig. 2c).
Cross-linking
All the cross-linking reactions were performed using purified solutions of complex I at a concentration of 1 mg ml−1. Following experimental optimisation, ten separate experiments were performed. Experiments varied in relation to the detergent added to the buffer (DDM, LMNG or LDAO/DDM), the cross-linking reagent (targeting lysine or acidic residues) and the protease used to digest the samples (trypsin or endoproteinase Glu-C) (Supplementary Table 1).
Isotopically labelled cross-linking reagents were purchased from Creative Molecules (Canada). 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) was purchased from Sigma. Homobifunctional, isotopically-coded N-hydroxysuccinimide (NHS) esters disuccidinimidyl suberate (DSS H12/D12), bis-sulfodisuccinimidyl suberate (BS3 H12/D12) and disuccinimidyl adipate (DSA 12C6/13C6) were used at a final concentration of 50 μM as cross-linking reagents to target lysine residues. The reactions were incubated for 45 min at 37 °C and quenched by adding NH4HCO3 to a final concentration of 50 mM and incubating for further 15 min. Isotopically labelled adipic acid dihydrazide (ADH H8/D8) and suberic acid dihydrazide (SDH H12/D12) were used to target the acidic residues, using DMTMM as catalyst. The cross-linking reaction was initiated by adding ADH or SDH and DMTMM to final concentrations 5 mg ml−1, 6 mg ml−1 and 12 mg ml−1, respectively. The samples were incubated at 37 °C for 60 min and the reactions stopped using gel filtration (Zeba Spin Desalting columns 7K MWCO).
The cross-linked samples were freeze-dried and then resuspended in 50 mM NH4HCO3, 8 M urea and 0.1% SDS to a final concentration of 1 mg ml−1. Size exclusion protein fractionation was performed through a Superdex 200 Increase 3.2/300 column (GE Healthcare) with 50 mM NH4HCO3, 8 M urea and 0.1% SDS as mobile phase at a flow rate of 25 μl min−1. Two-minute fractions were collected and their protein content evaluated by SDS–PAGE. Fractions of similar content were pooled into 4–5 main fractions and concentrated to 1 mg ml−1 using Amicon Ultra-0.5 mL Centrifugal Filters (Millipore).
The filtered cross-linked samples were then enzymatically digested. Samples were freeze-dried and resuspended in 50 mM NH4HCO3 and 8 M urea to a final protein concentration of 1 mg ml−1, reduced with 10 mM DTT and alkylated with 50 mM iodoacetamide. Following alkylation, samples were diluted with 50 mM NH4HCO3 to 1 M urea before trypsin digestion (or 2 M for Glu-C digestion). Trypsin and Glu-C were added at an enzyme-to-substrate ratio of 1:20 and 1:100, respectively. Digestions were carried out overnight at 37 °C and 25 °C for trypsin and Glu-C respectively. After digestion, the samples were acidified with formic acid to a final concentration of 2% (v/v) and the peptides fractionated by peptide size exclusion chromatography, using a Superdex Peptide 3.2/300 (GE Healthcare) with 30% (v/v) acetonitrile/0.1% (v/v) TFA as mobile phase and at a flow rate of 50 μl min−1. Fractions were collected every 2 min over the elution volume 1.0 ml to 1.7 ml. Before LC–MS/MS analysis fractions were freeze dried and resuspended in 2% (v/v) acetonitrile and 2% (v/v) formic acid.
The digests were analysed by nano-scale capillary LC–MS/MS using an Ultimate U3000 HPLC (ThermoScientific Dionex) to deliver a flow of approximately 300 nl min−1. A C18 Acclaim PepMap100 5 μm, 100 μm × 20 mm nanoViper (ThermoScientific Dionex), trapped the peptides before separation on a C18 Acclaim PepMap100 3 μm, 75 μm × 250 mm nanoViper (ThermoScientific Dionex). Peptides were eluted with a gradient of acetonitrile. The analytical column outlet was directly interfaced via a nano-flow electrospray ionisation source, with a hybrid dual pressure linear ion trap mass spectrometer (Orbitrap Velos, ThermoScientific). Data-dependent analysis was carried out, using a resolution of 30,000 for the full mass spectrometry spectrum, followed by ten MS/MS spectra in the linear ion trap. Mass spectrometry spectra were collected over a m/z range of 300–2000. MS/MS scans were collected using threshold energy of 35 for collision-induced dissociation.
For data analysis, Xcalibur raw files were converted into the open mzXML format through MSConvert (Proteowizard) with a 32-bit precision. mzXML files were directly used as input for xQuest searches on a local xQuest installation53. The selection of cross-linked precursor MS/MS data was based on the following criteria: a mass difference among the heavy and the light cross-linker of: 12.07532 Da for BS3, DSS and SDH, 6.02016 Da for DSA and 8.05016 Da for ADH; precursor charge ranging from 3+ to 8+; maximum retention time difference 2.5 min. Searches were performed against an ad hoc database containing all the sequences of ovine complex I subunits together with their reverse used as decoy database. The following parameters were set for xQuest searches: maximum number of missed cleavages (excluding the cross-linking site) 3; peptide length 4–50 amino acids; fixed modifications carbamidomethyl-Cys (mass shift 57.02146 Da); mass shift of the light cross-linker 138.06808 Da for DSS and BS3, 138.0906 Da for ADH, 110.03675 for DSA and 166.1218 for SDH; mass shift of mono-links 156.0786 and 155.0964 Da for DSS and BS3, 138.0906 Da for ADH, 127.0628 Da and 128.0468 Da for DSA, and 184.1324 Da for SDH; MS1 tolerance 10 ppm, MS2 tolerance 0.2 Da for common ions and 0.3 for cross-link ions; search in enumeration mode (exhaustive search). Search results were filtered according to the following criteria: MS1 mass tolerance window −3 to 7 ppm. Finally each MS/MS spectra was manually inspected and validated.
In total 218 unique cross-linked peptides were identified, of which 87 were between residues of different subunits (inter-subunit, Supplementary Table 2), 73 were between residues within the same subunit (intra-subunit, Supplementary Table 3), and 58 were clear false positives (Supplementary Table 4). False positives were identified by comparison to all known biochemical and structural information on complex I and the cross-links that are considered false positives are either between residues that are too distant from each other (>32 Å after allowing for exposed side chain flexibility from their modelled position), located on opposite sides of the membrane or the reactive residues are buried and not solvent accessible in the intact structure. Many of the false positive cross-links are found on unstructured coils at the edges of the complex I structure indicating that they probably result from transient interactions between different complexes I during the reaction (inter-complex cross-links). True positive cross-links were more likely to be observed in more than one experiment. Some high-scoring cross-links were observed between disordered termini or loops of subunits that could not be modelled in our structure; hence the accurate determination of distance for these cross-links was not possible. Nonetheless in cases where cross-linking residues are adjacent to the modelled regions, the cross-links were considered true and are included in Extended Data Fig. 5 and Supplementary Tables 2 and 3. No cross-links were observed for any of the mitochondrially encoded core subunits, which are buried in the membrane and coated with a layer of supernumerary subunits. Good quality cross-links were observed for all supernumerary subunits expect for B14.7, KFYI and AGGG. These data in conjunction with our electron microscopy maps allowed us to unambiguously assign all supernumerary subunits. Previous assignments were confirmed and importantly, subunits that previously had no known position in the complex (10 kDa, B14.5a, MWFE, B9, MNLL, SGDH, ASHI, B17, AGGG and B12) have now been assigned and built (see Supplementary Discussion for more details).
References
Sazanov, L. A. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 16, 375–388 (2015)
Moser, C. C., Farid, T. A., Chobot, S. E. & Dutton, P. L. Electron tunneling chains of mitochondria. Biochim. Biophys. Acta 1757, 1096–1109 (2006)
Letts, J. A. & Sazanov, L. A. Gaining mass: the structure of respiratory complex I-from bacterial towards mitochondrial versions. Curr. Opin. Struct. Biol. 33, 135–145 (2015)
Fassone, E. & Rahman, S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J. Med. Genet. 49, 578–590 (2012)
Walker, J. E. The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Q. Rev. Biophys. 25, 253–324 (1992)
Vinothkumar, K. R., Zhu, J. & Hirst, J. Architecture of mammalian respiratory complex I. Nature 515, 80–84 (2014)
Galkin, A., Dröse, S. & Brandt, U. The proton pumping stoichiometry of purified mitochondrial complex I reconstituted into proteoliposomes. Biochim. Biophys. Acta 1757, 1575–1581 (2006)
Wikström, M., Sharma, V., Kaila, V. R., Hosler, J. P. & Hummer, G. New perspectives on proton pumping in cellular respiration. Chem. Rev. 115, 2196–2221 (2015)
Baradaran, R., Berrisford, J. M., Minhas, G. S. & Sazanov, L. A. Crystal structure of the entire respiratory complex I. Nature 494, 443–448 (2013)
Sazanov, L. A. & Hinchliffe, P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311, 1430–1436 (2006)
Efremov, R. G. & Sazanov, L. A. Structure of the membrane domain of respiratory complex I. Nature 476, 414–420 (2011)
Zickermann, V. et al. Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science 347, 44–49 (2015)
Zhu, J. et al. Structure of subcomplex Iβ of mammalian respiratory complex I leads to new supernumerary subunit assignments. Proc. Natl Acad. Sci. USA 112, 12087–12092 (2015)
DiMaio, F. et al. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365 (2015)
Zhu, J., Vinothkumar, K. R. & Hirst, J. Structure of mammalian respiratory complex I. Nature 536, 354–358 (2016)
Letts, J. A., Fiedorczuk, K. & Sazanov, L. A. Architecture of respiratory supercomplexes. Nature http://dx.doi.org/10.1038/nature19774 (2016)
Carroll, J., Fearnley, I. M., Shannon, R. J., Hirst, J. & Walker, J. E. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol. Cell. Proteomics 2, 117–126 (2003)
Chouchani, E. T. et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 19, 753–759 (2013)
Vinogradov, A. D. & Grivennikova, V. G. The mitochondrial complex I: progress in understanding of catalytic properties. IUBMB Life 52, 129–134 (2001)
Sánchez-Caballero, L., Guerrero-Castillo, S. & Nijtmans, L. Unraveling the complexity of mitochondrial complex I assembly: A dynamic process. Biochim. Biophys. Acta 1857, 980–990 (2016)
Zickermann, V., Angerer, H., Ding, M. G., Nübel, E. & Brandt, U. Small single transmembrane domain (STMD) proteins organize the hydrophobic subunits of large membrane protein complexes. FEBS Lett. 584, 2516–2525 (2010)
Cronan, J. E. The chain-flipping mechanism of ACP (acyl carrier protein)-dependent enzymes appears universal. Biochem. J. 460, 157–163 (2014)
Angerer, H. Eukaryotic LYR proteins interact with mitochondrial protein complexes. Biology (Basel) 4, 133–150 (2015)
Oteiza, P. I. Zinc and the modulation of redox homeostasis. Free Radic. Biol. Med. 53, 1748–1759 (2012)
Kmita, K. et al. Accessory NUMM (NDUFS6) subunit harbors a Zn-binding site and is essential for biogenesis of mitochondrial complex I. Proc. Natl Acad. Sci. USA 112, 5685–5690 (2015)
Sharpley, M. S., Shannon, R. J., Draghi, F. & Hirst, J. Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248 (2006)
Brandt, U. Energy converting NADH:quinone oxidoreductase (complex I). Annu. Rev. Biochem. 75, 69–92 (2006)
Belevich, G., Knuuti, J., Verkhovsky, M. I., Wikström, M. & Verkhovskaya, M. Probing the mechanistic role of the long α-helix in subunit L of respiratory Complex I from Escherichia coli by site-directed mutagenesis. Mol. Microbiol. 82, 1086–1095 (2011)
Efremov, R. G., Baradaran, R. & Sazanov, L. A. The architecture of respiratory complex I. Nature 465, 441–445 (2010)
Palmisano, G., Sardanelli, A. M., Signorile, A., Papa, S. & Larsen, M. R. The phosphorylation pattern of bovine heart complex I subunits. Proteomics 7, 1575–1583 (2007)
Berrisford, J. M. & Sazanov, L. A. Structural basis for the mechanism of respiratory complex I. J. Biol. Chem. 284, 29773–29783 (2009)
Sazanov, L. A., Peak-Chew, S. Y., Fearnley, I. M. & Walker, J. E. Resolution of the membrane domain of bovine complex I into subcomplexes: implications for the structural organization of the enzyme. Biochemistry 39, 7229–7235 (2000)
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012)
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015)
Li, X. et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013)
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016)
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003)
Scheres, S. H. Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife 3, e03665 (2014)
Scheres, S. H. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012)
Bai, X. C., Rajendra, E., Yang, G., Shi, Y. & Scheres, S. H. Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 4, e11182 (2015)
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004)
Allegretti, M., Mills, D. J., McMullan, G., Kühlbrandt, W. & Vonck, J. Atomic model of the F420-reducing [NiFe] hydrogenase by electron cryo-microscopy using a direct electron detector. eLife 3, e01963 (2014)
Meents, A., Gutmann, S., Wagner, A. & Schulze-Briese, C. Origin and temperature dependence of radiation damage in biological samples at cryogenic temperatures. Proc. Natl Acad. Sci. USA 107, 1094–1099 (2010)
Krivov, G. G., Shapovalov, M. V. & Dunbrack, R. L. Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77, 778–795 (2009)
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protocols 10, 845–858 (2015)
Biasini, M. et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 (2014)
Rost, B., Yachdav, G. & Liu, J. The PredictProtein server. Nucleic Acids Res. 32, W321–W326 (2004)/
McGuffin, L. J., Bryson, K. & Jones, D. T. The PSIPRED protein structure prediction server. Bioinformatics 16, 404–405 (2000)
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001)
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004)
Shinzawa-Itoh, K. et al. Bovine heart NADH-ubiquinone oxidoreductase contains one molecule of ubiquinone with ten isoprene units as one of the cofactors. Biochemistry 49, 487–492 (2010)
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010)
Leitner, A., Walzthoeni, T. & Aebersold, R. Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat. Protocols 9, 120–137 (2014)
Kahraman, A., Malmström, L. & Aebersold, R. Xwalk: computing and visualizing distances in cross-linking experiments. Bioinformatics 27, 2163–2164 (2011)
Acknowledgements
We thank the ETH Zurich ScopeM Center for access to Titan Krios EM. Data processing was performed at the IST high-performance computer cluster. K.F. is partially funded by the Medical Research Council UK PhD fellowship. J.A.L. holds a long-term fellowship from FEBS. This project has received funding from the European Union’s 2020 research and innovation programme under grant agreement No 701309.
Author information
Authors and Affiliations
Contributions
K.F. purified complex I for grid preparation, prepared cryo-EM grids, acquired and processed EM data, and co-built the models; J.A.L. purified complex I for cross-linking experiments, analysed cross-linking data and co-built the models; G.D. performed cross-linking/mass-spectrometry experiments, K.K. performed model re-building in Rosetta and sequence alignments; G.D. and M.S. analysed cross-linking data; L.A.S. designed and supervised the project, processed and analysed data and wrote the manuscript, with contributions from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information
Nature thanks P. Ädelroth, M. Ryan and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Figure 1 Image processing procedures.
a, Representative micrograph of 2.6 k micrographs collected that all varied in defocus, ice thickness and particle count, with good quality particles circled. Scale bar, 100 nm. b, Representative 2D class averages obtained from reference-free classification. c, Classification and refinement procedures used in this study.
Extended Data Figure 2 Image and model refinement procedures.
a, Radiation-damage weighting. Relative B-factors (Bf) and intercepts (Cf) from the Relion particle polishing procedure. b, Left, gold-standard (two halves of data refined independently) Fourier shell correlation (FSC) curves for the maps of the entire complex complete map (resolution at FSC = 0.143 is 3.9 Å), membrane-arm-focused refinement (4.1 Å resolution) and peripheral-arm-focused refinement (3.9 Å resolution). Right, FSC curve of the combined map versus final model shows good agreement of the model with the map (FSC = 0.5 at 4.0 Å resolution). FSC curve against the entire complex complete map, which was not used in refinement, is shown as a control. c, Statistics of refinement.
Extended Data Figure 3 Local resolution estimation and combination of maps for model building.
a–c, Local resolution estimation by Resmap of the entire complex I (a), peripheral-arm-focused refinement map (b) and membrane-arm-focused refinement map (c). Maps are coloured according to the shown resolution scale in Å. d. The final map was produced by combining maps with the best local resolution features; that is, for peripheral-arm-focused refinement map (orange), for the distal part of membrane-arm-focused refinement map (green), for 42-kDa subunit map from the selected homogenous complex I class (64 k particles; blue) and the rest of the complex from the best map of the entire complex (magenta).
Extended Data Figure 4 Examples of cryo-EM density.
a, b, Coils and α-helices from core (a) and supernumerary (b) subunits. c, d, Example β-sheets from core PSST subunit (c) and supernumerary 39-kDa subunit (d). Cryo-EM density is shown with the model represented as sticks and coloured by atom with carbon in grey, oxygen in red, nitrogen in blue and sulfur in yellow.
Extended Data Figure 5 Identified cross-links.
a, Solvent-accessible surface (SAS) representation of cross-links. Surfaces for complex I subunits are shown transparent and coloured as in Fig. 1. Shortest SAS paths calculated using Xwalk54 are shown for cross-links as coloured worms with inter-subunit lysine reactive cross-links in blue, inter-subunit acid reactive cross-links in red, intra-subunit lysine reactive cross-links in light blue, intra-subunit acid reactive cross-links in light red. b, Inter-subunit cross-link schematic. Complex I subunits are shown in a similar orientation as in a. Left panel with core subunits cyan, previously assigned supernumerary subunits in magenta, newly assigned or newly built regions of supernumerary subunits in green, poly-alanine regions in orange and unmodelled regions in red. Observed cross-links are indicated by dashed black lines between either blue circles (lysine reactive cross-links) or red circles (acid reactive cross-links). No cross-links were observed to the core subunits of the membrane arm and hence they were omitted for clarity. The horizontal black lines indicate the approximate boundaries of the inner mitochondrial membrane. Subunits B14.7, B15 and ASHI are shown as being behind the membrane boundaries as they are found on the opposite (far) side of the membrane arm.
Extended Data Figure 6 Folds of supernumerary subunits.
Subunits are shown in cartoon representation, coloured blue to red from N to C terminus. Disulfide bridges are shown as sticks with sulfur in yellow.
Extended Data Figure 7 Examples of supernumerary subunits interactions.
a, Side view of complex I showing surfaces for subunits B14.5a and B16.6. b, IMS view of complex I showing surfaces for subunits SGDH and PDSW. The point at which the two subunits are intertwined is marked with a star. c, View of the hydrophilic arm looking from above the membrane arm. The surface of the 18-kDa subunit that spans the hydrophilic arm is shown. d, Matrix view of the tip of the membrane arm with the surface of supernumerary subunit B22 shown. e, Close up of the centre of the membrane arm on the IMS side. This region contains many interactions between supernumerary subunits and the side chains of residues involved are shown. The region is also a hot spot for cross-links, the side chains involved are shown and cross-links are indicated with dashed lines (acid cross-links: red; basic cross-links: blue). f, Close up of the C-terminal helix of supernumerary subunit PDSW at the centre of the membrane arm on the IMS side. This helix extends away from complex I and is encircled by the C termini of supernumerary subunits B14.5b, ESSS and B15. The side chains of residues involved in stabilizing interactions are shown. A possible disulfide bond between PDSW (Cys154) and ESSS (Cys112) and stabilizing salt bridges are indicated by dashed lines. Subunits are coloured as in Fig. 1.
Extended Data Figure 8 Comparison of ‘open’ and ‘closed’ 3D class structures.
a, b, Side (a) and top (b) view from the matrix for the alignment of the open class structure (in cyan) and closed class structure (in grey). To generate the closed class structure, the final structure of the open class was refined in real space in Phenix (5 macro cycles with morphing at each cycle) against 4.6 Å map of the closed class (Extended Data Fig. 1). All of the α-helices were well fit into density, but owing to low resolution of the closed class no further refinement was performed and the comparison of structures involves only the relative positions of secondary structure elements. The two structures were aligned via transmembrane core subunits and are displayed as cartoon models. In the closed class the peripheral arm undergoes a hinge-like motion around the Q site towards the tip of the membrane arm, with the direction of shift indicated by the arrow in b. As a result, subunit B13 moves ~3 Å closer to the 42-kDa subunit, allowing for direct contacts. The shift is larger at the periphery, reaching 7 Å at the tip of the peripheral arm. Additionally, subunit ND5 and its matrix bulge move about 3 Å towards peripheral arm.
Supplementary information
Supplementary Information
This file contains Supplementary Discussion and Supplementary Tables 1-4. (PDF 677 kb)
Rights and permissions
About this article

Cite this article
Fiedorczuk, K., Letts, J., Degliesposti, G. et al. Atomic structure of the entire mammalian mitochondrial complex I. Nature 538, 406–410 (2016). https://doi.org/10.1038/nature19794
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature19794
- Springer Nature Limited
This article is cited by
-
Progress in diagnosis and treatment of Leber’s hereditary optic neuropathy
Journal of Molecular Medicine (2024)
-
A lethal mitonuclear incompatibility in complex I of natural hybrids
Nature (2024)
-
Mutation at the entrance of the quinone cavity severely disrupts quinone binding in respiratory complex I
Scientific Reports (2023)
-
H2O2 selectively damages the binuclear iron-sulfur cluster N1b of respiratory complex I
Scientific Reports (2023)
-
An engineered variant of MECR reductase reveals indispensability of long-chain acyl-ACPs for mitochondrial respiration
Nature Communications (2023)