

Abstract
Human mitochondrial DNA (mtDNA) shows extensive within-population sequence variability1. Many studies suggest that mtDNA variants may be associated with ageing or diseases2,3,4, although mechanistic evidence at the molecular level is lacking5,6. Mitochondrial replacement has the potential to prevent transmission of disease-causing oocyte mtDNA. However, extension of this technology requires a comprehensive understanding of the physiological relevance of mtDNA sequence variability and its match with the nuclear-encoded mitochondrial genes. Studies in conplastic animals7,8,9 allow comparison of individuals with the same nuclear genome but different mtDNA variants, and have provided both supporting and refuting evidence that mtDNA variation influences organismal physiology. However, most of these studies did not confirm the conplastic status, focused on younger animals, and did not investigate the full range of physiological and phenotypic variability likely to be influenced by mitochondria. Here we systematically characterized conplastic mice throughout their lifespan using transcriptomic, proteomic, metabolomic, biochemical, physiological and phenotyping studies. We show that mtDNA haplotype profoundly influences mitochondrial proteostasis and reactive oxygen species generation, insulin signalling, obesity, and ageing parameters including telomere shortening and mitochondrial dysfunction, resulting in profound differences in health longevity between conplastic strains.
Similar content being viewed by others


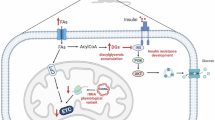
Main
The mtDNAs of C57BL/6 and NZB/OlaHsd mice differ by 12 missense mutations, 4 transfer RNA (tRNA) mutations, 8 ribosomal RNA (rRNA) mutations, and 10 non-coding-region mutations (Extended Data Fig. 1b, Extended Data Table 1 and Supplementary Data 1), a level of divergence comparable to that between human Eurasian and African mtDNAs. We developed a conplastic mouse strain (Extended Data Fig. 1a) with the C57BL/6 nuclear genome and the NZB/OlaHsd mtDNA, hereafter referred to as BL/6NZB. The original C57BL/6 strain with the C57BL/6 nuclear genome and C57BL/6 mitochondrial genome is hereafter referred to as BL/6C57. Nuclear genome purity was confirmed (Extended Data Fig. 1c–e). Conplastic animals, BL/6C57 and BL/6NZB, were fertile, and on a chow diet there were no weight differences throughout the growth period (Extended Data Fig. 2a, b), with marginal differences in activity observed in metabolic chambers (Extended Data Fig. 2c–e).
Unexpectedly BL/6NZB extended median lifespan by 16% without modifying maximum lifespan (Fig. 1a). After 9 months, BL/6NZB mice gained less weight (Fig. 1b). Two-year-old BL/6C57 animals manifested more signs of ageing10 than age-matched BL/6NZB mice (Fig. 1c, Extended Data Fig. 3a, b); including ovary preservation, with less lipofuscin accumulation at 12 months in BL/6NZB mice (Extended Data Fig. 3e, f). Telomeres in one-year-old animals were on average approximately 11% longer in BL/6C57 mice, whereas in two-year-old animals this was inverted, with BL/6NZB telomeres slightly longer (Fig. 1d). This is due to a BL/6C57 telomere-length-reduction rate that is double than that of BL/6NZB mice (Fig. 1e). Liver necropsy revealed a higher incidence of tumours in BL/6C57 mice at death (Fig. 1f and Extended Data Fig. 3c, d), suggesting greater genome instability.

a, Survival curves (n = 31 BL/6C57 and n = 29 BL/6NZB); Gehan–Breslow–Wilcoxon test *P < 0.05). b, Weight gain (chow diet, n = 19 males per genotype, mean ± s.e.m., *P < 0.05, two-tailed t-test). c, Mice that were ~2 years old were scored for ageing parameters (n = 5 for BL/6NZB, n = 4 for BL/6C57). For more information on ageing parameters see Extended Data Fig. 3a. a.u., arbitrary units. d, Telomere length in hair follicle cells of 1- (n = 6 per group) and 2-year-old mice (n = 5 for BL/6NZB, n = 4 for BL/6C57). Red lines indicate mean length (***P < 0.001, Wilcoxon’s rank sum test). e, Telomere length reduction during second year of life (mean ± s.e.m. *P < 0.05, two-tailed t-test). f, Tumour incidence in animals dying from natural causes (n = 14 for BL/6NZB, n = 20 for BL/6C57; **P < 0.01, Fisher’s exact test). g, Relative O2 consumption (coupled) and ATP synthesis rates in liver mitochondria (n = 4 per genotype and age; mean ± s.d.). h, O2 consumption (coupled) respiration in liver mitochondria simultaneously isolated from 22-, 30-, 120- and 300-day-old males (n = 8).
Four days after birth, respiration and ATP synthesis were lower in liver mitochondria from BL/6NZB mice. Both variables subsequently equalized, but from 100-days-old onwards, the rates in BL/6C57 liver mitochondria decreased progressively (Fig. 1g). These changes were due to a steady decline in BL/6C57 respiration, with BL/6NZB respiration remaining constant between days 20 and 300 (Fig. 1h). Liver mtDNA sequencing revealed no age-related increase in mtDNA mutations in either mtDNA backgrounds (Extended Data Table 2, Supplementary Data 1). Later metabolic deterioration, lower tumour incidence, slower telomere attrition11, and sustained ovarian function indicate that the NZB mtDNA promotes healthier ageing in the BL/6 nuclear background.
Gene expression signatures of 12-week-old mice (Fig. 2a and Supplementary Data 2) revealed differences in molecular functions related to ‘lipid and carbohydrate metabolism’ in liver and heart and ‘free radical scavenging’ specifically in liver (Fig. 2b). Consistent with increased PPARα activity, BL/6NZB mice had higher expression of lipid metabolism and lower expression of carbohydrate metabolism and inflammation pathways (Extended Data Fig. 4 and Supplementary Data 2).

a, Multidimensional scaling (MDS) representation of RNA-seq analysis (10-weeks-old, n = 3 pools per genotype, 4 mice per pool). b, Heatmap and hierarchical clustering of enrichment (−log10 Benjamini–Hochberg (BH)-adjusted P values) of the functions defined by the ingenuity pathway analysis (IPA) in set of genes differentially expressed between conplastic mice in heart (H) (top) and liver (L) (bottom). Yellow, non-significant (BH P = 1); red, highly significant (BH P ≤ 10−4). c, Partial least squares regression discriminant analysis (PLS-DA) plot for plasma and liver metabolites (10-weeks-old, n = 6 per genotype). d, MSEA of all detected liver (L) and plasma (P) GSH metabolites regardless of relevance to PLS-DA (red text denotes PLS-DA relevant), and of selected oxidative stress metabolic biomarkers. e, f, Heat maps and hierarchical clustering of enrichment (Zq differences between indicated genotypes and/or ages) of the proteins quantified by iTRAQ involved in ROS metabolism in either liver (e) or heart (f). See Supplementary Data 4 for identity of different proteins and Zq values. Purple text denotes mitochondrial location (n = 3 per genotype and age).
Plasma and liver metabolomics of 12–week-old mice confirm the mtDNA haplotype effect in three main groups: glutathione metabolism; digestion and absorption; and lipid and membrane metabolism (Fig. 2c, Extended Data Fig. 5 and Supplementary Data 3). Metabolite set enrichment analysis (MSEA) of glutathione metabolism revealed enhanced glutathione turnover12 and reactive oxygen species (ROS) defence in BL/6NZB animals (Fig. 2d and Extended Data Fig. 5b). In parallel, we detected lower levels of the hepatic lipid peroxidation product 12,13-hydroxyoctadec-9(Z)-enoate. Other affected metabolites included a number of fatty acids and lysophospholipids, consistent with the differences in membrane metabolism (Extended Data Fig. 5d).
Quantitative proteomics revealed a differential profile for the ROS defence proteome (Fig. 2e, f and Supplementary Data 4). In liver, the major mitochondria-located redox regulators Glrx2, Glrx5 and Txnrd2 were increased in young and old BL/6NZB mice, whereas cytosolic gamma-glutamyltranspeptidase 1 (involved in glutathione pool maintenance) and four mitochondrial redox regulators (Prdx3, Txnrd2, Sod2 and Nnt) were decreased in young BL/6NZB mice but then strongly increased on ageing (Fig. 2e). Ageing has profound but different effects on the ROS handling proteomes of the two conplastic mouse lines (Fig. 2e).
Measurement of mitochondrial ROS in vivo using MitoB13 revealed elevated ROS levels in young BL/6NZB mice in both liver and heart (Fig. 3a). As shown in cultured cells14, high levels of mitochondrial ROS correlated with mtDNA copy number (Fig. 3b). Chronic treatment of mice with N-acetyl cysteine (NAC) reduced the BL/6NZB MitoP:MitoB ratio (Fig. 3a); equalized mtDNA copy number (Fig. 3b); and reduced respiration and ATP synthesis rates in isolated liver mitochondria (Fig. 3b). The higher rate of mitochondrial ROS in young BL/6NZB animals (Fig. 3a) seems to conflict with their superior functional preservation with age. However, mitochondrial ROS production was inverted in animals above 30 weeks of age, with BL/6NZB liver mitochondria from middle-aged mice producing similar levels of ROS to young animals, whereas BL/6C57 mitochondria showed a sharp, age-related increase in ROS (Fig. 3c).

a, Relative amount of H2O2 ± NAC. b, Mitochondrial performance ± NAC. Liver: relative mtDNA amount; oxygen-dependent (coupled) respiration; pyruvate + malate-driven ATP production in isolated mitochondria. Heart: relative mtDNA amount. (12-week-old animals; n = 6 per genotype). c, Relative liver mitochondrial H2O2 generation (126 days old, n = 2 per genotype; 210 days old, n = 6 per genotype). d, Blood glucose after the indicated period of starvation (14 months old, n = 7 per genotype). e, The homeostasis model for insulin resistance (HOMA-IR) in overnight-starved mice (14 months old, n = 12 per genotype). f, Glucose tolerance test in starved mice (14 months old, n = 7 per genotype) g, Longitudinal quantification of lipid content by 1H MRS for the indicated tissue, age and genotype (number of individuals in brackets). h, Representative heart TEM images illustrating differences in mitochondria and lipid droplets between 20-week-old conplastic mice (n = 3 per genotype). i, Quantification of lipid content by 1H MRS in skeletal muscle (soleus) of 80-week-old animals (n = 4 per genotype). Data are means ± s.d., *P < 0.05, two-tailed t-test.
There were no dramatic differences in mitochondrial complex or supercomplex assembly (Extended Data Fig. 6a), and both strains showed the expected loss of supercomplex CIII:CIV due to the mutant form of SCAF1 characteristic of the BL/6 nuclear background15. However, BL/6NZB mice of all ages showed a slightly but consistently lower level of CI:CIII superassembly (Extended Data Fig. 6a, b).
Quantitative proteomics confirmed consistent behaviour patterns and between-genotype differences for structural proteins of all respiratory complexes, independently of whether they are encoded by nuclear or mtDNA genes (Extended Data Fig. 6c). In agreement with the differences in respiration (Fig. 3b), CI was less abundant in liver of young BL/6NZB mice (Extended Data Fig. 6c). In parallel with the age-related decline in respiration in BL/6C57 mice (Fig. 1h), the amounts of respiratory complexes in liver decrease in BL/6C57 but not in BL/6NZB mice (Extended Data Fig. 6c). An increase in the proportion of free CIII compared to that superassembled with CI is associated with adaptation to a fuel switch from glucose to fatty acids16. Consistent with this, the proteomic analysis revealed liver content changes in glycolytic and β-oxidization enzymes (Extended Data Fig. 6d).
Despite the influence of mtDNA haplotype on glucose and fatty acid metabolism, young mice showed no inter-strain differences in plasma glucose or in weight gain. In contrast, in one-year-old mice, the fasting-induced reduction in plasma glucose was significantly lower in BL/6C57 mice (Fig. 3d). This difference correlated with a lower capacity of BL/6C57 mice to regulate insulin levels (Fig. 3e) and to clear plasma glucose (Fig. 3f). Furthermore, on a high-fat diet (HFD), young BL/6C57 mice showed greater increases in body weight and white adipocyte size (Extended Data Fig. 7b, c). BL/6C57 mice showed an age-related increase in blood cholesterol, whereas levels in BL/6NZB remained steady (Extended Data Fig. 7a)
The mtDNA haplotype influence in lipid and glucose metabolism was confirmed by in vivo magnetic resonance spectroscopy (MRS) analysis of lipid content in liver, heart and skeletal muscle. Conplastic animals showed no differences in lipid content at 20 weeks (Fig. 3g); however, cardiac fibres of BL/6NZB mice had a significantly higher content and size of lipid droplets, suggesting a more active mobilization of lipids to the heart (Fig. 3h). The MRS analysis revealed that mature BL/6NZB adults (40 weeks) had higher tissue lipid content despite their lower body weight (Fig. 3g). At 80 weeks, lipid content in both strains was similar in liver and heart but was significantly higher in BL/6C57 muscle (Fig. 3g, i). This may reflect the accelerated deterioration of older BL/6C57 mice, with replacement of muscle mass by fat.
The mitochondrial degradome (mtDg) is involved in protein synthesis regulation, quality control, biogenesis, dynamics, mitophagy, and apoptosis17. Quantitative proteomics revealed between-strain differences in the mitochondrial degradome, especially in the heart, that were strongly influenced by age (Fig. 4a, b). Matrix proteases and chaperones involved in the mitochondrial unfolding response (mtUPR)18 were elevated in BL/6NZB animals, suggesting more active proteostasis (Fig. 4c, d). This was especially prominent in the liver, where elevation in stress chaperones was accompanied by elevation of proteases characteristic of the matrix (LonP1 and Clpp) and implicated in mtUPR (Fig. 4a, c).

See also Supplementary Data 4. a, b, Proteomic analysis of the mitodegradome. c, d, Detected proteases involved in the mitochondrial UPR and mitochondrial dynamics. Data are expressed as Zq differences between BL/6NZB and BL/6C57 mice aged 12 weeks (w) or 2 years (y) (mtDNA) or between old and young animals of the same genotype (age). e, Representative TEM images of mouse left ventricle from 12-week-old (3 animals per genotype) and 80-week-old mice (2 animals per genotype). f, Representative TEM of cardiomyocytes from 12-week-old animals treated with MitoQ (n = 3 per genotype). g, Morphometric analysis of mitochondria in TEM images of left ventricular cardiomyocytes obtained from 3 animals per group. Data are given as means ± s.e.m., non-parametric Kruskal–Walls test (not normal distribution), *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.00001; NS, not significant.
The conplastic strains showed differences in critical components of the apparatus involved in mitochondrial dynamics (Fig. 4c, d). Heart transmission electron microscopy (TEM) revealed between-strain differences in mitochondrial aspect and dynamics, affecting density, morphology and cristae area (Fig. 4e–g, Extended Data Fig. 8); with more mitochondrial fragmentation in hearts of old BL/6C57 mice (Fig. 4e). When young mice were treated with the mitochondria-specific antioxidant MitoQ19, heart mitochondria were less rounded in BL/6NZB mice (Fig. 4f) and had a decreased mitochondrial area (Fig. 4g). MitoQ did not affect lipid droplet-accumulation between BL/6C57 and BL/6NZB cardiomyocytes, but increased droplet size in BL/6C57 animals (Fig. 4g).
By highlighting the influence of mtDNA and nuclear (nDNA) interplay, our results demonstrate that substitution with a different wild-type mtDNA variant is sufficient to promote differences in mitochondrial function and a cellular adaptive response. This phenomenon is orchestrated by the complex network of mitochondrial stress response pathways in which proteostasis, mtUPR and ROS signalling lead to adaptive responses that impact on the organism’s metabolic performance and ageing. The potential for non-pathological mtDNA variants to shape organismal metabolism has implications for the emerging new field of mitochondrial replacement therapy in human oocytes20,21. Thus, consideration should be given to the use of donor mtDNA that is a close genetic match to the original mtDNA of the acceptor oocyte22.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Generation of conplastic mice and genotyping methodologies
Parental C57BL/6JOlaHsd and NZB/OlaHsd strains were purchased from Harlan Laboratories. The C57BL/6JOlaHsd strain was derived in 1974 from the C57BL/6J strain from Jackson Laboratories but differs from the parental strain. The C57BL/6JOlaHsd strain does not harbour the nicotinamide nucleotide transhydrogenase (NNT) spontaneous mutation that renders the encoded enzyme undetectable, and which is characteristic of the C57BL/6J strain provided by Jackson Laboratories. To avoid confusion, we call C57BL/6JOlaHsd mice C57BL/6 throughout the manuscript. Conplastic strains were obtained by backcrossing females (mitochondrial donors) with males of the parental recipient strain over 20 generations to create a new line harbouring the nuclear genome of one strain and the mtDNA of the other. Conplastic strains were maintained by backcrossing females (mitochondrial donors) of the conplastic strain with males of the parental recipient strain. Offspring after the tenth generation of backcrossing were considered conplastic. Total DNA extracted from tissue homogenates was genotyped by PCR restriction-fragment length polymorphism analysis (RFLP) and by Affymetrix Mouse Diversity Genotyping Array, which interrogates 623,000 single nucleotide polymorphisms (SNPs) covering the entire mouse genome at a resolution of 4.3 kb.
mtDNA next-generation sequencing
Liver samples were taken from 12- (n = 5 per genotype), 40- (n = 4 per genotype) and 80-week-old (n = 5 per genotype) conplastic mice. Total DNA was extracted with the DNeasy Blood and Tissue kit (Qiagen). mtDNA was amplified in two independent reactions using primers 7309Fw (CATAGGGCACCAATGATACTG) — 16265Rv (GAGTTTTGGTTCACGGAACAT) and 15358Fw (AAGAAGAAGGAGCTACTCCCC) — 8257Rv (CAGCTCATAGTGGAATGGCTA). Long-range PCRs were carried out using Takara Taq DNA Polymerase (Clontech). Products were cleaned using the QiAquick PCR Purification kit (Qiagen), quantified with the Qubit dsDNA BR Assay kit (Invitrogen) and mixed in equimolar amounts. mtDNA was then enzymatically sheared using the Ion Shear Plus Reagent (Ion Torrent) following the manufacturer’s instructions. To yield fragments with a median size of 200 bp, the shearing reaction was adjusted to 8 min at 37 °C, and fragments were size selected using Agencourt AMPure XP Reagent (Beckman Coulter). Both ends of each fragment were ligated with Ion P1 Adapters and the Ion Xpress Barcode Adapters. The ligated DNA was purified using Agencourt AMPure XP Reagent. Quantification and qualification for the amplified libraries were quantified and their quality assessed with Agilent 2200 TapeStation System and the Agilent High Sensitivity D1000 Screen Tape (Agilent Technologies) and with the Ion Library TaqMan Quantitation Kit (Ion Torrent). All barcoded libraries were pooled in equimolar amounts (50 pM) to ensure equal representation of each barcoded library in the sequencing run. Template preparation and Ion PI Chip loading were performed on the Ion Chef Instrument. NGS was performed on Ion Proton platform (Ion Torrent) with Ion PI Chip.
All raw reads were aligned to the C57BL/6J mouse mtDNA reference sequence (AY172335.1) using Torrent Mapping Alignment Program (TMAP) version 5.0.7. Variants were called using the Torrent Variant Caller (TVC) version 5.0.7, applying a minimum allele frequency threshold of 0.001 and reducing the coverage to 10,000 in over-sampled locations (keeping default values for somatic mutations for the rest of parameters) and then filed into variant call format (VCF) reports. Additionally, the relative frequency of the alternative allele was calculated for each individual and position. Cases deviating more than five standard deviations from the average frequency in that position for the 28 individuals were compared with variants obtained from TVC. In this way false positives (positions identified as variants in TVC but not clearly deviating from the mean value) were identified and discarded from the final variant set.
At the same time, variants found in the complete set of NZB individuals but lacking in all C57 individuals were identified as sequence differences between NZB mtDNA and C57 mtDNA. Extended Data Fig. 1b was generated with Geneious version 9.1.2, created by Biomatters.
Mouse experimentation
All animal procedures conformed to EU Directive 86/609/EEC and Recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes, enforced in Spanish law under Real Decreto 1201/2005. Approval the different experimental protocols require the estimation of the adequate sample size as well as the definition of the randomization and blinding criteria. Mice under endpoint criteria were excluded from analysis in long-term experiments. The mice were fed a standard chow diet (5K67 LabDiet) or, when indicated, a high-fat diet (D12492 Open Source Diets). Unless specified, experiments were carried out with males.
Growth curves
To calculate growth curves, mice were weighed at the starting age and every week/month thereafter for the period indicated in each experiment. Three independent groups of animals were used for Fig. 1b and Extended Data Figs 2a and 6b.
Survival study
Mice found dead at each daily inspection were considered as censored deaths, and were necropsied to tumour inspection. Criteria for euthanasia were based on an independent assessment made by a veterinarian, and only cases where the condition of the animal was considered incompatible with continued survival are represented as deaths in the curves. Survival curves, weight gain and tumour incidence analysis (Fig. 1a, b, f) were done in the same group of animals.
Behaviour and metabolism analysis
To analyse food and water intake, an independent group of 10–12 males per strain were individually housed, and weekly intake was calculated as the difference between the supplied weight of food and drink and the weight remaining after one week. Intake was calculated in males because females were grouped and had to compete for food and drink. Mouse activity was evaluated in 10-week-old mice using the PHYSIOCAGE system (Panlab). Animals were acclimatized to the room and cages in the week before the experiment, and measurements were made on four consecutive days (five days and four nights). Data were analysed with Metabolism v2.2 (Panlab). The first diurnal phase (12 h) was considered part of the acclimatization period and was eliminated from the final result. Indirect calorimetry measurements were made of individual 10-week-old mice (10 per sex and strain) housed in Oxylet System metabolic chambers (Panlab Harvard Apparatus). Before experiments, mice were acclimatized in the cages for 1 week. Throughout the 24 h measurement period, oxygen consumption and carbon dioxide production rates were measured at 24-min intervals, with a 4 min sample period and a 1 min purge; simultaneous measurements were made in four metabolic chambers. Respiratory exchange rate (RER, or respiratory quotient, RQ), a measure of metabolic substrate choice, was calculated as the ratio between CO2 production and O2 consumption. Energy expenditure (EE) was calculated as (3.815 + (1.232 × RQ)) × VO2 × 1.44). Data were analysed with Metabolism v2.2 (Panlab). Investigators were not blinded to the group allocation during the experiment and the sub-groups of animals included in repeated studies was randomly organized.
Glucose metabolism and blood biochemistry analysis
Experiments were performed in an independent group of 12 mice per genotype. Mice fed the chow diet were fasted for 6 or 16 h with free access to water. For intraperitoneal (IP) glucose tolerance tests (GTT), the mice received glucose injections of 1 or 2 mg per kg. Mice were bled from a tail clip. Blood glucose was measured with a handheld glucometer before injection (time 0) and at 15, 30, 60, 90 and 120 min after injection. Insulin was measured by an enzyme-linked immunosorbent assay (ELISA) (Millipore), 3 technical replicates per sample. The homeostasis model for insulin resistance (HOMA-IR) was calculated from the fasting blood glucose (mmol per litre) × fasting plasma insulin (μU ml−1) divided by 22.5.
Levels of serum cholesterol (ChoT), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) were determined by biochemical analysis of submandibular blood samples. Serum biochemistry was conducted in a Dimension RxL Max automated analyser. Investigators were not blinded to the group allocation when assessing the outcome.
Histological analysis
Tissue samples were fixed in 4% paraformaldehyde (24 h), processed and embedded in paraffin. Sections (5 μm) were prepared and mounted on coverslips for staining with haematoxylin and eosin and tissue images were captured using a NanoZoomer 2.0RS digital slide scanner (Hamamatsu). Sections of epididymal fat pad stained for white adipose tissue were examined under a Nikon 90i microscope fitted with a 10× objective. Adipocyte cross-sectional area was determined from epididymal fat pads using ImageJ software (4 sections per animal). Slides from liver necropsies were digitalized and analysed with NDP.view2 viewing (Hamamatsu). Lipofuscin area was determined from digitalized haematoxylin and eosin histological ovary sections (3 sections per ovary) using NDP.view2. Measurements were obtained by applying colour thresholds with ImageJ 1.48v (NIH). Investigators were blinded to the group allocation when assessing the outcome.
In vivo ROS determination
In vivo mitochondrial hydrogen peroxide was measured using the MitoB mass spectrometric probe as described previously13,23. Briefly, 25 nmol MitoB was injected into the tail vein between 8 a.m. and 10 a.m. After 6 h, mice were euthanized by cervical dislocation and tissues were harvested and flash frozen in liquid nitrogen. Tissues were stored at −80 °C and transported to Cambridge, UK on dry ice for analysis. When indicated, mice were treated with 1% NAC in drinking water for 8 weeks before MitoB injection. Investigators were blinded to the group allocation when assessing the outcome.
OXPHOS function
Mitochondria were isolated from mouse liver samples as described15, and oxygen consumption and ATP production were measured15. For mtDNA copy number quantification, total DNA was isolated from tissue homogenate preparation (liver and heart) and was quantified by real-time PCR using 7900HT Fast Real-Time PCR System (Applied Biosystem) as described14. When indicated, mice were treated with 1% NAC in drinking water for 8 weeks before sample collection. Each data represent mean of 4 technical replicates. Investigators were not blinded to the group allocation when assessing the outcome.
Blue native electrophoresis (BNE)
Supercomplex levels and composition were analysed in isolated mitochondria from liver and heart by BNE as described24 using the following antibodies: core1, ndufa9 (Abcam); FpSDH, COXI (Novex) and Tom20 (Santa Cruz Biotech). Investigators were not blinded to the group allocation when assessing the outcome.
Telomere length analysis
Telomere length was measured in tail skin samples by quantitative fluorescence in situ hybridization (Q-FISH). Paraffin-embedded tissue sections (7 μm) were hybridized with a Cy3-labelled PNA probe containing the telomere repeat sequence25. DAPI and Cy3 signals were acquired in separate channels using a confocal microscope (Leica SP5) fitted with a 60× objective. All slides were stained at the same time, and images were acquired consecutively, with lasers kept at the same intensity to avoid day-to-day variations. Maximum intensity projections from 12-bit image stacks (15 sections at 1.0 μm steps) were generated for image quantification.
The DAPI images were signal-intensity thresholded and segmented using ImageJ. Quantitative image analysis was performed using the Metamorph platform (Molecular Devices)25. After conversion to a 1-bit binary mask, the DAPI image was used to define the nuclear area. The binary DAPI mask was applied to the Cy3 image to obtain a combined image with the telomere fluorescence information for each nucleus. Cy3 fluorescence intensity was measured in arbitrary fluorescence units (au) as the mean grey value in each nucleus. Only cells from the bulge of the hair follicle were analysed. Telomere intensity values were exported to Excel. The analysis was conducted on one-year-old BL/6C57 (n = 6), one-year-old BL/6NZB (n = 6), two-year-old BL/6C57(n = 4), and two-year-old BL/6NZB (n = 5) mice, and two images were acquired per animal. The number of cells analysed per group is indicated in the frequency histograms (Fig. 1i).
The statistical significance of differences in telomere length distributions was calculated with a Wilcoxon’s rank sum test. A two-tailed t-test was used to compare the reduction in mean telomere length and the percentages of short and long telomeres. Calculations were made in Microsoft Excel and Prism. Differences were considered significant at *P < 0.05, at **P < 0.01, and at ***P < 0.001. Investigators were not blinded to the group allocation when assessing the outcome.
Phenotype scoring
Mice aged ~2 years old were scored for a variety of ageing markers (reduced hair density, greying hair, kyphosis, body weight reduction and development of systemic neoplasias) as previously described10. Investigators were blinded to the group allocation during the experiment.
Transcriptomic analysis
Twelve 10-week-old males per genotype were killed by cervical decapitation, and liver (median lobe) and heart (left ventricle) tissues were dissected and immediately frozen in liquid nitrogen. Total RNA was extracted with TRIzol reagent and then purified on RNeasy spin columns (Qiagen). RNA integrity (RNA Integrity Score ≥ 7.9) and quantity were determined with an Agilent 2100 Bioanalyzer. Equal RNA amounts from individual animals were combined at 4 samples per pool, and 3 pools per genotype were analysed in the Illumina Genome Analyser IIx. Reads were pre-processed with Cutadapt 1.2.1 to remove the TruSeq adaptor. To focus on nuclear genes, we identified and removed mitochondrial reads by aligning all reads to mitochondrial DNA using Tophat. Resulting reads were mapped on the mouse transcriptome (Ensembl gene-build GRCm38.v70) and genome, using RSEM v1.2.3 and Bowtie2 v2.0.6. Genes with at least one count per million in at least three samples were considered for downstream analysis. As data were generated in two different sequencing runs, we used the R function Combat (http://www.bu.edu/jlab/wp-assets/ComBat/Download.html) to adjust for potential batch effects. Statistical analysis was performed on the remaining set of genes using the bioconductor package EdgeR26. Raw reads and TMM-normalized batch corrected counts have been deposited in GEO with the accession number GSE56933. Sets of genes differentially expressed across conditions were analysed for functional associations using IPA (Ingenuity Knowledge Database, http://www.ingenuity.com). Benjamini–Hochberg adjusted P values are reported for differential gene expression and IPA functional analysis. Gene Set Enrichment Analysis (GSEA27) identified sets of co-expressed genes that are functionally linked based on different ontologies and/or pathways. Gene sets with a FDR <0.25 will be considered to functionally related. Investigators were not blinded to the group allocation when assessing the outcome.
Metabolomic analysis
Samples of liver and plasma from 10-week-old mice (n = 6 per genotype) were collected, frozen in liquid nitrogen and sent to Metabolon (Durham, NC, USA) for metabolomics analysis according to their proprietary methodology. The analysis profiled 313 metabolites in liver samples and 287 in plasma, with 197 common to both sources.
For the multivariate model analysis, between-group differences in the abundance of metabolites identified by mass spectrometry (LC–MS and GC–MS) were assessed by Welch’s two-sample t-test. Differences were considered statistically significant at P < 0.05. Chemometric statistical analysis was performed using R scripts (http://cran.r-project.org/) and the mixOmics package (v. 5.0.1) executed in RStudio (v. 0.98.501) for MacosX. Most metabolomic analysis employs unsupervised principal components analysis (PCA) to explore the quality of data and detect possible outliers and a supervised partial least squares-discriminant analysis (PLS-DA) to determine the number of metabolites responsible. PLS-DA is a supervised extension of PCA used to distinguish two or more classes by searching for original metabolic variables (X matrix) that are correlated to class membership (that is, liver, plasma, and mouse model) (Y matrix). Results of the PCA and PLS-DA were visualized by a scatter score plot of different PCs (PCA) and the first two latent variables (PLS-DA). The original X matrix contains the metabolic variables, while the Y matrix contains the class variable for which values are chosen to be the class descriptor. We performed the analysis by comparing BL/6C57 samples with BL/6NZB samples, and liver samples with plasma samples. False discovery rate and overfitting were avoided by using a leave-one-out cross-validation, performed with the calibration data set, to determine the number of latent variables for each fold and to construct a model. The resulting model was applied to the test data set, including the 287 plasma metabolites and the 313 liver metabolites, to compute the predicted Y values. Due to the small number of samples, this process was repeated the number of times needed in each analysis so that every sample served as an independent test sample once and only once. The predicted Y values produced by the cross-model validation were used to examine the classification error and determine the number of most discriminant metabolites. Based on this, we selected only those metabolites that appear in at least 50% of the repeated analysis. For metabolomic set enrichment analysis (MSEA)27, raw data from the metabolic analysis were normalized by quantiles using the Bioconductor LIMMA package. Metabolites with missing values were excluded. A moderated t-test, also implemented in the LIMMA package, was used to calculate log fold-differences between male BL/6NZB and BL/6C57 mice for liver and plasma samples separately, together with raw and Benjamini–Hochberg adjusted P values. MSEA was then run on the normalized samples and the signal-to-noise ratio statistic was used to rank metabolites in each pathway of interest. A false discovery rate (FDR) <0.25 was considered significant. We considered sets of at least 10 metabolites in all analyses, except for bile acid, which required at least 5 metabolites to be in the same class for a result to be considered significant.
Proteomic analysis
Heart and liver protein extracts from 12- or 80-week-old BL/6C57 and BL/6NZB mice (n = 3 per genotype and age) were reducted, alkylated with iodoacetamide and digested with trypsin and the resulting peptides were labelled with iTRAQ8-plex, according to the manufacturer’s instructions. Labelled peptides were injected onto a C-18 reversed phase (RP) nano-column (75 μm I.D. and 50 cm, Acclaim PepMap, Thermo Fisher, San José, CA, USA) and analysed in a continuous acetonitrile gradient consisting of 8–31% B for 240 min, and 50–90% B for 1 min (B = 0.5% formic acid in acetonitrile). Peptides were eluted from the RP nano-column at a flow rate of ~200 nl min−1 to an emitter nanospray needle for real-time ionization and peptide fragmentation in a Q-Exactive HF mass spectrometer (Thermo Fisher). Mass spectra were acquired in a data-dependent manner, with an automatic switch between MS and MS/MS using a top 20 method.
The raw files were analysed with Proteome Discoverer (version 1.4, Thermo Fisher Scientific), using a Uniprot database containing a joint Mouse + Rat Swissprot database (Uniprot release 57.3 May 2009; 26.885 entries). For database searching, parameters were selected as follows: trypsin digestion with 2 maximum missed cleavage sites, precursor mass tolerance of 800 p.p.m., fragment mass tolerance of 30 mmu, variable methionine oxidation and fixed cysteine carbamidomethylation, lysine and N-terminal modification of + 304.2054 (iTRAQ label). The same collections of MS/MS spectra were also searched against inverted databases constructed from the same target databases. Peptide identification from MS/MS data was performed using the probability ratio method28. FDR of peptide identifications were calculated using the refined method29; 1% FDR was used as the criterion for peptide identification. Each peptide was assigned only to the best protein proposed by the Proteome Discoverer algorithm. Quantitative information was extracted from iTRAQ sample MS/MS spectra using an in-house developed program (QuiXoT) and protein abundance changes were analysed using the Generic Integration Algorithm (GIA), as described in ref. 30. The GIA was also used to integrate the quantitative protein information from several experiments into a unique protein value. Calculation of statistical weights of each quantitation at the spectrum level was performed according to the WSPP model. The validity of the null hypothesis at each one of the levels (spectrum, peptide, protein within an experiment and protein) was carefully checked by plotting the cumulative distributions. Three mice for each genetic background and age were used as biological replicates for heart experiments, and two mice were used for liver experiments. Changes in relative protein abundance are expressed using the standardized log2 ratio (Zq) variable30. Investigators were not blinded to the group allocation when assessing the outcome.
1H magnetic resonance spectroscopy (MRS)
Male BL/6NZB and BL/6C57 mice (80 weeks old (n = 4–5), 20 weeks old (n = 6–8)) weighing 30–50g each were housed in groups of 4 with unrestricted access to food and water. Spectroscopy examinations were performed with a 7T preclinical system (Agilent Varian, Palo Alto, USA) equipped with a DD2 console and an active shielded 205/120 gradient insert coil with a 130 mT per m maximum strength. A double-tuned circular transmit/receive coil was used for phosphorus/proton (20 mm), located mainly on the soleus muscle, heart and liver (Rapid Biomedical GmBH, Rimpar Germany). Investigators were blinded to the group allocation during the experiment.
Proton spectra were acquired by 128 transients with 2,048 complex points with a spectral bandwidth of 10 kHz and a repetition time of 1.2 ms. Spectra were acquired with adiabatic radiofrequency pulses to improve sensitivity and minimize spectral distortions with an Ernst flip angle. Proton chemical shifts were expressed relative to the water signal (4.7 p.p.m.). Signals in nuclear magnetic resonance spectra were determined quantitatively on a Macintosh computer by integration after automatic or manual baseline correction, with fitting spectrum peak (after phase and baseline correction) to a Lorentzian function using the Mestrenova program (Mestrelab Research, Santiago de Compostela, Spain; released 2015-02-04 version:10.0.1-14719). An exponential line broadening (3 Hz for proton) was applied before Fourier transformation.
Transmission electron microscopy (TEM)
Mice were euthanized by cervical dislocation and the left ventricle apex was immediately dissected (12 weeks old, n = 3 per genotype and 80 weeks old, n = 2 per genotype). Heart samples were fixed in 2.5% glutaraldehyde, 4% formaldehyde in 0.1 M HEPES buffer for 4–5 h. After buffer washes, samples were post-fixed for 1 h at room temperature in a 1:1 solution of 1% osmium tetroxide and 3% aqueous potassium ferrocyanide. Samples were rinsed in distilled H2O. Tissues were dehydrated through a graded acetone series and embedded in Spurr’s low viscosity embedding mixture (Electron Microscopy Sciences). Ultra-thin sections (60 nm) were then mounted on copper grids and stained with lead citrate. Samples were examined on a JEOL 10-10 electron microscope through 1,500×, 5,000×, 40,000× and 80,000× objectives. Mitochondrial morphometry, cristae area (2 images, 5,000×, per animal) and lipid droplets (50 images, 5,000×, per animal) were segmented manually and analysed using Fiji (http://fiji.sc/Fiji) and ImageJ 1.48v software. The investigator was blinded to the group allocation when assessing the outcome.
Statistical analysis
Unless specified, statistical analyses and graphics were produced with GraphPad Prism 6 software. Data sets were compared by unpaired two-tailed t-tests. Differences were considered statistically significant at P values below 0.05. *P < 0.05; **P < 0.005; ***P < 0.0005. All results are presented as mean ± s.d. or mean ± s.e.m.
References
Wallace, D. C. Mitochondrial DNA variation in human radiation and disease. Cell 163, 33–38 (2015)
Ruiz-Pesini, E. et al. Human mtDNA haplogroups associated with high or reduced spermatozoa motility. Am. J. Hum. Genet. 67, 682–696 (2000)
Brown, M. D. et al. The role of mtDNA background in disease expression: a new primary LHON mutation associated with Western Eurasian haplogroup J. Hum. Genet. 110, 130–138 (2002)
Baudouin, S. V. et al. Mitochondrial DNA and survival after sepsis: a prospective study. Lancet 366, 2118–2121 (2005)
Amo, T. & Brand, M. D. Were inefficient mitochondrial haplogroups selected during migrations of modern humans? A test using modular kinetic analysis of coupling in mitochondria from cybrid cell lines. Biochem. J. 404, 345–351 (2007)
Amo, T., Yadava, N., Oh, R., Nicholls, D. G. & Brand, M. D. Experimental assessment of bioenergetic differences caused by the common European mitochondrial DNA haplogroups H and T. Gene 411, 69–76 (2008)
Roubertoux, P. L. et al. Mitochondrial DNA modifies cognition in interaction with the nuclear genome and age in mice. Nature Genet. 35, 65–69 (2003)
Yu, X. et al. Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains. Genome Res. 19, 159–165 (2009)
Sharpley, M. S. et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343 (2012)
Ross, J. M. et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501, 412–415 (2013)
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013)
Soga, T. et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 281, 16768–16776 (2006)
Cochemé, H. M. et al. Using the mitochondria-targeted ratiometric mass spectrometry probe MitoB to measure H2O2 in living Drosophila. Nature Protocols 7, 946–958 (2012)
Moreno-Loshuertos, R. et al. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nature Genet. 38, 1261–1268 (2006)
Lapuente-Brun, E. et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570 (2013)
Guarás, A. et al. The CoQH2/CoQ ratio serves as a sensor of respiratory chain efficiency. Cell Rep. 15, 197–209 (2016)
Quirós, P. M., Langer, T. & López-Otín, C. New roles for mitochondrial proteases in health, ageing and disease. Nature Rev. Mol. Cell Biol. 16, 345–359 (2015)
Houtkooper, R. H. et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457 (2013)
Kelso, G. F. et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596 (2001)
Tachibana, M. et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 461, 367–372 (2009)
Craven, L. et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 465, 82–85 (2010)
Chinnery, P. F. et al. The challenges of mitochondrial replacement. PLoS Genet. 10, e1004315 (2014)
Logan, A. et al. Using exomarkers to assess mitochondrial reactive species in vivo. Biochim. Biophys. Acta 1840, 923–930 (2014)
Acín-Pérez, R., Fernández-Silva, P., Peleato, M. L., Pérez-Martos, A. & Enriquez, J. A. Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539 (2008)
Flores, I. et al. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 22, 654–667 (2008)
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010)
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005)
Martínez-Bartolomé, S. et al. Properties of average score distributions of SEQUEST: the probability ratio method. Mol. Cell. Proteomics 7, 1135–1145 (2008)
Navarro, P. & Vázquez, J. A refined method to calculate false discovery rates for peptide identification using decoy databases. J. Proteome Res. 8, 1792–1796 (2009)
García-Marqués, F. et al. A novel systems-biology algorithm for the analysis of coordinated protein responses using quantitative proteomics. Mol. Cell. Proteomics 15, 1740–1760 (2016)
Acknowledgements
We thank C. López-Otín for critical reading; C. Jimenez for management; the Transgenesis Unit of the CNIC and M. Cueva for mouse work, A. Molina and R. Doohan for histology, the Genomics Unit of the CNIC for RNA-seq; ICTS-CNME (UCM) for technical support; J. Amigo and X. Bello for NGS bioinformatics support; and S. Bartlett for English editing. Supported by grants from the MEyC (SAF2015-65633-R, CSD2007-00020, SAF2012-38449, BIO2012-37926 and BIO2015-67580-P), the Madrid (CAM/P2010/BMD-2402), the EU (PCIG10-GA-2011-304217 and FP7-PEOPLE-2012-ITN-GA-317433), the ISC III (PI09-00946, PI12/01297, PI13-01136 and PI11-00078, PRB2 (IPT13/0001, ProteoRed), RIC ((RD12/0042/0045) and RETICS (RD12/0042/00056)). The CNIC is supported by the MEyC and the Pro-CNIC Foundation and is a SO-Center of Excellence (SEV-2015-0505).
Author information
Authors and Affiliations
Contributions
A.L.-P., R.M.-L., A.V.L.-V., R.A.-P. and S.C. performed experimental work. F.S.-C. and C.T. analysed genomic and transcriptomic data. E.A. and I.F. performed the telomere analysis. A.G.-G., M.L.B.-M. and E.R. performed the mouse phenotyping. A.L. and M.P.M. did the assessment of ROS levels. R.C., B.S. and A.C. analysed the microarrays and mtDNA next-generation sequencing. A.V.L.-V. and J.R.-C. analysed the metabolomics. E.C. and J.V. performed the proteomics. J.A.E., A.L.-P., R.M.-L., A.P.-M. and P.F.-S. contributed to design and analysis. A.L.-P. and J.A.E. wrote the manuscript. J.A.E. directed and designed the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Conplastic mouse generation and nuclear genome identity.
a, Schematic of the conplastic animal concept. b, Sequence differences between NZB and C57 mtDNAs by next-generation sequencing. See also Extended Data Table 1. c, Cluster dendrogram based on the expressed genome of the indicated mouse strain and the RNA-seq-derived expressed genome of conplastic animals. d, Comparison of nuclear single nucleotide polymorphism (SNP) differences between the indicated mouse lines by Affimetrix Mouse Diversity Genotyping Array analysis. e, Percentage of genotype differences between the mouse nuclear DNA SNPs identified by RNA-seq; the number of SNPs compared is indicated.
Extended Data Figure 2 Young conplastic mouse phenotyping.
a, Weight-gain during early life (BL/6NZB, BL/6C57, and NZBNZB separated by sex, n = 10 per group). b, Organ-weight to body-weight ratios separated by sex (n = 10 per group, ANOVA test). c, Activity profiles (BL/6NZB, BL/6C57, and NZBNZB separated by sex, 10-week-old mice, n = 10 per group). d, Area under the curve analysis of the activities shown in c, providing estimated activity intensities of the different strains. e, Ratios of activity to respiratory parameters in the different mouse strains. Data are means ± s.d., *P < 0.05, two-tailed t-test.
Extended Data Figure 3 Ageing phenotype of conplastic mice.
a, Two-year-old mice were scored for conspicuous signs of the indicated ageing parameters (0 (none) to 3 (high))10. b, Representative image of 2-year-old animals. c, Tumours incidence detected upon liver necropsy of animals dying from natural causes (BL/6NZB, n = 14; BL/6C57, n = 20 mice). d, Representative haematoxylin and eosin stained paraffin sections of liver adenoma, carcinoma, histiocytic sarcoma and hemangiosarcoma. e, Representative histological preparations of ovaries from animals of the indicated age and genotype. f, Lipofuscin accumulation over the reproductive period of conplastic females (n = 5 females per genotype and age) *P < 0.05, two-tailed t-test.
Extended Data Figure 4 Additional transcriptomic data.
a, Chord plot comparing liver expression of PPARα-regulated genes in conplastic mice. The chord plot shows gene changes (logFC) and relates them to their functional categories. b, Gene set enrichment analysis (GSEA), showing false discovery rate (FDR) estimates of significant and non-significant differences between conplastic mice for liver gene expression linked to the indicated processes.
Extended Data Figure 5 Differentially regulated metabolites and metabolic pathways in liver and plasma.
a, PLS-DA-identified metabolites responsible for differential metabolic signatures (P, plasma; L, liver). The metabolite list was cross-validated, and only metabolites present in 50% or more iterations were considered. Colour highlighting indicates metabolites as potential markers of the indicated metabolic route. b, Glutathione metabolic pathway and heat map showing MSEA assessment of all detected metabolites implicated in GSH metabolism, regardless of whether they were identified as relevant in PLS-DA (red text indicates PLS-DA relevant), and of selected oxidative stress metabolic biomarkers. These data are also shown in Fig. 2d. c, Heat map of detected metabolites implicated in bile acid and xenobiotic metabolism. d, Heat map of detected metabolites implicated in phospholipid metabolism. Heat maps are accompanied by MSEA assessment of significant (red) or nonsignificant differences (FDR). Colour-coding in all heat maps represents the log fold difference between BL/6NZB and BL/6C57 mice (black, not detected; white, no change (log fc = 1); red, trend for enrichment in BL/6NZB samples (log fc >1); yellow, trend for depletion in BL/6NZB samples (log fc <1).
Extended Data Figure 6 Quantitative proteomics analysis of mitochondrial complexes and functional categories.
a, Blue native electrophoresis of liver samples from 8-, 12-, 16- and 50-week-old conplastic mice. Top: complex I, (Ndufa9); middle: complex III (core-2); bottom: complex IV (Cox5b) and Tom20. b, Ratio of supercomplexed to free complex I, determined from the CI western blot in a. c, Relative abundance of component proteins of CI, CIV and CIII detected by iTRAQ quantitative proteomics in heart and liver of the indicated mouse strain at 12 weeks and 2 years of age. Each dot represents a single protein; red dots represent mtDNA-encoded proteins. d, Relative abundance of proteins of the TCA cycle, glycolysis, or β-oxidation detected by iTRAQ quantitative proteomics in heart and liver of the indicated mouse strain at 12 weeks old and 2 years of age. The four succinate dehydrogenase (complex II) subunits are highlighted in green. The iTRAQ-detected proteins and Zq values represented in c and d are compiled in Supplementary Data 4. Data are means ± s.d., ANOVA test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.00001.
Extended Data Figure 7 Lipid metabolism phenotype.
a, Serum cholesterol levels (ChoT), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) in young (2 months old, n = 8 per genotype) and adult (12 months old, n = 12 per genotype). b, Three-month body-weight gain in males fed chow (n = 4 per genotype) or a HFD (n = 8 per genotype). c, Adipocyte size in epididymal fat pads of HFD-fed conplastic males (n = = 3 per genotype and diet: BL/6C57 chow diet = 368 cells, HFD = 60 cells; BL/6NZB chow diet = 353 cells, HFD = 209 cells).
Extended Data Figure 8 Mitochondrial cristae and density.
a, Representative masks of individual heart mitochondria from 12-week-old mice (n = 3 per genotype). b, The cristae area per mitochondria estimated from masks. Significance was assessed by nonparametric Mann–Whitney U-test. Bars and whiskers represent means ± s.d.; each point represents the percentage of cristae area in an individual mitochondria; (BL/6NZB, n = 33; BL/6C57, n = 31) c, d, Aspect and electro-density of subsarcolemmal (SBS) or intermyofibrillar (IMF) mitochondria from left heart ventricular apex of 12-week-old mice.
Supplementary information
Supplementary Data 1
Data from the Next generation sequencing of mtDNA from both conplastic strains at three different ages. (XLSX 116 kb)
Supplementary Data 2
Data compiling the transcripts differentially expressed in liver and heart between conplastic mouse strains. (XLSX 165 kb)
Supplementary Data 3
Compilation of the metabolomic analysis of plasma and liver from complastic mice strains. (XLSX 234 kb)
Supplementary Data 4
Compilation of the iTRAQ proteomic analysis. (XLSX 71 kb)
Supplementary Information
This zipped file contains the original gel data for Extended Data Figure 5a. (ZIP 2111 kb)
Rights and permissions
About this article

Cite this article
Latorre-Pellicer, A., Moreno-Loshuertos, R., Lechuga-Vieco, A. et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565 (2016). https://doi.org/10.1038/nature18618
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature18618
- Springer Nature Limited
This article is cited by
-
Eight Strategies to Engineer Acceptance of Human Germline Modifications
Journal of Bioethical Inquiry (2024)
-
Mitochondrial Genetics and Function as Determinants of Bone Phenotype and Aging
Current Osteoporosis Reports (2023)
-
Peripheral modulation of antidepressant targets MAO-B and GABAAR by harmol induces mitohormesis and delays aging in preclinical models
Nature Communications (2023)
-
Sex-specific genetic regulation of adipose mitochondria and metabolic syndrome by Ndufv2
Nature Metabolism (2021)
-
Mitochondrial DNA impact on joint damaged process in a conplastic mouse model after being surgically induced with osteoarthritis
Scientific Reports (2021)