

Abstract
Electric-field-induced charge separation (polarization) is the most fundamental manifestation of the interaction of light with matter and a phenomenon of great technological relevance. Nonlinear optical polarization1,2 produces coherent radiation in spectral ranges inaccessible by lasers and constitutes the key to ultimate-speed signal manipulation. Terahertz techniques3,4,5,6,7,8 have provided experimental access to this important observable up to frequencies of several terahertz9,10,11,12,13. Here we demonstrate that attosecond metrology14 extends the resolution to petahertz frequencies of visible light. Attosecond polarization spectroscopy allows measurement of the response of the electronic system of silica to strong (more than one volt per ångström) few-cycle optical (about 750 nanometres) fields. Our proof-of-concept study provides time-resolved insight into the attosecond nonlinear polarization and the light–matter energy transfer dynamics behind the optical Kerr effect and multi-photon absorption. Timing the nonlinear polarization relative to the driving laser electric field with sub-30-attosecond accuracy yields direct quantitative access to both the reversible and irreversible energy exchange between visible–infrared light and electrons. Quantitative determination of dissipation within a signal manipulation cycle of only a few femtoseconds duration (by measurement and ab initio calculation) reveals the feasibility of dielectric optical switching at clock rates above 100 terahertz. The observed sub-femtosecond rise of energy transfer from the field to the material (for a peak electric field strength exceeding 2.5 volts per ångström) in turn indicates the viability of petahertz-bandwidth metrology with a solid-state device.
Similar content being viewed by others
Main
Matter responds to electromagnetic radiation by a displacement of its electrons with respect to the nuclei, turning its atomic constituents into dipole antennas. The overall strength of these dipoles per unit volume is characterized by the polarization vector, P. Its dependence on the incident electric field, E(t), describes the macroscopic material response. Its nonlinear component, PNL, constitutes the basis for manipulating the electronic and optical properties with the electric field of light1,2. The energy transferred from the electromagnetic field to the medium per unit volume can be expressed as:

Here we assume that the contribution of linear polarization to W(t) is negligible. This is a prerequisite for ultrahigh-rate signal manipulation, which relies on low dissipation. In fact, it is this dissipation that has limited the clock rate in contemporary integrated digital electronics to several gigahertz15 for more than a decade16,17.
A substantial increase of the electronic processing speed requires a new paradigm that is capable of greatly reducing the dissipation per switching cycle. Recent experiments indicated a possible way of advancing contemporary microwave electronics to the frequency of visible light by manipulating the electronic and optical properties of wide-bandgap materials with strong visible light fields at photon energies much smaller than the bandgap of the material18,19. However, the crucial question of how the energy density deposited irreversibly per switching cycle, Wirreversible, relates to the reversible energy exchange per unit volume, Wreversible, could not be answered. Pushing the frontiers of information processing to optical frequencies requires minimizing Wirreversible while keeping Wreversible high enough for reliable signal processing. Insight into field–matter energy exchange at optical frequencies requires access to W(t) on a sub-femtosecond scale.
To this end, we propagated a strong, linearly polarized field E(t) and its strongly attenuated replica Eref (t) = βE(t) through a thin sample of a transparent wide-bandgap material, in our case fused silica, of thickness  . The attenuation factor β is sufficiently small to prevent any observable nonlinear material response to Eref(t). Both transmitted waveforms are recorded in a measurement sequence as outlined in Fig. 1, once attenuated after and once attenuated before the sample by the same attenuation factor β. We show in Supplementary Information section 1 how a difference between these transmitted waves, ΔE(t) = E(
. The attenuation factor β is sufficiently small to prevent any observable nonlinear material response to Eref(t). Both transmitted waveforms are recorded in a measurement sequence as outlined in Fig. 1, once attenuated after and once attenuated before the sample by the same attenuation factor β. We show in Supplementary Information section 1 how a difference between these transmitted waves, ΔE(t) = E( , t) − β−1Eref(
, t) − β−1Eref( , t), directly yields the nonlinear polarization PNL(t) induced by the strong field E(t); see Supplementary equation (13).
, t), directly yields the nonlinear polarization PNL(t) induced by the strong field E(t); see Supplementary equation (13).

To induce a nonlinear material response PNL(t), the incident strong field E(0, t) is transmitted through the sample, and subsequently its amplitude is decreased by the attenuation factor β before sampling the transmitted electric field waveform in a streak camera setup (I). The nonlinear polarization response is ‘deactivated’ by attenuating the incident field before the sample and transmitting the weak reference field Eref(0, t) = βE(0, t) through the medium under scrutiny (II). The difference between the output waveforms, ΔE(t) = E( , t) − β−1Eref(
, t) − β−1Eref( , t), directly yields the nonlinear polarization of the medium, PNL(t), see Supplementary Information section 1. The false-colour plot shows a typical attosecond streaking spectrogram of the transmitted waveform used in the experiments.
, t), directly yields the nonlinear polarization of the medium, PNL(t), see Supplementary Information section 1. The false-colour plot shows a typical attosecond streaking spectrogram of the transmitted waveform used in the experiments.
In our experiments, we focus few-cycle near-infrared waveforms carried at a wavelength of  = 750 nm into thin fused silica samples (
= 750 nm into thin fused silica samples ( = 10 μm); for details of the experimental setup and procedures see Supplementary Information section 2. The focus of the transmitted waveform (that is, the interaction region) is imaged into an attosecond streak camera20,21. Here the temporal evolution of the transmitted electric fields E(z =
= 10 μm); for details of the experimental setup and procedures see Supplementary Information section 2. The focus of the transmitted waveform (that is, the interaction region) is imaged into an attosecond streak camera20,21. Here the temporal evolution of the transmitted electric fields E(z =  , t) and Eref(z =
, t) and Eref(z =  , t) (henceforth referred to as E(t) and Eref(t)) is sampled with sub-250-as extreme-ultraviolet pulses. The peak intensities Ipeak of the strong and attenuated waves have been set to (1.3 ± 0.1) × 1014 W cm−2 and (6.7 ± 0.3) × 1012 W cm−2, respectively. Figure 2a compares the transmitted fields and reveals the evolution of the nonlinear phase shift, ΔφNL(t), induced by the strong field. ΔφNL(t) increases towards the pulse peak, tapers off on its tail and finally vanishes; see insets to Fig. 2a. For Epeak ≈ 2.6 ± 0.1 V Å−1, the induced phase shift at the field maximum amounts to Δφmax = 0.7 ± 0.1 rad, which translates into a change of the refractive index by Δn ≈ (0.9 ± 0.1) × 10−2.
, t) (henceforth referred to as E(t) and Eref(t)) is sampled with sub-250-as extreme-ultraviolet pulses. The peak intensities Ipeak of the strong and attenuated waves have been set to (1.3 ± 0.1) × 1014 W cm−2 and (6.7 ± 0.3) × 1012 W cm−2, respectively. Figure 2a compares the transmitted fields and reveals the evolution of the nonlinear phase shift, ΔφNL(t), induced by the strong field. ΔφNL(t) increases towards the pulse peak, tapers off on its tail and finally vanishes; see insets to Fig. 2a. For Epeak ≈ 2.6 ± 0.1 V Å−1, the induced phase shift at the field maximum amounts to Δφmax = 0.7 ± 0.1 rad, which translates into a change of the refractive index by Δn ≈ (0.9 ± 0.1) × 10−2.

a, After passage through a 10-μm-thick fused silica sample, the electric field E(t) of the few-cycle near-infrared pulse with a peak intensity of 1.3 × 1014W cm−2, approximately 10% below the threshold for optical damage, is modified as a result of the nonlinear light–matter interaction, as revealed by its comparison to a low-intensity (Ipeak = 7 × 1012 W cm−2) reference waveform Eref(t) (for β = 0.27). This comparison yields a transient positive phase shift induced by the strong field, as anticipated from the dynamic increase of the refractive index owing to the optical Kerr effect. The two insets show close-ups of the comparison near the centre and at the end of the pulse, revealing the full reversibility of the effect. E(t) and Eref(t) are obtained from averaging a set of three recordings performed under identical conditions on individual samples. b, The phase shift Δφpeak evaluated at the peak of the field envelope for different peak intensities Ipeak of E(t) is found to exhibit a linear dependence on the field intensity. Each data point represents the mean value of three individual recordings under identical conditions; the error bars indicate the standard deviation.
The field-induced phase shift evaluated at the pulse centre, Δφpeak, is depicted in Fig. 2b and exhibits a linear scaling with the applied peak intensity. In contrast to previous research22,23,24, our time-resolved study reveals the absence of saturation of the optical Kerr effect up to Epeak ≈ 2.7 V Å−1, close to the threshold for dielectric breakdown for few-cycle laser pulses. The Kerr nonlinearity therefore appears to be potentially suitable for petahertz-scale signal manipulation and metrology beyond critical fields19 Ecrit ≈ Δg/(ea) (where Δg denotes the bandgap, e = |e| is the elementary charge, and a is the lattice period; for silica, Ecrit ≈ 2 V Å−1), provided that dissipation originating from carriers promoted into the conduction band during the nonlinear interaction remains low. Although no lasting negative phase shift indicative of residual conduction band population is observable at the trailing edge of the waveform (where the Kerr effect vanishes), an accurate determination of the resultant Wirreversible and the related Wreversible requires evaluation of PNL(t).
The difference ΔE(t) = E(t) − β−1Eref(t) yields PNL(t) via Supplementary equation (13) (for details, see Supplementary Information section 1). Figure 3 depicts PNL(z =  /2, t) along with E(z =
/2, t) along with E(z =  /2, t), both numerically propagated to the middle of the sample where their relative timing can be most precisely determined (see Supplementary Information section 1) for Epeak = 2.6 ± 0.1 V Å−1. PNL(t) oscillates almost perfectly in phase with E(t), indicating a dominant role of bound electrons. This is in strong contrast to the response of free electrons appearing in the ionization of neon atoms in the gas phase25, exhibiting a 90° phase shift with respect to the driving field (Supplementary Information section 3). A closer inspection reveals that PNL(t) lags slightly behind E(t) on the front edge and the peak of the pulse, indicating—according to equation (1)—energy transfer from the field to the electronic system of fused silica, both of which become of opposite sign on the trailing edge of the pulse.
/2, t), both numerically propagated to the middle of the sample where their relative timing can be most precisely determined (see Supplementary Information section 1) for Epeak = 2.6 ± 0.1 V Å−1. PNL(t) oscillates almost perfectly in phase with E(t), indicating a dominant role of bound electrons. This is in strong contrast to the response of free electrons appearing in the ionization of neon atoms in the gas phase25, exhibiting a 90° phase shift with respect to the driving field (Supplementary Information section 3). A closer inspection reveals that PNL(t) lags slightly behind E(t) on the front edge and the peak of the pulse, indicating—according to equation (1)—energy transfer from the field to the electronic system of fused silica, both of which become of opposite sign on the trailing edge of the pulse.

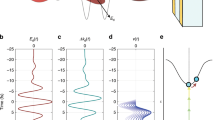
a, The strong electric field numerically back-propagated to the centre of the fused silica sample (z =  /2 = 5 μm) is contrasted with the nonlinear polarization PNL(t) evaluated from E(t) and Eref(t) (Fig. 2a) at the same position (see Supplementary Information section 1). The response time of the nonlinear polarization near optical breakdown is found to be about 100 attoseconds at the pulse peak (close-up in the top inset). The other two insets display the computed spatial rearrangement of the electron density distribution for two extrema of the electric field at instants t1 and t2 in false-colour representation (red indicates an increase and blue a decrease relative to the unperturbed state). Electrons located in the vicinity of the oxygen atoms appear to dominate the polarization response, whereas the electron cloud around the silicon centres remains largely unaffected. b, The response time of the nonlinear polarization is evaluated near the pulse peak as a function of the peak intensity of the applied field (circles) and compared to the results of a perturbation theory calculation (solid line; for details see Supplementary Information section 4). The field-induced change in refractive index Δn is evaluated from PNL(t) at the pulse peak as a function of the applied peak intensity Ipeak. The nonlinear index n2 determined from a linear regression (dashed line) is approximately one-third of the values acquired from time-integrated measurements using multi-cycle pulses29. All data points and error bars represent the average and the standard deviation, respectively, of the evaluation of three individual data sets recorded under identical conditions.
/2 = 5 μm) is contrasted with the nonlinear polarization PNL(t) evaluated from E(t) and Eref(t) (Fig. 2a) at the same position (see Supplementary Information section 1). The response time of the nonlinear polarization near optical breakdown is found to be about 100 attoseconds at the pulse peak (close-up in the top inset). The other two insets display the computed spatial rearrangement of the electron density distribution for two extrema of the electric field at instants t1 and t2 in false-colour representation (red indicates an increase and blue a decrease relative to the unperturbed state). Electrons located in the vicinity of the oxygen atoms appear to dominate the polarization response, whereas the electron cloud around the silicon centres remains largely unaffected. b, The response time of the nonlinear polarization is evaluated near the pulse peak as a function of the peak intensity of the applied field (circles) and compared to the results of a perturbation theory calculation (solid line; for details see Supplementary Information section 4). The field-induced change in refractive index Δn is evaluated from PNL(t) at the pulse peak as a function of the applied peak intensity Ipeak. The nonlinear index n2 determined from a linear regression (dashed line) is approximately one-third of the values acquired from time-integrated measurements using multi-cycle pulses29. All data points and error bars represent the average and the standard deviation, respectively, of the evaluation of three individual data sets recorded under identical conditions.
The response time of the polarizing electronic system, τresponse, can be evaluated from the central zero-crossing of the fields (Fig. 3, upper left panel) as τresponse ≈ 80 as for Epeak = 2.6 ± 0.1 V Å−1. This is smaller than estimates from the Bohr orbit time1 and from χ(3) measurements in the range of 0.1–1 fs and decreases further with decreasing intensity, to well below 40 as for Epeak < 2.2 V Å−1, as displayed in Fig. 3b. This can be understood by connecting τresponse to the nonlinear (field-induced) absorption coefficient, αNL. For τresponse much smaller than the laser period, equation (1) yields a simple linear relationship, αNL ∝ τresponse. For multi-photon absorption, αNL scales highly nonlinearly with the intensity and, according to this relationship, so does τresponse. Supplementary Information section 4 presents detailed modelling of the intensity scaling of τresponse as well as a derivation of αNL(τresponse).
Rendering PNL(t) an experimental observable, attosecond polarization spectroscopy allows to explore the intricate dynamic exchange of energy during nonlinear light–matter interactions. Inserting the measured values of PNL(t) and E(t) into equation (1) provides direct experimental access to the work W(t) done on the electrons by the laser field per unit volume, that is, the energy density transferred from the field to the electronic system. Figure 4a plots the measured W(t) for several different peak intensities and the results of time-dependent density functional theory (TD-DFT) modelling (see inset to Fig. 4, ref. 26 and Supplementary Information section 5). We find very good qualitative agreement between theory and experiment regarding all observables analysed, including the behaviour of the maximum phase shift, the change in refractive index and the evaluated amount of dissipated energy. Quantitative agreement is achieved only when the theoretically employed peak electric field is adjusted to values approximately 20% larger. This discrepancy can be attributed to inaccuracies of the exchange-correlation potential used in the TD-DFT calculations.

a, The amount of energy the few-cycle near-infrared laser field transfers into a unit volume of silica is obtained from the measured E(t) and PNL(t) via equation (1). W(t) shows signatures of a substantial transient virtual conduction-band population (which is proportional to Wmax − Wirreversible) oscillating in synchrony with the driving electric field. In the steepest of these oscillations, energy is transferred into the material within less than 650 as at Epeak = 2.7 V Å−1. The amount of energy irreversibly dissipated in the sample Wirreversible depends critically on the maximum applied field strength Epeak. Shown are the results of recordings for three different field amplitudes with Epeak = 2.5 V Å−1, 2.6 V Å−1 and 2.7 V Å−1, as indicated, and a measurement closest to the average of five recordings with Epeak set equal to 2.1 V Å−1 (the uncertainty in the stated values of Epeak is ± 0.1 V Å−1). At this field strength, Wirreversible becomes immeasurably small (with its error exceeding its nominal value). In the inset, W(t) is computed from the nonlinear polarization at z =  /2 = 5 μm obtained by the TD-DFT calculations outlined in Supplementary Information section 5 for a set of three different values of the peak electric field26, as indicated. The spectrum of the computed nonlinear polarization shows the emergence of odd harmonics of the fundamental radiation (high harmonic generation). The results shown here are computed from the low-pass filtered nonlinear polarization to mimic the frequency transfer characteristics of the optical setup employed for the experiments. b, The squared electric field evolution of the reference wave and its envelope (black line) in comparison to the squared field and its envelope of the wave transmitted at a peak field strength of 2.5 V Å−1, showing clear indications of energy redistribution and consequent reshaping of the pulse envelope caused by the nonlinear polarization (see text). c, The dissipated energy density, equal to the irreversibly transferred energy density shown in a, as a function of the relative refractive index change, as extracted from the results of the TD-DFT simulation (circles) and experimental data taken at Epeak = 2.6 ± 0.1 V Å−1 (square). The excellent agreement between theory and experiment verifies the simulation results, permitting reliable prediction of the relevant quantities for much lower field strengths. The dashed line marks the dissipated energy density of a state-of-the-art MOSFET; for details see Supplementary Information Fig. 16. All error bars indicate the standard deviation of the evaluation of three sets of recordings performed under identical conditions.
/2 = 5 μm obtained by the TD-DFT calculations outlined in Supplementary Information section 5 for a set of three different values of the peak electric field26, as indicated. The spectrum of the computed nonlinear polarization shows the emergence of odd harmonics of the fundamental radiation (high harmonic generation). The results shown here are computed from the low-pass filtered nonlinear polarization to mimic the frequency transfer characteristics of the optical setup employed for the experiments. b, The squared electric field evolution of the reference wave and its envelope (black line) in comparison to the squared field and its envelope of the wave transmitted at a peak field strength of 2.5 V Å−1, showing clear indications of energy redistribution and consequent reshaping of the pulse envelope caused by the nonlinear polarization (see text). c, The dissipated energy density, equal to the irreversibly transferred energy density shown in a, as a function of the relative refractive index change, as extracted from the results of the TD-DFT simulation (circles) and experimental data taken at Epeak = 2.6 ± 0.1 V Å−1 (square). The excellent agreement between theory and experiment verifies the simulation results, permitting reliable prediction of the relevant quantities for much lower field strengths. The dashed line marks the dissipated energy density of a state-of-the-art MOSFET; for details see Supplementary Information Fig. 16. All error bars indicate the standard deviation of the evaluation of three sets of recordings performed under identical conditions.
In all cases, the energy transferred from the field to the material increases up to the pulse peak and slightly beyond. This is because the field, while growing, needs to do ever more work to remove the electrons ever farther from their field-free location. The field amplitude decreasing after the pulse peak allows the displaced electrons to return gradually to their equilibrium position and radiate a part of the absorbed energy back into the driving laser field. This results in a negative slope for W(t). The positive and negative slope are connected to the phase lag and phase advance of PNL(t) with respect to its driving field E(t) before and after the pulse peak, respectively; these are clearly discernible in Fig. 3.
The energy density Wirreversible = W(t → ∞) irreversibly deposited in the system defines the charge carrier density promoted from the valence band into the conduction band according to Ncarrier ≈ Wirreversible/Δg (assuming population of the lowest-energy states of the conduction band). For Epeak = 2.6 V Å−1 a residual relative carrier concentration of Ncarrier/NVB = 2.6 × 10−4 is found, where NVB = 1.4 × 1023 cm−3 is the density of the valence-band electrons. This small residual carrier concentration is pivotal for future ultrafast signal processing and can hardly be determined with similar sensitivity by any other experimental method. By analogy, we can define the reversibly exchanged energy density as the difference between the maximum transferred energy, Wmax, and Wirreversible. Wreversible = Wmax − Wirreversible can be interpreted in terms of a virtual conduction-band population with a number of virtual carriers of Nvirtual ≈ Wreversible/Δg. Nvirtual is the result of a projection of the laser-dressed and fully occupied valence-band states onto conduction-band states and hence it fully returns the energy density associated with it to the field upon its disappearance27. In contrast, the real population, Ncarrier, survives the field and—upon its subsequent decay—causes dissipation.
Although Nvirtual seems rather elusive, attosecond metrology presents a very direct manifestation of the underlying reversible field–matter energy exchange. The initial energy flow dW/dt into the electronic system first extracts energy from the field on the leading edge of the pulse. This is returned by a reversed energy flow on the trailing edge, resulting in a temporal shift of the pulse peak, Δτpeak ≈ 1 fs, as shown in Fig. 4b. The phenomenon is widely known as self-steepening or optical shock wave formation1. Our study reveals that this phenomenon is an inherent consequence of the reversible field–matter energy transfer accompanying the field-induced change in the phase of the pulse. Hence, a field-induced change in the group index, Δng, is inextricably linked to that of the refractive index, Δn, implying a group delay and a phase shift, respectively.
Signal manipulation relies on the change of refractive index Δn (and Δng), which is characterized by Wreversible; in contrast, dissipation is detrimental to signal manipulation and is determined by Wirreversible. Hence, the scaling of Wirreversible and Wreversible (or, equivalently, Δn) with the applied field strength is of key importance for future signal-processing applications. We evaluated the dissipated energy per unit volume and per several-femtosecond optical switching/modulation cycle versus Δn from our ab initio TD-DFT calculations, which we verified against measurement at the highest field strength, near optical breakdown (see Fig. 4c and the discussion in Supplementary Information section 6). At measurable levels of Δn, Wirreversible in a silica optical switch can be some four orders of magnitude smaller than the heat dissipation of a state-of-the-art metal oxide semiconductor field-effect transistor (MOSFET) operating at up to 10 GHz in integrated circuits. This very much reduced dissipation per switching cycle should therefore allow the operation of a dielectric switch/modulator at 100 THz or beyond.
An equally important discovery is the sub-femtosecond rise time of the transferred energy at Epeak ≥ 2.5 V Å−1 within each optical cycle. With slightly shorter pulses than those used in these experiments28, more than 90% of Wmax will be transferred within a single sub-femtosecond rise depicted in Fig. 4. The resultant buildup of carriers in the conduction band within less than 1 fs will permit sampling of electric-field waveforms beyond the petahertz frontier in the simple setting demonstrated recently18.
Our proof-of-principle study on silica shows that careful choice of the peak electric field strength at E < Ecrit may open a route towards 100-THz-rate signal processing. The observed sub-femtosecond gradient in nonlinear energy transfer and the related change in electronic/optical properties at E > Ecrit may pave the way towards sampling optical fields (from the infrared to the ultraviolet) with a compact, cost-effective solid-state device. A petahertz solid-state oscilloscope should enable signal processing and metrology at visible light frequencies.
Traditional pump–probe spectroscopy makes use of the cycle-averaged amplitude envelope to resolve dynamics. In contrast, attosecond polarization spectroscopy uses the oscillating field as a probe, providing direct access to the full (linear and nonlinear) oscillating polarization and hence to the (reversible and irreversible) energy exchange between visible light and matter, as well as a delay in the system response. Hence, attosecond polarization spectroscopy is a generalization of pump–probe spectroscopy, yielding complete information about the dynamic electronic response of matter to strong visible light fields with attosecond resolution and, thanks to the intense attosecond field gradients, with a signal-to-noise ratio orders of magnitude better than that of any other attosecond technique demonstrated so far. Implemented with a probe waveform of sufficiently broad spectral coverage, the approach allows, in principle, complete retrieval of the nonlinear polarization and hence of the entire response of the electronic system to strong-field excitation.
References
Boyd, R. W. Nonlinear Optics (Academic Press, Elsevier, 2008)
Wegener, M. Extreme Nonlinear Optics: an Introduction (Springer, 2005)
Valdmanis, J. A., Mourou, G. & Gabel, C. W. Picosecond electro-optic sampling system. Appl. Phys. Lett. 41, 211–212 (1982)
Wu, Q. & Zhang, X. C. Free-space electro-optic sampling of terahertz beams. Appl. Phys. Lett. 67, 3523 (1995)
Sell, A., Leitenstorfer, A. & Huber, R. Phase-locked generation and field-resolved detection of widely tunable terahertz pulses with amplitudes exceeding 100 MV/cm. Opt. Lett. 33, 2767–2769 (2008)
Hebling, J., Lo Yeh, K., Hoffmann, M. C. & Nelson, K. A. High-power THz generation, THz nonlinear optics, and THz nonlinear spectroscopy. IEEE J. Sel. Top. Quantum Electron. 14, 345–353 (2008)
Huber, R. et al. Switching ultrastrong light–matter coupling on a subcycle scale. J. Appl. Phys. 109, 102418 (2011)
Leitenstorfer, A., Nelson, K. A., Reimann, K. & Tanaka, K. Focus on nonlinear terahertz studies. New J. Phys. 16, 045016 (2014)
Kuehn, W. et al. Terahertz-induced interband tunneling of electrons in GaAs. Phys. Rev. B 82, 075204 (2010)
Junginger, F. et al. Nonperturbative interband response of a bulk InSb semiconductor driven off resonantly by terahertz electromagnetic few-cycle pulses. Phys. Rev. Lett. 109, 147403 (2012)
Somma, C., Reimann, K., Flytzanis, C., Elsaesser, T. & Woerner, M. High-field terahertz bulk photovoltaic effect in lithium niobate. Phys. Rev. Lett. 112, 146602 (2014)
Ulbricht, R., Hendry, E., Shan, J., Heinz, T. F. & Bonn, M. Carrier dynamics in semiconductors studied with time-resolved terahertz spectroscopy. Rev. Mod. Phys. 83, 543–586 (2011)
Kampfrath, T., Tanaka, K. & Nelson, K. A. Resonant and nonresonant control over matter and light by intense terahertz transients. Nature Photon. 7, 680–690 (2013)
Krausz, F. & Ivanov, M. Attosecond physics. Rev. Mod. Phys. 81, 163–234 (2009)
Taur, Y. & Ning, T. H. Fundamentals of Modern VLSI Devices (Cambridge Univ. Press, 2009)
Markov, I. L. Limits on fundamental limits to computation. Nature 512, 147–154 (2014)
Ionescu, A. M. & Riel, H. Tunnel field-effect transistors as energy-efficient electronic switches. Nature 479, 329–337 (2011)
Schiffrin, A. et al. Optical-field-induced current in dielectrics. Nature 493, 70–74 (2012)
Schultze, M. et al. Controlling dielectrics with the electric field of light. Nature 493, 75–78 (2012)
Itatani, J. et al. Attosecond streak camera. Phys. Rev. Lett. 88, 173903 (2002)
Kienberger, R. et al. Atomic transient recorder. Nature 427, 817–821 (2004)
Pati, A. P., Wahyutama, I. S. & Pfeiffer, A. N. Subcycle-resolved probe retardation in strong-field pumped dielectrics. Nature Commun. 6, 7746 (2015)
Loriot, V., Hertz, E., Faucher, O. & Lavorel, B. Measurement of high order Kerr refractive index of major air components. Opt. Express 17, 13429–13434 (2009); erratum 18, 3011–3012 (2010)
Brée, C., Demircan, A. & Steinmeyer, G. Saturation of the all-optical Kerr effect. Phys. Rev. Lett. 106, 183902 (2011)
Geissler, M. et al. Light propagation in field-ionizing media: extreme nonlinear optics. Phys. Rev. Lett. 83, 2930–2933 (1999)
Yabana, K., Sugiyama, T., Shinohara, Y., Otobe, T. & Bertsch, G. Time-dependent density functional theory for strong electromagnetic fields in crystalline solids. Phys. Rev. B 85, 045134 (2012)
Yablonovitch, E., Heritage, J. P., Aspnes, D. E. & Yafet, Y. Virtual photoconductivity. Phys. Rev. Lett. 63, 976–979 (1989)
Cavaleri, A. L. et al. Intense 1.5-cycle near infrared laser waveforms and their use for the generation of ultra-broadband soft-x-ray harmonic continua. New J. Phys. 9, 242 (2007)
Milam, D. Review and assessment of measured values of the nonlinear refractive-index coefficient of fused silica. Appl. Opt. 37, 546–550 (1998)
Acknowledgements
We acknowledge discussions with M. Stockman and V. Apalkov. This work was supported by the Max Planck Society and the Deutsche Forschungsgemeinschaft Cluster of Excellence: Munich Centre for Advanced Photonics (http://www.munich-photonics.de). M.S. was supported by a Marie Curie International Outgoing Fellowship (FP7-PEOPLE-2011-IOF). E.M.B. acknowledges funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement number 290605 (PSI-FELLOW/COFUND) and from the Swiss National Science Foundation through NCCR MUST. This research is based upon work supported by the US Air Force Office of Scientific Research under award number FA9550-16-1-0073 and used computational resources of the K computer provided by the RIKEN Advanced Institute for Computational Science through the HPCI System Research project (Project ID: hp140103).
Author information
Authors and Affiliations
Contributions
F.K. and M.S. initiated, conceived and supervised the study. A.S. and E.M.B. developed the experimental method. A.S., E.M.B. and C.J. (in close cooperation with T.L., O.R., M.J., W.S. and V.S.) prepared and performed the experiment. S.A.S., H.F., K.Y. and N.K. accomplished the theoretical modelling. A.S., E.M.B., V.S.Y., R.K., N.K., M.S. and F.K. analysed and interpreted the experimental data. All authors discussed the results and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary text and Data, Supplementary Figures 1-16 and Supplementary References. (PDF 2139 kb)
Rights and permissions
About this article

Cite this article
Sommer, A., Bothschafter, E., Sato, S. et al. Attosecond nonlinear polarization and light–matter energy transfer in solids. Nature 534, 86–90 (2016). https://doi.org/10.1038/nature17650
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature17650
- Springer Nature Limited
This article is cited by
-
Sub-cycle multidimensional spectroscopy of strongly correlated materials
Nature Photonics (2024)
-
Dynamic optical response of solids following 1-fs-scale photoinjection
Nature (2023)
-
Synthesis, Surface Morphology, Gas Sensor, DSC Technique and Third-Order Behavior of Conducting Polymer
Journal of Fluorescence (2023)
-
Size-controlled quantum dots reveal the impact of intraband transitions on high-order harmonic generation in solids
Nature Physics (2022)
-
Attosecond clocking of correlations between Bloch electrons
Nature (2022)