

Abstract
The discovery of four new Xenoturbella species from deep waters of the eastern Pacific Ocean is reported here. The genus and two nominal species were described from the west coast of Sweden1,2, but their taxonomic placement remains unstable3,4. Limited evidence placed Xenoturbella with molluscs5,6, but the tissues can be contaminated with prey7,8. They were then considered deuterostomes9,10,11,12,13. Further taxon sampling and analysis have grouped Xenoturbella with acoelomorphs (=Xenacoelomorpha) as sister to all other Bilateria (=Nephrozoa)14,15, or placed Xenacoelomorpha inside Deuterostomia with Ambulacraria (Hemichordata + Echinodermata)16. Here we describe four new species of Xenoturbella and reassess those hypotheses. A large species (>20 cm long) was found at cold-water hydrocarbon seeps at 2,890 m depth in Monterey Canyon and at 1,722 m in the Gulf of California (Mexico). A second large species (~10 cm long) also occurred at 1,722 m in the Gulf of California. The third large species (~15 cm long) was found at ~3,700 m depth near a newly discovered carbonate-hosted hydrothermal vent in the Gulf of California. Finally, a small species (~2.5 cm long), found near a whale carcass at 631 m depth in Monterey Submarine Canyon (California), resembles the two nominal species from Sweden. Analysis of whole mitochondrial genomes places the three larger species as a sister clade to the smaller Atlantic and Pacific species. Phylogenomic analyses of transcriptomic sequences support placement of Xenacoelomorpha as sister to Nephrozoa or Protostomia.
Similar content being viewed by others

Main
Xenoturbellida Bourlat et al., 2006
Genus Xenoturbella Westblad, 1949
Xenoturbella monstrosa sp. nov.
Etymology. Latin for extraordinary size.
Material examined. Holotype, Scripps Institution of Oceanography Benthic Invertebrate Collection SIO-BIC BI1037, sex unknown (Extended Data Fig. 1c). Paratype SIO-BIC BI1038, from type locality, sex unknown. Paratype SIO-BIC BI1039, female, from Mexican locality (Fig. 1b and Extended Data Fig. 1d–h).

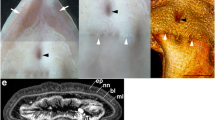
a, X. monstrosa (arrows) in a field of A. diagonalis (Ad) and Ectenagena elongata (Ee) clams at 2,890 m depth in Monterey Bay, California. Scale estimated from the average size of A. diagonalis27. b, X. monstrosa (arrow at upper left) and X. churro (asterisk at lower right) at 1,722 m depth near a methane seep in the Guaymas Basin, Mexico. Red spots (lasers) are 29 cm apart. Shell fragments in sediment, but no living clams observed in vicinity. Both specimens contained C. pacifica DNA in their tissues. c, X. profunda female in an A. gigas (Ag) clam field near a carbonate-hosted hydrothermal vent at ~3,700 m depth in Pescadero Basin, Mexico. d. X. hollandorum on sediment adjacent to whale bones (b). Abbreviations: a, anterior; rf, ring furrow; h, hemichordate; o, bone-eating Osedax worms; p, polynoid scaleworm; s, shrimp.
Locality. Monterey Submarine Canyon, California, 36° 36.8′ N, 122° 26.0′ W, ~2,890 m depth, vesicomyid clam field (Extended Data Fig. 1a, b). Collected via slurp system, research vessel (R/V) Western Flyer/remote operated vehicle (ROV) Tiburon dive 610, 25 October 2004. Also from Guaymas Transform Fault, Gulf of California, Mexico, 27° 34.659′ N, 111° 27.036′ W, 1,722 m depth, near a cold-water methane seep, ‘Pinkies Vent’. Collected via slurp, R/V Western Flyer/ROV Doc Ricketts dive 385, 13 April 2012.
Description. Body ~20 cm long in life, purple or pale pink. Dorsal body wall with two deep, longitudinal furrows. Ring furrow and side furrow found. Body in front of ring furrow rounded; posterior tapers gradually. Mouth oval-shaped (when relaxed), lying ventrally midway between ring furrow and anterior end. Epidermal network over two-thirds of ventral surface. Body wall with gametes dorsally and ventrally. Exogenous DNA present in tissue from co-occurring vesicomyid bivalves Archivesica diagonalis (in holotype) or Calyptogena pacifica (in Mexican paratype).
Xenoturbella churro sp. nov.
Etymology. Noun used in apposition. Resembles fried-dough pastry called churro.
Material examined. Holotype (SIO-BIC BI1040), female (Fig. 1b and Extended Data Fig. 2a–e).
Locality. Guaymas Transform Fault, Gulf of California, Mexico, 27° 34.659′ N, 111° 27.036′ W, 1,722 m depth, near ‘Pinkies Vent’. Collected via slurp, R/V Western Flyer/ROV Doc Ricketts dive 385, 13 April 2012.
Description. Body 10 cm long in life, uniformly orange/pink. Dorsal body wall with four deep, longitudinal furrows. Ring furrow and side furrow present. Anterior end rounded, posterior tapers sharply. Mouth oval (when relaxed), lying ventrally midway between ring furrow and anterior end. Epidermal network over two-thirds of ventral surface. Body wall with gametes dorsally and ventrally. Exogenous DNA from co-occurring vesicomyid bivalve C. pacifica present in tissue.
Xenoturbella profunda sp. nov.
Etymology. Latin for deep.
Material examined. Holotype SIO-BIC BI1041, male (Extended Data Fig. 3b, c, g, h). Paratypes (SIO-BIC BI1042-46, UNAM ICML-EMU-11010), a female and five males or juveniles (Fig. 1c and Extended Data Fig. 3a, d–f), from type locality.
Locality. Pescadero Basin, Mexico, 23° 57.23′ N, 108° 51.73′ W, 3,700 m depth on sediment near hydrothermal vent. Collected via slurp, R/V Western Flyer/ROV Doc Ricketts over several dives in April 2015.
Description. Body to 15 cm in life, uniformly pale pink, with epidermis, circular and longitudinal muscles, parenchyma and gastrodermis. Dorsal body with a pair of deep, longitudinal furrows. Ring furrow and side furrow found. Anterior end rounded, posterior tapering gradually. Mouth oval (when relaxed), ventral, anterior to ring furrow. Epidermal network over two-thirds of ventral surface. Gametes dorsally and ventrally. Oocytes reach 450 μm in diameter, sperm with spherical heads. Exogenous DNA of vesicomyid bivalve A. gigas present in tissue.
Xenoturbella hollandorum sp. nov.
Etymology. Named for Linda and Nicholas Holland for their contributions to biology.
Material examined. Holotype SIO-BIC BI1036, sex unknown (Fig. 1d and Extended Data Fig. 4a–d).
Locality. Monterey Submarine Canyon, California, 36° 48.132′ N, 121° 59.647′ W; ~631 m depth, on sediment adjacent to bones of a grey whale. Collected via push core, R/V Western Flyer/ROV Tiburon dive 1,160, 18 December 2007.
Description. Body short (2.5 cm) and uniformly bright pink. Dorsal body with a pair of longitudinal furrows. Ring furrow and side furrow found. Ventral mouth present, diamond-shaped (when relaxed), just anterior to ring furrow. Epidermal network over ventral surface anterior to ring furrow. Exogenous cytochrome-c-oxidase subunit I (COI) sequences of bivalves not detected.
While we have markedly increased the diversity of the group, the morphological similarities of the new species leads us to keep them in Xenoturbella1. Live specimens of all four new species exhibited an epidermal branching network ventrally that has not been previously recognized. This network was inconspicuous in X. hollandorum and consequently may have been overlooked previously in X. bocki. X. profunda is gonochoric, as opposed to the hermaphroditism reported for X. bocki, which also has much smaller eggs1,4. X. monstrosa was seen in abundance once (Fig. 1a, Extended Data Fig. 1a, b and Supplementary Video 1) in a vesicomyid clam field (mostly Ectenagena extenta, some A. diagonalis) in Monterey Canyon, but repeated visits to this site failed to reveal more specimens. The X. monstrosa collected from Monterey were in a patch of mainly A. diagonalis that were dying or spawning, producing large amounts of mucus (Supplementary Video 1). The Gulf of California specimen extends the range of X. monstrosa to >2,500 km and depth from ~1,700 to ~3,000 m. The single specimen of X. churro was found only 30 cm from the Mexican specimen of X. monstrosa (Fig. 1b and Supplementary Video 2). Xenoturbella profunda lives deepest of the known species, and all seven collected specimens were observed on sediments (showing bacterial mats and various fauna, including vesicomyids) near a hydrothermal vent (Supplementary Video 3).
Mitochondrial COI was sequenced for all specimens examined and compared with other Xenoturbella. The nominal species from Sweden, X. bocki and X. westbladi, comprised a single haplotype network with several shared sequences (Extended Data Fig. 5a), which strongly suggests it is a single species, although this has previously been dismissed17. Here we treat X. westbladi as a junior synonym of X. bocki. The two specimens of X. monstrosa from California differed by one base pair (bp) and from the Mexican specimen by up to 7 bp (Extended Data Fig. 5b). Seven specimens of X. profunda exhibited four haplotypes, differing by one to three bp (Extended Data Fig. 5c).
Whole mitochondrial genomes of the four new species (Extended Data Fig. 6) exhibited the same gene order as X. bocki9,18. Phylogenetic analysis of these genomes placed the smaller, shallower-dwelling species, X. hollandorum and X. bocki, as sister taxa differing at ≥6.4% of the nucleotide sites and 59 amino acids. The distinction of X. hollandorum as a new species is also supported by nuclear sequence data (Supplementary Table 1). Among all other Xenoturbella species, mitochondrial genomes differed by 10–20% (Supplementary Data Table 1), with the deep-water species forming a clade with respect to X. hollandorum and X. bocki (Fig. 2). Phylogenetic comparison of mitochondrial proteins placed Xenacoelomorpha with deuterostomes, as previously reported9,16,18 (Extended Data Fig. 7), although poorly supported. Notably, the PhyloBayes19 analysis recovered Xenacoelomorpha as sister group to Chordata, also with poor support, and we question the utility of such data at deep phylogenetic scales.

Support assessed via 100 pseudo-replicates of bootstrapping. Scale bar, substitutions per nucleotide position. All nodes were highly supported as indicated by bootstrap support of 100% (asterisk). Rooting based on the topology recovered from the analysis of mitogenomes of Metazoa (Extended Data Fig. 7). Scale bar, substitutions per nucleotide position.
Phylogenomic analyses of Xenoturbella compared our transcriptomic data from X. profunda with limited transcriptomic (expressed sequence tag) data from X. bocki11, and genomic and transcriptomic data from other animals. The main data set included 26 terminals and 1,178 genes with 70% average gene occupancy (Supplementary Table 5). Maximum likelihood analyses using RAxML20 with either the WAG, LG or GTR amino-acid substitution models and gamma (Γ) correction for rate heterogeneity produced identical tree topologies and similar support values, grouping Xenoturbella with the acoel Hofstenia (=Xenacoelomorpha) as sister to Nephrozoa (deuterostomes + protostomes; Fig. 3). All key nodes were well supported. Alternative hypotheses assessed using SOWH21 constraining Xenacoelomorpha as sister group to Ambulacraria (n = 100, Δ-likelihood = 1,313.9, P = 0.027), or to Deuterostomia (n = 100, Δ-likelihood = 1,042.07, P = 0.031), were rejected as significantly worse. A species tree22 generated from the consensus of 393 individual gene-trees (average occupancy = 80%) (Extended Data Fig. 8) placed Xenacoelomorpha as sister to Nephrozoa, although there was poor support for the latter clade. We also used PhyloBayes under CAT + GTR + Γ, as recommended in a previous assessment of the position of Xenoturbella16. The result placed Xenacoelomorpha as sister to protostomes with moderate support, a placement not previously recovered (Extended Data Fig. 9). To explore if elevated substitution rates in some loci could be influencing our analyses23, we removed the ribosomal proteins (51 loci) from the 1,178-locus data set and then split this into slowest- and fastest-evolving matrices. Maximum likelihood analyses revealed no major differences between the two data sets or incongruence with the complete data set (Extended Data Fig. 10).

Within Bilateria (B), Xenacoelomorpha is sister group to Nephrozoa (N), which includes Protostomia and Deuterostomia. Key nodes were highly supported, as indicated by bootstrap support of 100% (asterisks). Data were analysed using RAxML with GTR, LG and WAG models, + Γ. Support values of less than 100 are shown for GTR + Γ; the same nodes had higher support with LG + Γ and WAG + Γ. Scale bar, substitutions per amino-acid position.
We sampled four new Xenoturbella species from cold-water hydrocarbon seeps, a hydrothermal vent, and from the vicinity of a whale-fall, all environments that host dense communities of animals. These discoveries highlight the possibility of further Xenoturbella discoveries in deep-sea environments that sustain their likely food source, bivalve molluscs. Although the placement of Xenoturbella has been a challenge in metazoan phylogenetics, the present application of transcriptomic data resulted in much larger sampling of genes, which has helped to resolve deep phylogenetic relationships24. Our analyses showed no support for the hypothesis that Xenacoelomorpha lies inside deuterostomes7,9,16, a result also corroborated by another phylogenomic study on the position of Xenacoelomorpha25. The placement of Xenacoelomorpha as sister group to Nephrozoa (Fig. 3 and Extended Data Figs 8 and 10), or to Protostomia (Extended Data Fig. 9), suggests the suite of features previously proposed to be character losses in Xenoturbella13 appear instead to be plesiomorphic absences.
Methods
No statistical methods were used to predetermine sample size.
Collecting details
Once on board the ship, specimens were maintained in chilled seawater in a 4 °C cold-room. Xenoturbella was photographed under a Leica MZ8 stereomicroscope. Larger animals were photographed with a macro lens. Animals were then relaxed in 7% MgCl in freshwater before further photography. Pieces of tissue were then taken for molecular sequencing and frozen or placed into RNAlater or 95% ethanol. Other pieces of tissue were fixed in 4% paraformaldehyde in 0.2 M sodium phosphate buffer overnight before rinsing and being placed in buffer with sodium azide or processed further and kept in 70% ethanol. One specimen of X. monstrosa was fixed and preserved in 95% ethanol and the other in 10% formalin in seawater. All other specimens were fixed in 4% paraformaldehyde in buffer and preserved in 70% ethanol. All specimens are lodged at the Scripps Institution of Oceanography Benthic Invertebrate Collection (SIO-BIC), La Jolla, California, except for a paratype of X. profunda, which is lodged at the Instituto de Ciencias del Mar y Limnologia (UNAM), Mazatlán, Mexico, and catalogued as ICML-EMU11010. Taxonomic acts are registered on Zoobank (http://zoobank.org/) as urn:lsid:zoobank.org:pub:135BCEF9-A1E4-4926-B346-3711A6485690.
Amplification and sequencing of mitochondrial and nuclear genes and mitochondrial genomes
We extracted DNA from Xenoturbella individuals using a DNeasy Blood & Tissue Kit following the manufacturer’s specifications (Qiagen). COI for all Xenoturbella individuals and their bivalve prey was amplified with the primers listed in Supplementary Table 2. Histone H3 (HH3) was also amplified with the primers listed in Supplementary Table 2. NCBI accession numbers are listed in Supplementary Table 3. To acquire Xenoturbella mitochondrial genomes, we initially amplified portions of three genes: 16S rRNA (16S), cytochrome B (CytB) and cytochrome-c-oxidase subunit III (COIII). Primer sequences and thermocycling conditions are provided in Supplementary Table 2. All amplifications were done using illustra PuReTaq Ready-To-Go PCR Beads (GE Life Sciences) following the manufacturer’s protocol. PCR products were cleaned using USB ExoSAP-IT, sequenced by Eurofins MWG Operon. Geneious R7 (ref. 28) was used to inspect and trim sequences.
We then used the 16S, CytB and COIII sequences and the Primer3 (ref. 29) algorithm in Geneious to design Xenoturbella-specific primers for long PCR amplifications. The mitochondrial genomes of X. monstrosa, X. hollandorum and X. churro were amplified in two overlapping fragments that were each 8,000 bp long using primers listed in Supplementary Table 2. All long PCR products were amplified using Platinum Taq DNA Polymerase High Fidelity (Invitrogen) following the manufacturer’s specifications. The PCR products were visualized on 0.9% agarose gels run at 80 V for 90 min. PCR products were cleaned using USB ExoSAP-IT or GelElute Extraction kit (5 Prime). The X. hollandorum mitochondrial genome was outsourced to Engencore (Selah Genetics) for sequencing and assembly with the Roche 454 platform and Newbler version 2.3. The X. monstrosa and X. churro mitochondrial genomes were sequenced by Macrogen using Illumina HiSeq2000. The reads were assembled de novo after low-quality reads and adaptor sequences were removed using Geneious. The X. profunda mitochondrial genome was obtained from the transcriptome data. X. profunda and X. churro had fragments of the mitochondrial genome that were not sequenced. These missing ‘fill-in’ fragments were recovered via direct sequencing using primers listed in Supplementary Table 2 (refs 12, 30, 31, 32).
RNA extraction and transcriptome sequencing
X. profunda tissue was finely chopped and placed in RNAlater (Ambion) for 24 h at 4 °C and subsequently at −80 °C for long-term storage. RNA was extracted using Direct-zol RNA Kits (Zymo Research) following the manufacturer’s protocol. The optional DNase I step was performed during the RNA extraction to remove residual genomic DNA. RNA extractions were then purified using RNA Clean & Concentrator kits (Zymo Research) following the manufacturer’s protocol. Libraries were prepared from the purified mRNA using KAPA Stranded mRNA-Seq Kits (Kapa Biosystems) following the manufacturer’s protocol. Libraries were sequenced (150 bp paired-end) using a 300 cycle Miseq kit (Illumina) by the IGM Genomics Center (University of California, San Diego). 9,941,202 read pairs were generated from this run, which were then assembled using Trinity into 31,374 transcripts after low-quality, adaptor-contaminated or ribosomal rRNA reads were removed.
Mitochondrial data analysis
We used TCS33 to construct COI haplotype networks for the Xenoturbella species with multiple sequences. The mitochondrial phylogenetic analysis was based on the taxon sampling used previously16. All 13 protein coding regions of the Xenoturbella mitochondrial genomes were extracted and translated into amino-acid sequences in Geneious using translation table 5 (invertebrate mitochondrial). Translated amino-acid sequences from the other taxa were downloaded directly from GenBank (Supplementary Table 4). The complete mitochondrial genomes of the four new Xenoturbella species were uploaded to the MITOS web server for annotation using translation table 5 (http://mitos.bioinf.uni-leipzig.de).
The translated amino-acid sequences of each protein-coding gene were aligned independently using MAFFT34 and the FFT-NS-i ×1000 algorithm. Each of these 13 alignments was then run on the GBLOCKS server with the least stringent settings to remove poorly aligned regions (http://molevol.cmima.csic.es/castresana/Gblocks_server.html)35. We used Geneious to concatenate the trimmed sequences into a single alignment of 2,519 positions.
The amino-acid data were analysed using RAxML 8.1.22 (ref. 20) with the data partitioned by gene (13 partitions) and with the GTR + Γ model. Clade support was assessed using 100 bootstrap pseudo-replicates. A PhyloBayes analysis of the data was also run using CAT + GTR + Γ, as done previously with a similar data set16, although the files available from that study contained only 12 of the 13 protein coding genes (COIII was missing) and was also missing a further 361 bp from cytochrome b for Amphipholis.
For assessing the relationships among the five Xenoturbella species, the nucleotide data were analysed using RAxML with GTR + Γ. Clade support was assessed using 100 bootstrap pseudo-replicates. Rooting was based on the topology found in the analysis of the metazoan mitochondrial genomes (Extended Data Fig. 7).
Phylogenomic analyses
Taxa included in the transcriptome analysis are listed in Supplementary Table 5. Agalma36 is an analysis pipeline (https://bitbucket.org/caseywdunn/agalma) for phylogenetics that was used to construct alignments of orthologous gene sequences from raw sequence data.
Adaptor sequences, low quality reads and ribosomal RNA sequences were removed from the X. profunda raw Illumina and NCBI SRA data. The transcriptomes were assembled using Trinity (version r20140413p1)37 from the cleaned reads. The UCSC genome browser was used to download transcribed DNA sequences from the mrna.fa.gz file of the taxa with sequenced genomes. The assemblies and genome data were translated into amino-acid sequences by longest open reading frame. All of the translated transcriptomes were loaded into the Agalma database and then sent through the ‘post-assemble’ and ‘phylogeny’ pipelines. An all-by-all blast was used to find homologous sequences across the species based on sequence similarity. These blast results were aligned and run through RAxML 8.0 to make gene trees. These gene trees were used to identify orthologues; non-homologous sequences were removed. Two supermatrices were constructed using the homologous protein alignments. The complete matrix had 7,878 genes and 37% average gene occupancy. The matrix used in the phylogenetic analysis was trimmed down to 1,178 genes with 70% average gene occupancy (Supplementary Table 5).
The 70% matrix was used in all phylogenetic analysis. RAxML was used to calculate maximum likelihood trees. The -m PROTGAMMAAUTO option was called to automatically test for the best-fitting amino-acid substitution model. This selected the LG model. However, the more complex GTR model was not included in this test. For this reason, we ran rapid bootstrap analyses (100 replicates) and searches for the best scoring ML tree using both the LG and GTR amino-acid substitution models and the Γ model of rate heterogeneity (PROTLGGTR or PROTGAMMAGTR). We also ran the suboptimal model WAG + Γ, as used in previous studies involving Xenoturbella11.
The Swofford–Olsen–Waddell–Hillis (SOWH) test38 evaluates statistical support for incongruent phylogenetic topologies. We used SOWHAT (https://github.com/josephryan/sowhat)21 to perform SOWH tests by comparing the following best constrained trees: (1) with Xenacoelomorpha as sister to Deuterostomia (((Ciona, Gallus, Homo, Branchiostoma, Anneissia, Saccoglossus, Strongylocentrotus), (Hofstenia, X_bocki, X_profunda)), Peripatopsis, Capitella, Macrodasys, Baseodiscus, Priapulus, Lottia, Phoronis, Loxosoma, Gnathostomula, Adineta, Drosophila, Mnemiopsis, Trichoplax, Amphimedon, Nematostella, Schmidtea); or (2) with Xenacoelomorpha as sister to Ambulacraria within Deuterostomia ((((Saccoglossus, Strongylocentrotus, Anneissia), (Hofstenia, X_profunda, X_bocki)), Branchiostoma, Homo, Gallus, Ciona), Peripatopsis, Schmidtea, Capitella, Macrodasys, Baseodiscus, Priapulus, Lottia, Phoronis, Loxosoma, Gnathostomula, Adineta, Drosophila, Mnemiopsis, Trichoplax, Amphimedon, Nematostella); against the maximum likelihood tree topology recovered using GTR + Γ. SOWHAT was run with RaxML for 130 replicates using WAG + Γ. WAG was used instead of LG or GTR because of computational capacity restrictions. Astral II22 was used to estimate a species tree given a set of unrooted gene trees. A total of 393 gene alignments with an average occupancy of 80% were input into Astral II (https://github.com/smirarab/ASTRAL/). The Astral II species tree was generated from the 393 gene trees with 100 replicates of bootstrapping. PhyloBayes19 MPI was run on CIPRES (https://www.phylo.org/) for 4,460 generations using the CAT + GTR + Γ4 options shown previously to be optimal16. The first 2,200 trees were discarded as burn-in and the posterior consensus was computed on the remaining 2,260 trees. All PhyloBayes analyses were conducted using the CIPRES Science Gateway26 and all other phylogenetic analysis were conducted on Amazon Web services C4 Instances (AWS EC2).
To assess if the phylogenetic signal differed between slow- and fast-evolving proteins, submatrices were constructed from the 70% average gene occupancy matrix. First, ribosomal proteins were screened from the original 70% matrix by blasting to the Uniprot ribosomal protein database (http://www.uniprot.org/docs/ribosomp). This filtered data set consisted of 1,127 genes (51 ribosomal proteins were removed). A gene tree was made for each gene and ranked by evolutionary rate23 using a custom python script from (https://github.com/NathanWhelan/Order-genes-by-evolutioanry-rate). The ranked genes were divided into the fastest-evolving 50% and the slowest-evolving 50%. The alignments were concatenated and then used to construct a maximum likelihood tree using the PROTGAMMAGTR amino-acid substitution model in RAxML. Support was assessed with 100 bootstrap pseudo-replicates.
All alignments, SOWHAT constraint trees and partition information are available on the Dryad repository (http://datadryad.org/resource/doi:10.5061/dryad.79dq1).
Accession codes
Data deposits
Sequence data have been deposited in GenBank; accession numbers can be found in Supplementary Tables 3–5.
References
Westblad, E. Xenoturbella bocki n.g., n.sp. a peculiar, primitive turbellarian type. Ark. Zool. 1, 11–29 (1949)
Israelsson, O. New light on the enigmatic Xenoturbella (phylum uncertain): ontogeny and phylogeny. Proc. R. Soc. Lond. B 266, 835–841 (1999)
Reisinger, E. Was ist Xenoturbella? Z. Wiss. Zool. 164, 188–198 (1960)
Nakano, H. What is Xenoturbella? Zool. Lett. 1, 22 (2015)
Norén, M. & Jondelius, U. Xenoturbella’s molluscan relatives... . Nature 390, 31–32 (1997)
Israelsson, O. …and molluscan embryogenesis. Nature 390, 32 (1997)
Bourlat, S. J., Nielsen, C., Lockyer, A. E., Littlewood, D. T. J. & Telford, M. J. Xenoturbella is a deuterostome that eats molluscs. Nature 424, 925–928 (2003)
Bourlat, S. J. et al. Feeding ecology of Xenoturbella bocki (phylum Xenoturbellida) revealed by genetic barcoding. Mol. Ecol. Resour. 8, 18–22 (2008)
Bourlat, S. J. et al. Deuterostome phylogeny reveals monophyletic chordates and the new phylum Xenoturbellida. Nature 444, 85–88 (2006)
Bourlat, S. J., Nielsen, C., Economou, A. D. & Telford, M. J. Testing the new animal phylogeny: a phylum level molecular analysis of the animal kingdom. Mol. Phylogenet. Evol. 49, 23–31 (2008)
Dunn, C. W. et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452, 745–749 (2008)
Bourlat, S. J., Rota-Stabelli, O., Lanfear, R. & Telford, M. J. The mitochondrial genome structure of Xenoturbella bocki (phylum Xenoturbellida) is ancestral within the deuterostomes. BMC Evol. Biol. 9, 107 (2009)
Telford, M. J., Budd, G. E. & Philippe, H. Phylogenomic insights into animal evolution. Curr. Biol. 25, R876–R887 (2015)
Hejnol, A. et al. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. R. Soc. B 276, 4261–4270 (2009)
Ryan, J. F. et al.; NISC Comparative Sequencing Program. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 342, 1242592 (2013)
Philippe, H. et al. Acoelomorph flatworms are deuterostomes related to Xenoturbella . Nature 470, 255–258 (2011)
Israelsson, O. & Budd, G. E. Eggs and embryos in Xenoturbella (phylum uncertain) are not ingested prey. Dev. Genes Evol. 215, 358–363 (2005)
Perseke, M. et al. The mitochondrial DNA of Xenoturbella bocki: genomic architecture and phylogenetic analysis. Theory Biosci. 126, 35–42 (2007)
Lartillot, N., Lepage, T. & Blanquart, S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288 (2009)
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014)
Church, S. H., Ryan, J. F. & Dunn, C. W. Automation and evaluation of the SOWH Test with SOWHAT. Syst. Biol. 64, 1048–1058 (2015)
Mirarab, S. & Warnow, T. ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31, i44–i52 (2015)
Whelan, N. V., Kocot, K. M., Moroz, L. L. & Halanych, K. M. Error, signal, and the placement of Ctenophora sister to all other animals. Proc. Natl Acad. Sci. USA 112, 5773–5778 (2015)
Dunn, C. W., Giribet, G., Edgecombe, G. D. & Hejnol, A. Animal phylogeny and Its evolutionary implications. Annu. Rev. Ecol. Evol. Syst. 45, 371–395 (2014)
Cannon, J. T. et al. Xenacoelomorpha is the sister group to Nephrozoa. Nature http://dx.doi.org/10.1038/nature16520 (this issue)
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proc. Gateway Computing Environments Workshop (GCE), 14 November 2010, New Orleans, LA, 1–8 (IEEE, 2010)
Barry, J. P. & Kochevar, R. E. Calyptogena diagonalis, a new vesicomyid bivalve from subduction zone cold seeps in the Eastern North Pacific. Veliger 42, 117–123 (1999)
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012)
Koressaar, T. & Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291 (2007)
Boore, J. L. & Brown, W. M. Mitochondrial genomes of Galathealinum, Helobdella, and Platynereis: sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa. Mol. Biol. Evol. 17, 87–106 (2000)
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994)
von Nickisch-Rosenegk, M., Brown, W. M. & Boore, J. L. Complete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: gene arrangements indicate that Platyhelminths are Eutrochozoans. Mol. Biol. Evol. 18, 721–730 (2001)
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000)
Katoh, K. & Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 9, 286–298 (2008)
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000)
Dunn, C. W., Howison, M. & Zapata, F. Agalma: an automated phylogenomics workflow. BMC Bioinformatics 14, 330 (2013)
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnol. 29, 644–652 (2011)
Goldman, N., Anderson, J. P. & Rodrigo, A. G. Likelihood-based tests of topologies in phylogenetics. Syst. Biol. 49, 652–670 (2000)
Acknowledgements
We thank the crew of the R/V Western Flyer and pilots of the ROVs Tiburon and Doc Ricketts for their skill and patience during hunts for these ‘purple socks’. We also thank S. Johnson for verifying bivalve sequences obtained from Xenoturbella and L. Lundsten for hunting through many video files for imagery. We acknowledge the Cyberinfrastructure for Phylogenetic Research (CIPRES) Science Gateway26 for computing resources, and thank M. Miller for additional resources, S. Mirarab for discussions on species tree methods and N. Holland for comments on the manuscript. This work was supported by the David and Lucile Packard Foundation via the Monterey Bay Aquarium Research Institute, Scripps Institution of Oceanography and the National Science Foundation Assembling the Tree of Life program (DEB1036368 to G.W.R.).
Author information
Authors and Affiliations
Contributions
G.W.R., N.G.W. and R.C.V. collected the specimens. N.G.W. and J.I.C. generated and assembled mitochondrial data for the new Xenoturbella species. J.I.C. generated the Xenoturbella Illumina transcriptome and assembled the data. G.W.R. and J.I.C. performed phylogenetic analyses of mitochondrial genomes and transcriptome data. G.W.R. analysed the morphology of Xenoturbella spp. for the taxonomic descriptions. G.W.R. drafted the paper with J.I.C., N.G.W. and R.C.V. All authors commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 X. monstrosa sp. nov.
a, Photograph taken by ROV of a vesicomyid clam field at ~3,000 m depth, Monterey Bay, California. Most clams are E. extenta. Two specimens of Xenoturbella are visible (arrows). Scale bar estimated from size of E. extenta (5 cm average width). b, Supplementary Video 1 frame grab showing same clam field as a with A. diagonalis (Ad) and E. extenta clams. Numerous Xenoturbella (arrows) were observed, including two specimens sampled for this study. Scale bar estimated from size of E. extenta (5 cm average width). c, Ventral view of the holotype SIO-BIC BI1037. Although highly contracted and incomplete (the posterior end was removed and frozen and the ventral area removed for histology), the specimen is still over 10 cm long. The mouth (m), ring furrow (rf) and side furrow (sf) are visible. d, Frame grab of paratype SIO-BIC BI1039, a female in situ. e, Dorsal view of paratype SIO-BIC BI1039 (relaxed) showing ring furrow (rf), side furrow (sf), oocytes in body wall (oo) and part of the ventral surface (v). f, Ventral view of paratype SIO-BIC BI1039 showing the mouth (m), ring furrow (rf), oocytes in body wall (oo) and part of the epidermal ventral glandular network (vgn). g, Close-up of the ventral posterior of paratype SIO-BIC BI1039 showing the trailing off of the ventral glandular network (vgn) and oocytes clearly visible in the body wall. h, Close-up of the ventral anterior of paratype SIO-BIC BI1039 showing the mouth (m), ring furrow (rf) and the beginning of the ventral glandular network (vgn) near the anterior tip of the animal.
Extended Data Figure 2 X. churro sp. nov.
a, Frame grab of holotype SIO-BIC BI1040, a female, from the Guaymas Transform Fault, Gulf of California, Mexico, at ~1,700 m depth in situ. The ring furrow (rf) is visible towards the anterior end. b, Ventral view of the holotype SIO-BIC BI1040, (relaxed) showing mouth (m), ring furrow (rf), oocytes (oo), side furrow (sf) and epidermal ventral glandular network (vgn). Part of the dorsal side (d) is visible. c, Dorsal view of paratype SIO-BIC BI1039 showing ring furrow (rf) and oocytes (oo). d, Close-up of the ventral posterior of the holotype, showing the trailing off of the ventral glandular network (vgn), oocytes and the distinctively tapering posterior tip. e, Close-up of the anterior end of the holotype, showing the mouth (m), ring furrow (rf) and the beginning of the ventral glandular network (vgn) near the anterior tip of the animal.
Extended Data Figure 3 X. profunda sp. nov.
a, Frame grab of paratype SIO-BIC BI1044, a female, from the Pescadero Basin, Gulf of California, Mexico, at ~3,700 m depth in situ. The ring furrow (rf) is visible towards the anterior end. b, Ventral view of the holotype SIO-BIC BI1041, a male (relaxed) showing mouth (m), ring furrow (rf), side furrow (sf) and epidermal ventral glandular network (vgn). c, Dorsal view of the holotype showing the ring furrow (rf). The ventral glandular network (vgn) is visible where part of the ventral side is exposed. d, Close-up of the ventral anterior end of female paratype SIO-BIC BI1044, showing mouth (m), ring furrow (rf), side furrow (sf) and beginning of ventral glandular network (vgn) near the anterior tip of the animal. e, Close-up of the dorsal posterior of female paratype SIO-BIC BI1044, showing oocytes of different sizes distributed in the parenchyma. f, Parasagittal section (1 μm, stained with toluidine blue) through dorsal body of female paratype SIO-BIC BI1044 showing thick epidermis (e), subepidermal nerve net (n), basal lamina (bl), circular muscle layer (cm), longitudinal muscle layer (lm) and parenchyma with oocytes of various sizes (oo). The gastrodermis is not shown here. g, Parasagittal section (1 μm, stained with toluidine blue) through dorsal body wall of the male holotype SIO-BIC BI1041. Tissue layers as in the female, but a layer of developing (sp.) and mature sperm (s) instead of oocytes is present. The gastrodermis (g) lies beneath the parenchyma layer. h, Parasagittal section (1 μm, stained with toluidine blue) showing parenchyma layer with late spermatids or mature sperm with spherical heads and free flagella.
Extended Data Figure 4 X. hollandorum sp. nov.
a, Dorsal view of the live unrelaxed holotype SIO-BIC BI1036, showing ring furrow (rf). b, Ventral view of the holotype (relaxed) showing mouth (m), ring furrow (rf) side furrow (sf) and epidermal ventral glandular network (vgn). c, Close-up of the anterior end of the holotype, showing mouth (m), ring furrow (rf), side furrow (sf) and the ventral glandular network (vgn) near the anterior tip of the animal. d, Close-up of the diamond-shaped mouth.
Extended Data Figure 5 Mitochondrial COI haplotype networks.
a, X. bocki and X. westbladi haplotypes (GenBank accession numbers indicated). Sequences from the two nominal species are completely interconnected. This network suggests that there is only one species in Swedish waters, which should be recognized under the older name, X. bocki. b, X. monstrosa from Monterey Bay (California) and Gulf of California (Mexico) (SIO-BIC accession numbers indicated). c, X. profunda from the vicinity of a hydrothermal vent at 3,700 m in the Pescadero Basin, Mexico (SIO-BIC and UNAM specimens indicated). Holotypes are designated with asterisks in the figure. Networks generated with TCS33.
Extended Data Figure 6 Mitochondrial genomics of Xenoturbella.
a, Gene order for the four new species compared with that of X. bocki9,18. The gene order was consistent across all species with only minor variation in amino acids and length of the control region (also see Supplementary Table 1).
Extended Data Figure 7 Maximum likelihood phylogeny based on all 13 mitochondrial proteins.
Five Xenoturbella species formed a weakly supported sister clade to Acoela = Xenacoelomorpha. Xenacoelomorpha was sister to deuterostomes with weak support. Data were partitioned and analysed using RAxML and GTR + Γ. Analysis using PhyloBayes under CAT + GTR + Γ placed Xenacoelomorpha inside deuterostomes as a weakly supported sister group to Chordata (not shown), a result not recovered previously16 with the same program, model and terminals (except for X. profunda). Scale bar, substitutions per amino-acid position. The lower support for Xenacoelomorpha in its placement with deuterostomes, or its placement with Chordata, compared with previously shown, may be due to additional Xenoturbella data and/or the fact that all 13 mitochondrial proteins were used here.
Extended Data Figure 8 Species tree based on 393 gene trees using Astral II22.
Nodal support assessed via 100 pseudo-replicates of bootstrapping for each gene, combining both information from each gene tree and its respective bootstrap replicates. B, Bilateria; N, Nephrozoa.
Extended Data Figure 9 PhyloBayes analysis of 1,178 genes with 70% average gene occupancy.
CAT + GTR + Γ was used and the first 2,000 out of 4,460 generations for two chains were discarded as burn-in and the remainder used to generate a consensus tree. Scale bar, substitutions per amino-acid position. The largest discrepancy observed between chains was 0.164609 and the mean was 0.000989. Xenacoelomorpha grouped with protostomes. B, Bilateria.
Extended Data Figure 10 Maximum likelihood analyses of ‘fast’- and ‘slow’-evolving data sets.
The 70% occupancy data set was reduced to 1,127 genes (51 ribosomal proteins removed) and then divided into the fastest-evolving 50% and the slowest-evolving 50%. These were analysed using the PROTGAMMAGTR amino-acid substitution model in RAxML. a, Phylogeny based on the ‘fast-evolving’ data set with 216,066 amino acids. b, Phylogeny based on the ‘slow-evolving’ data set with 168,571 amino acids. Nodal support assessed via 100 pseudo-replicates of bootstrapping. Asterisks indicate 100% bootstrap support. B, Bilateria; N, Nephrozoa.
Supplementary information
Supplementary Information
This zipped file contains Supplementary Tables 1-5 and a Supplementary Table guide. (ZIP 310 kb)
Clam field at ~3,000 metres depth Monterey Bay, California.
ROV Tiburon dive 610, October 25, 2004. Collection of a X. monstrosa specimen (type) and images of other Xenoturbella in a field of vesicomyids (mainly Archivesica diagonalis in this clip). (MOV 17448 kb)
Near Pinkie’s Vent at ~1,700 m depth, a cold-water seep in the Guaymas Transform Fault, Gulf of California, Mexico.
ROV Doc Ricketts dive 385, April 13, 2012. The Mexican paratype of X. monstrosa is shown initially followed by the holotype of X. churro and then the collection of both specimens. (MOV 27879 kb)
Sediment near a hydrothermal vent at ~3,700 metres in the Pescadero Basin, Gulf of California, Mexico.
ROV Doc Ricketts dive 757, April 24, 2015. The video shows the female paratype of X. profunda gliding over the sediment in an S-pattern amongst polynoid scaleworms, sea anemones and vesicomyid clams. The video is speeded up to x6 times. (MP4 17706 kb)
Rights and permissions
About this article

Cite this article
Rouse, G., Wilson, N., Carvajal, J. et al. New deep-sea species of Xenoturbella and the position of Xenacoelomorpha. Nature 530, 94–97 (2016). https://doi.org/10.1038/nature16545
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature16545
- Springer Nature Limited
This article is cited by
-
Induced spawning with gamete release from body ruptures during reproduction of Xenoturbella bocki
Communications Biology (2023)
-
Mixotrophic chemosynthesis in a deep-sea anemone from hydrothermal vents in the Pescadero Basin, Gulf of California
BMC Biology (2021)
-
The sperm of Xenacoelomorpha revisited: implications for the evolution of early bilaterians
Zoomorphology (2019)
-
The digestive system of xenacoelomorphs
Cell and Tissue Research (2019)