

Abstract
Sexual reproduction requires recognition between the male and female gametes. In flowering plants, the immobile sperms are delivered to the ovule-enclosed female gametophyte by guided pollen tube growth. Although the female gametophyte-secreted peptides have been identified to be the chemotactic attractant to the pollen tube1,2,3, the male receptor(s) is still unknown. Here we identify a cell-surface receptor heteromer, MDIS1–MIK, on the pollen tube that perceives female attractant LURE1 in Arabidopsis thaliana. MDIS1, MIK1 and MIK2 are plasma-membrane-localized receptor-like kinases with extracellular leucine-rich repeats and an intracellular kinase domain. LURE1 specifically binds the extracellular domains of MDIS1, MIK1 and MIK2, whereas mdis1 and mik1 mik2 mutant pollen tubes respond less sensitively to LURE1. Furthermore, LURE1 triggers dimerization of the receptors and activates the kinase activity of MIK1. Importantly, transformation of AtMDIS1 to the sister species Capsella rubella can partially break down the reproductive isolation barrier. Our findings reveal a new mechanism of the male perception of the female attracting signals.
Similar content being viewed by others


Main
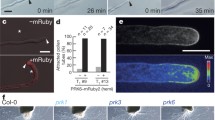
Peptides have recently been identified as female attractants, such as Zea mays EGG APPRATUS 1 (ZmEA1) in maize, defensin-like peptides LURE1 and LURE2 in Torenia fournieri (TfLURE1 and TfLURE2) and AtLURE1 in A. thaliana1,2,3. However, the receptor(s) in the pollen tube perceiving the female attractants is not known. To identify the male receptors, we selected receptor-like kinases (RLKs) preferentially expressed in Arabidopsis pollen (tubes)4,5,6,7 as candidates (Extended Data Fig. 1a). To investigate their function, the kinase-dead dominant negative (DN) forms were expressed in wild-type plants under the pollen-specific LAT52 (ref. 8) promoter. Micropylar targeting of the RLKDN-expressing pollen tubes was analysed under minimal pollination9. Second, we analysed the micropylar targeting of the pollen tubes of the corresponding knockout mutants. Third, we examined possible interactions between these RLKs for potential co-receptors by yeast two-hybrid analysis. Through this combinatory approach, two homologous leucine-rich-repeat RLKs clades, At5g45840 and At4g18640 (previously designated as MRH1 (ref. 10)), and At4g28650 and At4g08850, were identified and designated MALE DISCOVERER1 (MDIS1) and MDIS2, and MDIS1-INTERACTING RECEPTOR LIKE KINASE1 (MIK1) and MIK2, respectively (Extended Data Fig. 1b). MDIS1DN pollen tubes exhibit decreased micropylar guidance (Extended Data Fig. 1c–f) and fertilization efficiency (Extended Data Fig. 1g) in the T1 hemizygotes and T3 homozygotes compared to the wild type. The progenies of two single T-DNA insertion lines (MDIS1DN-1 and MDIS1DN-2) segregate at 2.3:1 and 2.2:1 for the transgenes. During reciprocal crosses, decreased male transmission was observed, but not reduced female transmission (Extended Data Table 1). This result indicates that MDIS1DN interferes with the pollen tube guidance. Furthermore, MDIS1 interacts with MIK1 and MIK2 in yeast (Extended Data Fig. 1h). Genomic-fused GUS and green fluorescent protein (GFP) reporters further confirmed the expression of MDIS1 and MDIS2 in pollen tubes and seedlings, and their localization in plasma membrane and endomembrane compartments, respectively (Fig. 1a–d, Extended Data Fig. 2a–c and Supplementary Videos 1 and 2). Corroboratively, MDIS1, MDIS2, MIK1, MIK2 and the close homologue of MIK1, PXY, were predominantly expressed in pollen tubes (Extended Data Fig. 2d). PXY has been shown to be the receptor of TDIF in vascular development11,12 and was detected at low level in pollen and pollen tubes. Genetic results showed that MIK1 may not be the receptor of TDIF12, but it cannot be excluded that MIK1 might be the receptor of other pistil-expressed CLE peptides. Immunostaining revealed the expression of MIK1 and MIK2 in pollen tubes (Fig. 1e–j and Extended Data Fig. 2e, f). These results suggest that they function in pollen tubes.

a–d, MDIS1–GUS (a), MDIS2–GUS (b), MDIS1–GFP (c) and MDIS2–GFP (d) in pollen tubes. e–j, Wild-type (e–h), mik1 (i) and mik2 (j) tubes stained with MIK1 (e, g) or MIK2 (f, h) antibody. Arrows denote tube tips. Scale bars, 5 μm. k–p, Phenotype of wild-type (k) and mutant (l–p, red arrows) pollen tubes at the micropyle (asterisks). Images are representative of 30 images captured. Scale bars, 50 μm. q, Statistical analysis. Error bars, s.e.m. of 3 independent replicates; n = 300 for each sample. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t-test). -1 and -2 are genetic complementation lines.
To investigate their roles in the pollen tube, knockout mutants mdis1-2, mdis2, mik1 and mik2, and a knockdown mutant mdis1-1 were obtained, and mdis1-2 was used for mdis1 analysis (Extended Data Fig. 3a, b). During reciprocal crosses with mdis1/+ mdis2/− or mdis1/− mdis2/+, we observed reduced male transmission and normal female transmission (Extended Data Table 2). Furthermore, the in vivo tube length and in vitro pollen germination ratio of mdis1 mdis2, mik1, mik2 and mik1 mik2 were normal (Extended Data Fig. 3c–e). When growing in the wild-type pistils, the wild-type pollen tubes enter the micropyle directly (Fig. 1k). The mutants, however, displayed two major types of defective pollen tube responses to the ovules (Fig. 1l–q), that is, type I is featured by failed pollen tube entry (Fig. 1l–n), type II is featured by one pollen tube failing but another tube entering (Fig. 1o) and occasionally mdis1 and mdis1 mdis2 pollen tubes branching at the micropyle (Fig. 1p). The type II phenotype may explain the lack of seed set defect under natural pollination. To confirm this hypothesis, we counted the number of earlier (appeared larger) and later fertilized wild-type ovules by the mik1 mik2 pollen under limited pollination. The ratio of later to earlier fertilized ovules by the mutant pollen tubes is higher than that by the wild-type pollen tubes (Extended Data Fig. 3f, g), indicating that the fertilization efficiency of mutant pollen tubes is decreased. The mdis1 mdis2 and mik1 mik2 double mutations exaggerate the guidance defect, but mdis1 mik1 did not (Fig. 1q), indicating that MDIS1/MDIS2 and MIK1/MIK2 probably act in the same pathway. The full-length genomic sequence of MDIS1-GFP and the MIK1 coding sequence driven by the LAT52 promoter alleviates the phenotype of mdis1 mdis2 and mik1 mik2 to the single mutant (Fig. 1q). These data indicate that both MDIS and MIK have a role in the tube perception of the female signal.
To verify if MDIS1, MDIS2, MIK1 and MIK2 are the receptors of LURE1, we examined the binding of AtLURE1.2 with the purified recombinant ectodomain (ECD) of MDIS1, MDIS2, MIK1 and MIK2. The glutathione S-transferase (GST)-tagged ECD of MDIS1, MIK1 and MIK2 binds His-tagged LURE1.2, but not to the His-tagged defensin-like peptide AtPDF1.2 (ref. 13), as shown by a pull-down assay (Fig. 2a). AtPRK3 (AT3G42880), a leucine-rich repeat RLK highly expressed in the pollen tube14, does not bind LURE1.2. Consistently, the purified proteins are properly folded demonstrated by mass spectrometry analysis that showed that the disulfide bonds between the two cysteine residues in the amino- or carboxy-terminal capping domains of the purified MDIS1ECD, MIK1ECD and MIK2ECD were properly formed (Extended Data Fig. 4). Furthermore, microscale thermophoresis (MST) analysis showed that LURE1.2 strongly interacts with MDIS1, MIK1 and MIK2, with a dissociation constant (Kd) of 1.76 ± 0.09 μM, 672 ± 42.4 nM and 464 ± 13.4 nM, respectively, but MDIS2 and PRK3 exhibit no binding with LURE1.2 (Fig. 2b). The ERECTA protein previously shown to bind TfLURE2 at a background affinity (279 ± 60 nM) using a microsome and Quartz crystal microbalance method15, also displayed a background affinity binding to TfLURE2 (94.6 ± 2.46 μM) using MST analysis (Extended Data Fig. 5a). The discrepancy in affinity probably resulted from different methods used and it is common that the affinity derived from a cell-based assay is much higher than an in vitro protein-based assay; presumably they differ in cellular context and other signalling components. Interestingly, the MIK1 homologue PXY also binds His–LURE1.2, with a dissociation constant of 704 ± 49.2 nM in the MST assay. The finding that MDIS2 does not bind LURE1 and the additive phenotype of mdis1 mdis2 suggest that MDIS2 may bind other unidentified female attractants, as suggested by the partial guidance defect of LURE1-knockdown plants3. We further confirmed the binding of MDIS1, MIK1 and MIK2 to LURE1.2 by co-immuno-precipitation (co-IP) in Arabidopsis leaf protoplasts. The haemagglutinin (HA)-tagged full-length MDIS1, MIK1 and MIK2 bind His–LURE1.2, but HA-tagged BRI1-ASSOCIATED KINASE1 (BAK1)16 does not (Fig. 2c). Furthermore, His–LURE1.2 is associated with the protoplasts expressing MDIS1–HA, MIK1–HA and MIK2–HA (Fig. 2d). The Flag-tagged LURE1.2 purified from LURE1.2-overexpressing plants was co-immunoprecipitated by protoplast-expressed MDIS1–, MIK1– and MIK2–HA, respectively (Fig. 2e). The binding of plant-purified LURE1.2–Flag to MDIS1, MIK1 and MIK2 was competitively replaced by an excess of His–LURE1.2, suggesting that the bindings are specific (Fig. 2f). Furthermore, we demonstrated that LURE1.2 triggers endocytosis of MDIS1–GFP in the pollen tube tip (Extended Data Fig. 5b–e). Consistently, the wild-type pollen tubes were attracted to the LURE1.2-embeded beads efficiently, while the mutant tubes show a significantly reduced response to the attractant (Fig. 2g–l), in the semi-in-vitro guidance assay3. The above data showed that MIDS and MIK bind LURE1 both in vitro and in vivo.

a, Pull-down (PD) assay as indicated. b, Binding affinity by MST. Error bars, s.e.m. of 3 independent measurements. ΔFLUO, change in fluorescence. c, Interactions between HA fusions and His–LURE1.2 in protoplasts. d, His–LURE1.2 binds the protoplasts expressing HA fusions. e, Co-IP between HA fusions and LURE1.2–Flag. f, Competition between LURE1.2–Flag and His–LURE1.2 to HA fusions. Full blots are shown in Supplementary Data. g–l, Growth of wild-type (g–i) and mutant (j, k) pollen tubes to the LURE1.2 beads. Red arrows, unattracted; white arrows, attracted. Images are representative of 30 images captured. Scale bars, 20 μm. l, Attraction frequency. n, number of pollen tubes scored. Error bars, s.e.m. of 3 independent replicates. **P < 0.01, ***P < 0.001 (Student’s t-test).
Next, we explored whether MIK1 and MIK2 might work synergistically with MDIS1. Direct interactions between MDIS1ECD and MIK1ECD or MIK2ECD were detected in pull-down and co-IP assays (Fig. 3a, b and Extended Data Fig. 5f, g). Importantly, exogenously applied LURE1.2 substantially enhanced the interaction between MDIS1–Flag and MIK1–HA in vivo (Fig. 3c). The MST result verified that LURE1.2 enhances the interaction between MDIS1ECD and MIK1ECD or MIK2ECD (Fig. 3d). Furthermore, bimolecular fluorescence complementation confirmed that LURE1.2 enhances the interaction between MDIS1 and MIK proteins (Extended Data Fig. 6a–f). An in planta co-IP assay with self-pollinated flowers of MDIS1-GFP transgenic plants confirmed that the MIK–MDIS1 complex perceives LURE1 (Fig. 3e and Extended Data Fig. 6g, h).

a, Pull-down assay as indicated. b, c, LURE1 enhances the interaction between MDIS1–Flag and MIK1–HA or MIK2–HA by co-IP in protoplasts. d, Affinity between GST–MDIS1ECD and His–MIK1ECD or MIK2ECD in the presence of His–LURE1.2 by MST. Error bars, s.e.m. of 3 independent measurements. e, MDIS1–GFP interacts with MIK1, MIK2 and LURE1 in planta. Arrows denote target proteins. f, GST–MIK1KD phosphorylated itself and His–MDIS1KD. Asterisks denote phosphorylated proteins. g, LURE1.2 induces MIK1–Flag self-phosphorylation. h, MIK1–Flag phosphorylates MDIS1–HA and itself after LURE1.2 treatment. i, LURE1.2 induces homodimerization of MIK1. Full blots are shown in Supplementary Data.
Ligand-induced heterodimerization of co-receptor complex transduces signals by transphosphorylation during pathogen and brassinosteroid perception17,18. We determined whether this is true for MIK and MDIS since MDIS1 and MDIS2 are atypical RLKs19. Using a Phos-tag mobility shift assay, we found that the kinase domain of MDIS1 (MDIS1KD) was phosphorylated by MIK1KD, which exhibits self-phosphorylation, whereas MDIS1KD shows no self-phosphorylation (Fig. 3f). By mass spectrometry, we found that MDIS1 is phosphorylated by MIK1 at Ser663, and MIK1 is auto-phosphorylated at eight sites (Thr741, Thr742, Thr862, Ser864, Thr710, Tyr879, Thr880 and Thr992) (Extended Data Fig. 7). When MDIS1–Flag and MIK1–HA were expressed in protoplasts separately, LURE1.2 induced the autophosphorylation of MIK1 but not of MDIS1 (Fig. 3g). When MDIS1–Flag and MIK1–HA were co-expressed in the presence of LURE1.2, both MDIS1 and MIK1 were phosphorylated (Fig. 3h). Furthermore, LURE1.2 induces dimerization of MIK1, whereas MDIS1 dimerizes constitutively (Fig. 3i).
The homologues of MDIS1, MIK1 and MIK2 exist in these closely related species. We detected transcripts of CrMDIS1 and EsMDIS1 in the pollen of C. rubella and Eutrema salsugineum, but not CrMIK1 and EsMIK2 (Extended Data Fig. 8). This suggests that expression of MIK1 and MIK2 in pollen evolved after the divergence between C. rubella and the ancestor of A. thaliana. This indicates that the MDIS1–MIK complex in the pollen tube was newly evolved, and MDIS1 may function as receptor of the attractants in the older species solely or synergistically with other RLKs. Thus, to explore whether AtMDIS1 is able to break down the reproduction isolation barrier, we transformed AtMDIS1 to C. rubella. Using a semi-in-vitro assay, the micropyle targeting efficiency of transgenic C. rubella pollen tubes to the A. thaliana ovules is substantially increased (Fig. 4). Since the discovery of LUREs as the female attractant, the search for its male receptor has been hampered by the redundancy of the receptors and LUREs. In this study, we provided strong biochemical, cytological and genetic evidences that the MIK1–MDIS1 complex functions as the LURE1 receptor and determined their activation mechanism. Nevertheless, our data and others also indicate that there are other LURE receptors that are yet to be identified.

a, b, C. rubella pollen tubes expressing LAT52:AtMDIS1 target the wild-type A. thaliana ovule in semi-in-vitro system (a), but not the wild-type C. rubella pollen tubes (b). Images are representative of 30 images captured. Scale bars, 20 μm. c, Reverse transcription PCR (RT–PCR ) showing the expression of AtMDIS1 in the pollen of transgenic C. rubella. d, Targeting efficiency of A. thaliana ovules by pollen tubes of AtMDIS1 transgenic C. rubella. Error bars, s.e.m. of 3 independent measurements. ***P < 0.01 (Student’s t-test). n, number of pollen tubes scored outside the micropyles.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Plant material
The Arabidopsis thaliana wild-type (Col-0), T-DNA insertion mutants mdis1-1 (GABI_463E06), mdis1-2 (GABI_090F03), mdis2 (SALK_004879) and Capsella rubella were obtained from ABRC stock centre. mik1 (SALK_095005) and mik2 (SALK_061769) were obtained from J. Zhou. The E. salsugineum seeds were obtained from Q. Xie. Plants were grown at 22 °C under long-day conditions (16-h light/8-h dark cycles). For C. rubella and E. salsugineum, the sterilized seeds were vernalized on the MS media at 4 °C for 30 days and then grown at 22 °C under long-day conditions.
In vitro pollen germination and in vivo tube growth
Pollen tubes were germinated on the germination media (1 mM CaCl2, 1 mM Ca(NO3)2, 1 mM MgSO4, 0.01% H3BO4, 18% sucrose and 0.5% agarose) and cultured for 5 h at 22 °C. The germination ratio was scored under light microscopy. Mean value was calculated from three independent experiments and for each experiment, more than 300 pollen were scored. For in vivo tube growth, pollen from the wild-type and mutants were pollinated on the emasculated pistil with mature stigma as reported20. The pistils were collected at 3, 6 and 8 h after pollination and fixed for aniline blue staining. The pollen tubes in the pistil were photographed with Leica M205 microscope. The length of pollen tubes was measured with Image J software (http://rsb.info.nih.gov/ij/).
Aniline blue staining and microscopy
Flowers at 12c stage were emasculated and left to grow for 12–24 h to achieve pistil maturation. Then about 20 pollen grains from wild-type or mutant plants, respectively, were dispersed on the stigma papillar cells with a tiny brush. After 24 h, pistils were excised and fixed in Carnoy’s fixative (75% ethanol and 25% acetic acid) as reported21,22. The pistils were washed in 50 mM PBS buffer (NaHPO4/NaH2PO4, pH 7.0) three times and immersed in 1 M NaOH overnight for softening. Then after three washes with PBS, the pistil was stained with 0.1% aniline blue (pH 8.0 in 0.1 M K3PO4) for 6 h. The stained pistils were observed under Axio Skop2 microscope (Zeiss) equipped with an ultraviolet filter set. Ovules with micropylar guidance defect and the ratio of fertilized ovules to the number of pollen tubes in the style were calculated and the mean values from three independent experiments were compared with that of the wild type.
Generation of constructs and plant transformation
For the dominant-negative constructions, the kinase domains were inactivated by replacing the conserved lysine residue in the intracellular ATP-binding domain with glutamic acid to generate dominant-negative constructs. For the atypical kinase, the intracellular domain was chimaerically replaced with that of BRASSINOSTEROID INSENSITIVE1 (BRI1)23 receptor kinase with an inactive kinase domain (K to E substitution). For GFP and GUS reporter expression, genomic sequences containing 2 kb native promoters and the genomic coding sequence for MDIS1 and MDIS2 were subcloned into the pCAMBIA1300-GFP binary vector. For complementation of mik mutants, full-length coding sequence driven by LAT52 promoter was cloned into pCAMBIA1300. Similarly, full-length LURE1.2 fused with a C-terminal Flag tag driven by the 35S promoter was cloned into the pCAMBIA1300. For complementation assay, the genomic fused GFP constructs were transformed into the mutant using Agrobacterium-mediated floral dip method24. To break down the reproductive isolation barrier, the full-length MDIS1 coding sequence under the LAT52 promoter was introduced into C. rubella by floral dip method.
Protein purification and pull-down assay
LURE1.2 and PDF2.1 lacking the putative N-terminal signal peptides (71 and 55 amino acids, respectively) were fused N-terminally with a His-tag using pET28a vector (Novagen). Similarly, the ectodomains of MDIS1, MDIS2, MIK1, MIK2 and PRK3 lacking the predicted signal peptides were fused with an N-terminal GST tag using a pGEX4T-2 vector. The fused proteins were expressed in Escherichia coli strain Rossetta DE3 (Stratagene). Cells were grown to an A600 nm value of 0.6 at 37 °C and then induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 6 h at 22 °C. The cells were lysed by sonication on ice in lysis buffer containing 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, Complete Protease Inhibitor Cocktail (Roche) and 1 mg ml−1 lysozyme (Wako). After centrifugation at 12,000 g for 20 min at 4 °C, the supernatants and pellets were collected separately; the pellet was washed three times with the lysis buffer. For LURE1, the insoluble His–LURE1.2 peptides in the inclusion bodies were solved in 1 M urea supplemented with 6 M guanidine-HCl (in Tris-HCl buffer, pH 8.0) for 1 h on ice. Then the peptides were diluted at 1:10 and refolded for 3 days at 4 °C using glutathione (reduced form: oxidized form = 10:1, MERCK) and l-arginine ethyl ester dihydrochloride (Sigma-Aldrich). The folded peptides were dialysed with 3-kDa centrifugal filter (Millipore) and eluted with 50 mM Tris-HCl (pH 8.0) and then used for pull-down, co-IP, protoplasts treatment, pollen tube guidance assays and antibody generation. For purification of GST-tagged ectodomain of MDIS1, MDIS2, MIK1, MIK2 and PRK3 proteins, cells from 2 l culture were collected and lysed respectively as described above. The supernatants were used for affinity purification by glutathione agarose beads (GE, 17-0756-01) to avoid extra folding process, although more fused proteins were in the pellets than the supernatant. For GST pull-down assay, the purified proteins were mixed and incubated for 3 h and then subjected to pull-down assay with glutathione agarose beads for 3 h at 4 °C. The beads were collected by centrifugation and then washed five times with buffer containing 25 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% Triton X-100 and 0.1% SDS. Finally, the proteins bound on the beads were boiled with 1× SDS sample buffer in 95–100 °C water bath and then subjected to SDS–PAGE and immunoblot with anti-GST (GE Healthcare, 27-4577-01) and anti-His (Santa Cruz) antibody. For mobility shift detection of phosphorylated proteins, phosphatase inhibitor phrostop (Roche) was added during purification and incubation. Moreover, 50 μM Phos-tag (AAL-107) and 50 μM MnCl2 was added to the gel according to the manufacturer’s procedure. After electrophoresis, the gel was treated with 10 mM EDTA, pH 8.0, for 10 min to remove the Mn2+ before immunoblot assay.
Co-IP
Seedlings of LURE1.2-Flag transgenic plants were ground to fine powder in liquid nitrogen and solubilized with extraction buffer (0.05 M HEPES-KOH, pH 7.5, 150 mM KCl, 1 mM EDTA, 0.1% Triton X-100 with freshly added proteinase inhibitor cocktail (Roche)). The extracts were centrifuged at 10,000g for 10 min, and the supernatant was incubated with pre-washed anti-Flag M2 magnetic beads (Sigma-Aldrich, M8823) for 3 h at 4 °C, and then the beads was washed six times with the extraction buffer. The immunoprecipitates were eluted with 3 × Flag peptides. For co-IP in protoplasts, the transformed protoplasts expressing MDIS1–HA, MIK–HA and BAK1–HA were incubated with the purified LURE1.2–Flag or the 200 nM folded His–LURE1.2 purified from E. coli for 10 min and lysed for co-IP with pre-washed anti-HA agarose beads (Sigma-Aldrich, A2095). The precipitates were diluted with SDS sample buffer, separated on a 10% SDS–PAGE gel and subjected to immunoblot with the corresponding antibodies (anti-Flag, Sigma-Aldrich, F1804; anti-HA, Santa Cruz, sc-7392; anti-His, Santa Cruz, sc-803). Arabidopsis protoplast transformation was performed as reported previously25. For the His–LURE1-protoplast binding assay, the protoplasts incubated with 10 μm LURE1.2 for 5 min, washed three times with the culture buffer and then lysed for SDS–PAGE and immunoblot. For the enhanced interaction between MDIS1 and MIK proteins by LURE1.2, the protoplasts co-transformed with MDIS1–HA and MIK1–Flag were divided into two equal volumes. One was incubated with 0.5 nM LURE1.2 and another with equal volume of 50 mM Tris-HCl (pH 8.0) as mock control for 10 min and subjected to anti-HA immunoprecipitation. For the phosphorylation test, the transformed protoplasts were divided equally into two and incubated for 10 min with 200 nM LURE1.2 or 50 mM Tris-HCl (pH 8.0), respectively. For competition assay, protoplasts expressing MDIS1–HA, MIK1–HA and MIK2–HA were each divided equally into four centrifuge tubes and incubated with purified LURE1.2–Flag. Then active His–LURE1.2 of different concentrations was added to the protoplasts and incubated for 10 min and subsequently co-immunoprecipated with anti-HA conjugated agarose beads. For co-IP in planta, the flowers opened in the morning were collected in the afternoon at the estimated time when the pollen tubes are approaching the ovules. Total proteins were subjected to co-IP with anti-GFP conjugated agarose (ChromoTek, gta-200) or anti-LURE1.2 and protein-A-conjugated magnetic beads (Bio-Rad, 161–4013). The immunoprecipitates was subjected to SDS–PAGE and immunoblot with the corresponding antibodies (anti-GFP-HRP, Miltenyi Biotec, 130-091-833). All the co-IP experiments were repeated at least three times.
Semi-in-vitro pollen germination and guidance assay
For A. thaliana, the same germination media as that for in vitro germination was used. For C. rubella, a modified media (4 mM CaCl2, 4 mM Ca (NO3)2, 0.01% H3BO4, 10% sucrose and 0.5% agarose) was used. Semi-in-vitro germination and ovule-pollen attraction assay were performed as reported in A. thaliana3. Pollen tubes entered the micropyle were scored as successful breakdown of the reproductive isolation and the pollen tubes bypass outside the micropyle within 20 μm were scored as failing to enter the micropyle. For the attraction assay, gelatin (Nacalai) beads containing 40 μM LURE1.2 were made and placed beside the pollen tube tip using a micro-manipulator (Narishige) equipped with an inverted microscope (Zeiss AxioVert. A1) as described previously26. Behaviour of pollen tubes was monitored and recorded with a CCD camera. Pollen tubes growing to the beads with >30° direction change were regarded as effective pollen tube attraction.
qPCR
Total RNA was extracted from pollen, in vitro germinated pollen tubes (3 h after pollination) and seedlings with TRIzol reagent (Invitrogen) and then treated with DNase I (RNase-free DNase kit, Qiagen) to remove DNA. SuperScript III Reverse Transcriptase (Invitrogen) was used for the reverse transcription reactions. qPCR was performed with Power SYBR Green PCR Master Mix on the Bio-RAD C1000 Thermal Cycler using Tubulin 2 as the internal control for quantitative normalization. The specificity of the primers was examined by running the PCR products on 2.5% agarose gels and sequencing.
MST assay
The affinity of the purified GST, GST–MDIS1ECD, MDIS2ECD, MIK1 ECD, MIK2 ECD, ERECTAECD and PXY ECD to His–LURE1.2 was measured using the Monolith NT.115 (Nanotemper Technologies). The GST-fusion proteins were fluorescently labelled according to the manufacturer’s procedure. The solution buffer was exchanged to labelling buffer and the protein concentration was adjusted to 2 μM. Then fluorescent dye NT-647-NHS was added and mixed and incubated for 30 min at 25 °C in the dark. Finally, the labelled proteins were dialysed with column B (Nanotemper L001) and eluted with 50 mM Tris-HCl (pH 8.0) supplemented with 0.02% Tween 20. For each assay, the labelled protein (about 1 μM) was incubated with the same volume unlabelled His–LURE1.2 of 12 different serial concentrations in 50 mM Tris-HCl (pH 8.0) supplemented with 0.02% Tween 20 at room temperature for 10 min. The samples were then loaded into silica capillaries (Polymicro Technologies) and measured at 25 °C by using 20%–40% LED power and 20% MST power. Each assay was repeated three times. Data analyses were performed using Nanotemper analysis software and OriginPro 9.0 software.
Bimolecular fluorescence complementation analysis in tobacco
The constructs containing MDIS1-NE (MDIS1 fused with the N-terminal YFP), MIK1-CE and MIK2-CE (MIK1 and MIK2 fused with the C-terminal YFP, respectively) were generated as described previously8. The Agrobacterium tumefaciens EHA105 strains carrying MDIS1-NE and MIK-CE were equally mixed with and without EHA105 strain carrying LURE1.2–Flag and transformed into half of the same tobacco leaf. The transformed leaves were photographed 2 days later with a confocal laser scanning microscope (Zeiss Meta 510). Images were acquired using the same optical setting and average total pixel intensity values were calculated by sampling images of different leaves using the ImageJ software as reported27. Mean values of three experiments, each with five transformed leaves, were compared using Student’s t-test for biological significance.
Determination of phosphorylation sites and disulphide bonds of MDIS1 and MIK1 in vitro
The E. coli cells expressing the fusion proteins were lysed and centrifuged at 4 °C. The affinity-purified fusion proteins from the supernatants were subjected to mass spectrometry. His–MDIS1KD was incubated with GST–MIK1KD in vitro in kinase assay buffer (25 mM Tris-HCl, pH 8.0, 10 mM MgCl2 and 100 mM ATP) for 1 h at 30 °C. The proteins were separated by 10% SDS–PAGE and the gel was stained with Coomassie blue G250. The corresponding proteins band were cut into slices and subjected to alkylation/tryptic digestion followed by LC–MS/MS as reported previously28. For disulfide bonds determination, GST–MDIS1ECD, GST–MIK1ECD and GST–MIK2ECD were affinity purified from the supernatants of the bacterial lysis and eluted with 50 mM Tris-HCl, pH 8.0. Then disulfide bonds were determined by mass spectrometry as previously reported29.
Phylogenetic analysis
Alignment of protein sequences were aligned using ClustalW2 program (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Phylogenetic tree of the alignment were drawn with MEGA5 (http://www.megasoftware.net/) using the neighbour-joining method with bootstrapping based on 1,000 replicates. The leucine-rich repeat domains were predicted with LRRfinder (http://www.lrrfinder.com/) and HHPREP program. The transmembrane domains were predicted with TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/). The signal peptides were predicted with SignalP 4.1 Server (http://www.cbs.dtu.dk/services/SignalP/).
Yeast two-hybrid assay
The coding sequences of MDIS1 or MIK1 and MIK2, respectively, were cloned into the pBT3-SUC bait or pPR3-N prey according to the manufacture’s procedure (DualsystemBiotech). Yeast strain NMY51 was co-transformed with the bait and prey constructs and grown on the selective medium lacking Trp, Leu, His and adenine.
RT–PCR
Total RNA was extracted from pollen, leaf, flower and total plant of C. rubella and E. salsugineum with TRIzol reagent (Invitrogen) and then treated with DNase I (RNase-free DNase kit, Qiagen) to remove any contaminating DNA. SuperScript III Reverse Transcriptase (Invitrogen) was used in reverse transcription reactions. ACTIN11 was used as the control for quantitative normalization. The specificity of the primers was confirmed by sequencing of the band after electrophoresis. The accession numbers for the amplified genes are as follows: CrMDIS1 (XM_006280043), EsMDIS1 (XM_006398206), CrMIK1 (XM_006285722), EsMIK1 (XM_006412864), CrMIK2 (XM_006286915), EsMIK2 (XM_006397188), CrACTIN11 (XM_006297859) and EsACTIN11 (XM_006407307).
GUS assay and GFP observation
The histochemical GUS activity assay was performed in the solution containing 2 mM X-Gluc (Sigma) in 50 mM PBS (pH 7.0) and 0.5 mM potassium/ferrocyanide. GUS solution was added to the samples and incubated at 37 °C overnight. Digital images were taken with a Zeiss Axio Skop2 plus microscope. For GFP observation, images were taken with Zeiss confocal laser scanning microscope with a setting of 488 nm excitation (Carl Zeiss, Meta 510 confocal microscope).
Endocytosis of MDIS1—GFP
The semi-in-vitro germinated MDIS1–GFP pollen tubes were treated with 500 nM LURE1.2 and photographed by CLSM 780 (Zeiss) after different times.
Antibody generation and immunostaining
The anti-MIK1 and anti-MIK2 antibodies were raised in mouse with the purified His-tagged extracellular domains lacking the predicted N-terminal signal peptide. Anti-LURE1.2 antibody was raised in mouse with the folded active His–LURE1.2 fusion protein. For MIK1 and MIK2, the specificity of antibodies was tested with the fusion proteins expressed in protoplasts and the total proteins of pollen from the wild-type and corresponding mutant plants. For LURE1.2, the antibody specificity was tested with the total protein from the leaves of LURE1.2–Flag-overexpressing plants. For immunostaining, the semi-in-vitro germinated pollen tubes were fixed in 3.7% paraformaldehyde (3.7% formaldehyde, 1 mM CaCl2, 1 mM MgSO4, 50 mM HEPES, 5% sucrose, pH 7.4) for 30 min, washed with PME buffer (50 mM PIPES, 1 mM MgCl2, 5 mM EGTA, pH 6.8) three times and then subjected to 1% Driselase and 1% cellulase for 10 min. The sample was sequentially washed with PBS buffer (pH 7.4) three times, NP40 buffer (0.5% Nonidet P-40, 1% BSA, in PBS, pH 7.4) and PBS buffer once. Antibodies diluted 1:500 (with PBS containing 3% BSA) were incubated with the sample overnight at 4 °C and then washed with PBS three times. The samples were incubated for 1 h at 4 °C with FITC-labelled goat anti-mouse secondary antibody (KBL, 202-1806) and washed with PBS three times. Anti-fade mounting medium (Invitrogen, P36934) was used for signal detection by confocal laser scanning microscopy (Zeiss Meta 510) with 488 nm excitation.
References
Marton, M. L., Cordts, S., Broadhvest, J. & Dresselhaus, T. Micropylar pollen tube guidance by egg apparatus 1 of maize. Science 307, 573–576 (2005)
Okuda, S. et al. Defensin-like polypeptide LUREs are pollen tube attractants secreted from synergid cells. Nature 458, 357–361 (2009)
Takeuchi, H. & Higashiyama, T. A species-specific cluster of defensin-like genes encodes diffusible pollen tube attractants in Arabidopsis. PLoS Biol. 10, e1001449 (2012)
Loraine, A. E., McCormick, S., Estrada, A., Patel, K. & Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant Physiol. 162, 1092–1109 (2013)
Qin, Y. et al. Penetration of the stigma and style elicits a novel transcriptome in pollen tubes, pointing to genes critical for growth in a pistil. PLoS Genet. 5, e1000621 (2009)
Zimmermann, P., Hirsch-Hoffmann, M., Hennig, L. & Gruissem, W. GENEVESTIGATOR. Arabidopsis microarraydatabase and analysis toolbox. Plant Physiol. 136, 2621–2632 (2004)
Wang, Y. et al. Transcriptome analyses show changes in gene expression to accompany pollen germination and tube growth in Arabidopsis. Plant Physiol. 148, 1201–1211 (2008)
Muschietti, J., Dircks, L., Vancanneyt, G. & McCormick, S. LAT52 protein is essential for tomato pollen development: pollen expressing antisense LAT52 RNA hydrates and germinates abnormally and cannot achieve fertilization. Plant J. 6, 321–338 (1994)
Li, H. J. et al. POD1 regulates pollen tube guidance in response to micropylar female signaling and acts in early embryo patterning in Arabidopsis. Plant Cell 23, 3288–3302 (2011)
Jones, M. A., Raymond, M. J. & Smirnoff, N. Analysis of the root-hair morphogenesis transcriptome reveals the molecular identity of six genes with roles in root-hair development in Arabidopsis. Plant J. 45, 83–100 (2006)
Fisher, K. & Turner, S. PXY, a receptor-like kinase essential for maintaining polarity during plant vascular-tissue development. Curr. Biol. 17, 1061–1066 (2007)
Hirakawa, Y. et al. Non-cell-autonomous control of vascular stem cell fate by a CLE peptide/receptor system. Proc. Natl Acad. Sci. USA 105, 15208–15213 (2008)
Sels, J., Mathys, J., De Coninck, B. M., Cammue, B. P. & De Bolle, M. F. Plant pathogenesis-related (PR) proteins: a focus on PR peptides. Plant Physiol. Biochem. 46, 941–950 (2008)
Chang, F., Gu, Y., Ma, H. & Yang, Z. AtPRK2 promotes ROP1 activation via RopGEFs in the control of polarized pollen tube growth. Mol. Plant 6, 1187–1201 (2013)
Lee, J. S. et al. Competitive binding of antagonistic peptides fine-tunes stomatal patterning. Nature 522, 439–443 (2015)
Li, J. et al. BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 110, 213–222 (2002)
Santiago, J., Henzler, C. & Hothorn, M. Molecular mechanism for plant steroid receptor activation by somatic embryogenesis co-receptor kinases. Science 341, 889–892 (2013)
Sun, Y. et al. Structural basis for flg22-induced activation of the Arabidopsis FLS2–BAK1 immune complex. Science 342, 624–628, (2013)
Castells, E. & Casacuberta, J. M. Signalling through kinase-defective domains: the prevalence of atypical receptor-like kinases in plants. J. Exp. Bot. 58, 3503–3511 (2007)
Chen, L. Y. et al. The Arabidopsis alkaline ceramidase TOD1 is a key turgor pressure regulator in plant cells. Nature Commun . 6, 6030 (2015)
Li, H. J. et al. Arabidopsis CBP1 is a novel regulator of transcription initiation in central cell-mediated pollen tube guidance. Plant Cell 27, 2880–2893 (2015)
Chen, Y. H. et al. The central cell plays a critical role in pollen tube guidance in Arabidopsis. Plant Cell 19, 3563–3577 (2007)
Li, J. & Chory, J. A putative leucine-rich repeat receptor kinase involved in brassinosteroid signal transduction. Cell 90, 929–938 (1997)
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998)
Yoo, S. D., Cho, Y. H. & Sheen, J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nature Protocols 2, 1565–1572 (2007)
Walter, M. et al. Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation. Plant J. 40, 428–438 (2004)
Palikaras, K., Lionaki, E. & Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528 (2015)
Thingholm, T. E., Jorgensen, T. J., Jensen, O. N. & Larsen, M. R. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nature Protocols 1, 1929–1935 (2006)
Lu, S. et al. Mapping native disulfide bonds at a proteome scale. Nature Methods 12, 329–331 (2015)
Acknowledgements
We thank L. Qu for technique assistance in the pollen tube guidance assay. We thank Y. Guo, Quantum Design Inc. China, and core facilities of public technology service centre of Institute of Microbiology and Institute of Genetics and Developmental Biology (Chinese Academy of Sciences) for the MST measurement. We thank J. Zhou and Q. Xie for sharing seeds. This work was supported by the Ministry of Science and Technology of China grants 2013CB945103 to W.-C.Y. and 2015CB910202 to H.-J.L. and the National Natural Science Foundation of China 31330053 and 31221063 to W.-C.Y.
Author information
Authors and Affiliations
Contributions
H.-J.L. and W.-C.Y. designed the study, interpreted the results and wrote the paper. T.W. performed most of the experiments. L.L. performed the guidance assay. Y.X. performed the mutants screening. M.-X.Z. performed the LURE1 construction. P.-F.J. performed the cell biology analysis and W.C. performed the qPCR experiments. Y.-C.W. performed mass spectrometry analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Pollen tubes expressing MDIS1DN shows micropylar guidance defect.
a, Phylogenetic tree of the analysed RLKs expressed in pollen (tubes). b, Protein structure of MIK1, MIK2 and MDIS1. Green box, leucine-rich repeats; red, signal peptide and transmembrane domain; yellow, kinase domain; blue, proline-rich domain; purple, linker region. c, Schematic diagram of dominant-negative construct of MDIS1 driven by the pollen-specific promoter LAT52. ECD, ectodomain; TM, transmembrane domain of MDIS1. The kinase domain of MDIS1 was replaced by the dead kinase domain of BRI1 with an AAG-to-GAG site mutation. d, The wild-type pollen tube (arrow) enters the micropyle opening directly. Images are representative of 30 images captured. e, The pollen tube (arrow) from the MDIS1DN transgenic plants exhibits defective micropylar guidance to the wild-type ovules. Images are representative of 30 images captured. Asterisks in d and e represent micropyles. Scale bars, 50 μm. f, Percentage of wild-type ovules with micropylar guidance defect minimally pollinated with pollen from six independent hemizygous and homozygous MDIS1DN transgenic lines. Error bars, s.e.m. of 3 independent replicates; **P < 0.01 (Student’s t-test); n = 300 for each sample. g, Fertilization efficiency of the pollen tubes from the six MDIS1DN hemizygous and homozygous lines. The ratio of numbers of successfully targeted pollen tubes to the pollen tubes in the styles was calculated from 30 minimally pollinated pistils. Error bars, s.e.m. of 3 independent replicates; **P < 0.01 (Student’s t-test); n = 200 for each sample. h, MDIS1 interacts with MIK1 and MIK2 as shown by dual membrane yeast two-hybrid system. Yeasts were co-transformed with bait construct MDIS1-Cub and prey construct MIK1-NubG or MIK2-NubG, and the transformants were grown on selective media.
Extended Data Figure 2 MDIS1, MDIS2, MIK1 and MIK2 are expressed in the pollen tubes.
a, Time-lapse images showing the dynamic distribution of MDIS1–GFP and MDIS2–GFP during pollen tube growth in vitro. Images are representative of 30 images captured. Scale bars, 10 μm. b, c, Histological GUS staining of seedlings transformed with MDIS1- and MDIS2-GUS under the native promoters, respectively. Images are representative of 20 images captured. Scale bars, 5 mm. d, Quantitative PCR (qPCR) showing the expression of MDIS1, MDIS1, MIK1, MIK2 and PXY in pollen, pollen tubes and seedlings. Error bars, s.e.m of 3 independent replicates. e, Specificity test of MIK1 and MIK2 antibodies with Arabidopsis protoplasts expressing Flag-tagged MIK1 and MIK2. Equal amount of Arabidopsis MIK–Flag-transformed (T) or wild-type (untransformed; UT) protoplasts were lysed and subjected to immunoblotting. Anti-MIK1 and anti-MIK2 recognize the corresponding protoplasts-expressed Flag fusion proteins specifically. f, The target protein was recognized by anti-MIK1 and anti-MIK2 in the wild-type pollen, but not in the corresponding mutants. Total protein of the same amount of pollen grains from the wild type and mutants were subjected to SDS–PAGE and immunoblot. Arrows denote target proteins.
Extended Data Figure 3 Pollen performance of the mutants.
a, Schematic representation of gene structure of MDIS1, MDIS2, MIK1 and MIK2 and the T-DNA insertion site. The T-DNA insertion positions are indicated by triangles. Filled boxes, exons; open boxes, untranslated region; lines, introns. b, Expression of the transcripts in the opening flowers of the wild-type and corresponding mutants. c, Representative images of pollen tube length of the corresponding mutants grown in the wild-type pistils at 3, 6 and 8 h after pollination (HAP). Arrows indicate the points the bulk of the pollen tubes reached. Images are representative of 60 images captured. Scale bars, 200 μm. d, Pollen tube length of mdis1 mdis2, mik1, mik2 and mik1 mik2 is comparable to the wild type. n = 60 pistils for each sample; P > 0.1 (Student’s t-test); n.s., not significant. Error bars, s.e.m. of 3 independent measurements. e, The in vitro pollen germination of mdis1 mdis2, mik1, mik2 and mik1 mik2 is normal. Error bars, s.e.m. of 3 independent replicates; P > 0.1 (Student’s t-test); n = 300 for each sample. f, g, The ratio of earlier to later fertilized wild-type ovules targeted by the mik1 mik2 and mik1 mik2/+ pollen tubes is higher than by the wild-type pollen tubes. Approximately 40 pollen tubes were hand-pollinated on the wild-type stigma, which was then subjected to aniline blue staining 30 HAP. Left panel in f represents image of the earlier fertilized ovules; right panel represents image of the later fertilized ovules in the same silique. Arrow denotes the enlarged ovule. Scale bars, 20 μm. g, Statistics of results shown in f. n, numbers of ovules scored. Error bars, s.e.m. of 3 independent replicates; *P < 0.05, ***P < 0.001 (Student’s t-test).
Extended Data Figure 4 Verification of the predicted disulfide bonds by mass spectrometry.
Disulfide bonds of the purified MDIS1ECD, MIK1ECD and MIK2ECD were identified at Cys193–Cys201 of MDIS1, Cys60–Cys67 of MIK1 and Cys683–Cys695 of MIK2. Cys64 of MDIS1, Cys609 and Cys616 of MIK1 were at the oxidized form.
Extended Data Figure 5 LURE1.2 induced the endocytosis and decrease of MDIS1–GFP in the pollen tube tip.
a, Binding affinity between ERECTA and TfLURE2 by MST. Error bars, s.e.m. of 3 independent measurements. b–e, Confocal images showing the distribution of MDIS1–GFP before LURE1.2 (0.5 μM) treatment (b), and at 0 min (c), 20 min (d) and 60 min (e) after treatment. Images are representative of 63 images captured. Intensity plots along the red lines of each image are shown below. Scale bars, 5 μm. The maximum y-axis values are the same for all intensity plots. The arrows indicate the signal accumulation at the plasma membrane. Scale bars, 5 μm. f, g, His–MIK1ECD and His–MIK2ECD specifically bind GST–MDIS1ECD, but not the GST affinity beads. Full blots are shown in Supplementary Data.
Extended Data Figure 6 LURE1.2 is perceived by the MDIS1–MIK complex.
a–f, Confocal images of tobacco leaf showing stronger bimolecular fluorescence complementation signal in the presence of LURE1.2–Flag (a, d) as compared with the weak signal in the absence of LURE1.2–Flag (b, e). c–f, Quantification of the total fluorescence signal of the same areas. Error bars, s.e.m. of 3 independent replicates; *P < 0.05 (Student’s t-test). Five leaves with positive signal were analysed for each experiment. Scale bars, 50 μm. g, Anti-LURE1 and anti-Flag antibodies recognize the LURE1–Flag fusion protein. h, Endogenous interaction between LURE and MIK1 or MIK2 by LURE antibody with the total crude proteins extracted from the wild-type pollinated flowers (8 HAP), but not with the mik1 mik2 mutant. Arrow denotes target proteins. Full blots are shown in Supplementary Data.
Extended Data Figure 7 Ion-trap MS/MS spectra identifying phosphorylation sites of the kinase domain of MDIS1 and MIK1.
Identification of one phosphorylation site for MDIS1 (Ser663) and eight for MIK1 (Thr741, Thr742, Thr862, Ser864, Thr710, Tyr879, Thr880 and Thr992) by ion-trap liquid chromatography tandem mass spectrometry (LC–MS/MS).
Extended Data Figure 8 Expression pattern of homologues of MDIS1, MIK1 and MIK2 in C. rubella and E. salsugineum by RT–PCR analysis.
a, CrMDIS1, but not CrMIK1 or CrMIK2, is expressed in pollen of C. rubella. b, EsMDIS1, but not EsMIK1 or EsMIK2, is expressed in pollen of E. salsugineum. ACTIN11 transcripts were amplified as controls. Genomic DNA was used as the control for primer specificity.
Supplementary information
Supplementary Information
This file contains full blots of the gels. (PDF 629 kb)
Video 1: Fluorescence dynamics of MDIS1-GFP in the pollen tube
This video shows the time-lapse imaging of the dynamic localization of MDIS1-GFP in the pollen tube. (WMV 700 kb)
Video 2: Fluorescence dynamics of MDIS2-GFP in the pollen tube
This video shows the time-lapse imaging of the dynamic localization of MDIS2-GFP in the pollen tube. (WMV 1427 kb)
Rights and permissions
About this article

Cite this article
Wang, T., Liang, L., Xue, Y. et al. A receptor heteromer mediates the male perception of female attractants in plants. Nature 531, 241–244 (2016). https://doi.org/10.1038/nature16975
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature16975
- Springer Nature Limited
This article is cited by
-
Large-scale analysis of the N-terminal regulatory elements of the kinase domain in plant Receptor-like kinase family
BMC Plant Biology (2024)
-
Synergid cell calcium oscillations refine understanding of FERONIA/LORELEI signaling during interspecific hybridization
Plant Reproduction (2024)
-
Two aspartic proteases, BnaAP36s and BnaAP39s, regulate pollen tube guidance in Brassica napus
Molecular Breeding (2023)
-
Does selection occur at the intermediate zone of two insufficiently isolated populations? A whole-genome analysis along an altitudinal gradient
Journal of Plant Research (2023)
-
Defense response changes in roots of oil palm (Elaeis guineensis Jacq.) seedlings after internal symptoms of Ganoderma boninense Pat. infection
BMC Plant Biology (2022)