

Abstract
Multiple sclerosis involves an aberrant autoimmune response and progressive failure of remyelination in the central nervous system. Prevention of neural degeneration and subsequent disability requires remyelination through the generation of new oligodendrocytes, but current treatments exclusively target the immune system. Oligodendrocyte progenitor cells are stem cells in the central nervous system and the principal source of myelinating oligodendrocytes1. These cells are abundant in demyelinated regions of patients with multiple sclerosis, yet fail to differentiate, thereby representing a cellular target for pharmacological intervention2. To discover therapeutic compounds for enhancing myelination from endogenous oligodendrocyte progenitor cells, we screened a library of bioactive small molecules on mouse pluripotent epiblast stem-cell-derived oligodendrocyte progenitor cells3,4,5. Here we show seven drugs function at nanomolar doses selectively to enhance the generation of mature oligodendrocytes from progenitor cells in vitro. Two drugs, miconazole and clobetasol, are effective in promoting precocious myelination in organotypic cerebellar slice cultures, and in vivo in early postnatal mouse pups. Systemic delivery of each of the two drugs significantly increases the number of new oligodendrocytes and enhances remyelination in a lysolecithin-induced mouse model of focal demyelination. Administering each of the two drugs at the peak of disease in an experimental autoimmune encephalomyelitis mouse model of chronic progressive multiple sclerosis results in striking reversal of disease severity. Immune response assays show that miconazole functions directly as a remyelinating drug with no effect on the immune system, whereas clobetasol is a potent immunosuppressant as well as a remyelinating agent. Mechanistic studies show that miconazole and clobetasol function in oligodendrocyte progenitor cells through mitogen-activated protein kinase and glucocorticoid receptor signalling, respectively. Furthermore, both drugs enhance the generation of human oligodendrocytes from human oligodendrocyte progenitor cells in vitro. Collectively, our results provide a rationale for testing miconazole and clobetasol, or structurally modified derivatives, to enhance remyelination in patients.
Similar content being viewed by others


Main
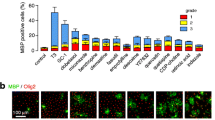
As repair of damaged myelin may provide therapeutic benefit in multiple sclerosis (MS) and other demyelinating disorders6,7,8,9,10,11,12, we set out to identify drugs that could be re-purposed as remyelinating therapeutics. We selected the US National Institutes of Health (NIH) Clinical Collection I and II libraries comprising 727 drugs with a history of safe use in clinical trials, to test for maturation of oligodendrocyte progenitor cell (OPCs) into myelinating oligodendrocytes. Using mouse epiblast stem cell (EpiSC)-derived OPCs, we developed an in vitro phenotypic screen that accurately quantified differentiation into mature oligodendrocytes by high content imaging of myelin protein expression (Fig. 1a).

a, Representative images of vehicle- and drug-hit-treated mouse EpiSC-derived OPCs from the primary screen. Nuclear (DAPI (4′,6-diamidino-2-phenylindole), blue) and MBP (red) staining along with HCA to identify oligodendrocyte (oligo.) nuclei (green) and MBP+ processes (yellow). Scale bar, 100 μm. b, Scatter plot of primary screen results displayed as normalized values of MBP process length and intensity for all 727 drugs with the 22 hits marked in red. Baseline (vehicle) was set at zero and thyroid hormone (positive control) was set at 100. c, Montaged images of whole postnatal day 7 mouse cerebellar slices treated with drug or vehicle for 5 days and stained for MBP (green). Insets show a representative example of the HCA script used to identify and quantify MBP+-aligned fibres (light blue). Scale bars, 1 mm for whole slices and 100 μm for insets. d, Relative quantification of HCA and western blot data from cerebellar slices treated for 5 days. For HCA screen, n = 1 with 6–12 slices averaged per group (also see Extended Data Fig. 2a). For western blot, n = 3 independent replicates of 12 slices per group. Values are mean for HCA and mean ± s.e.m. for western blot. e, Representative western blot of MBP isoforms and β-actin (loading control) of cerebellar slices treated for 5 days. Full blots are available in Supplementary Fig. 1. f, Chemical structures of clobetasol and miconazole. Source data are provided for Fig. 1b, d.
Two batches (>100 million cells each) of pure OPCs were generated from independent mouse pluripotent EpiSC lines of opposite sex (Extended Data Fig. 1a). EpiSC-derived OPCs shared virtually all defining molecular and cellular properties, including gene expression profiles with in vivo isolated OPCs, but provided the key advantage of being highly scalable (Extended Data Fig. 1b)3. For in vitro screening, the seeding density, endpoint assays, and dimethylsulphoxide (DMSO) (vehicle) tolerance were optimized in pilot studies to assure accurate and reproducible measurement of OPC differentiation in a 96-well format (Extended Data Fig. 1c).
For the primary screen, OPCs were treated with vehicle alone (0.05% (v/v) DMSO) as a negative control, thyroid hormone (a known OPC differentiation inducer) as a positive control13, or drug dissolved in DMSO at a concentration of 5 μM. After 72 h, cells were fixed and labelled with antibodies to myelin basic protein (MBP) and the length and intensity of MBP labelled oligodendrocyte processes measured (Fig. 1a). These features were reliable indicators of alteration in cellular phenotype, as indicated by consistency and high signal to background ratio of positive and vehicle controls across all screening plates (Extended Data Fig. 1d–g). We then normalized the experimental data for the tested drugs against thyroid hormone (set value of 100) on a per plate basis. On the basis of this analysis, we identified the 22 drugs that enhanced oligodendrocyte formation greater than five standard deviations above DMSO treatment and outperformed thyroid hormone in the measured parameters (Fig. 1b). Notably, one of the top 22 drugs was benztropine, a muscarinic receptor antagonist recently shown to induce OPC differentiation and remyelination8,9.
To validate and prioritize the 22 drug hits, the assay was repeated using alternative OPCs, reagents, and parameters to eliminate screen-specific artefacts (see Methods). Drugs were ranked by their dose-dependent ability to induce oligodendrocyte generation from OPCs without toxicity (Extended Data Fig. 2a). To demonstrate reproducibility, an independent laboratory tested selected drug hits using distinct equipment, plate format (1,536-well), personnel, and imaging/analysis scripts (see Methods). Of the 16 hits tested at the external screening site, 14 were validated as potent inducers of oligodendrocyte differentiation (Extended Data Fig. 2a, b).
We next tested whether the drug hits could promote the maturation of native OPCs in central nervous system (CNS) tissue. Cerebellar slices were generated from mice at postnatal day 7—a time that precedes widespread myelination—and treated ex vivo with drug or DMSO (vehicle) for 5 days and labelled with anti-MBP antibodies (Fig. 1c)6,7. We screened 11 of the top drugs and used a high content analysis (HCA) algorithm developed in house to rank them on the basis of their ability to increase the extent of MBP+ aligned fibres in whole cerebellar slices. The ‘high’ performing group consisted of four drugs that increased the number of MBP+ aligned fibres ∼150% or greater (Fig. 1d and Extended Data Fig. 2a). We validated the accuracy of our high content screen by semi-quantitative western blotting of MBP protein isoforms in independent slice culture experiments (Fig. 1d, e)14,15.
Analysis of structure–activity relationships revealed that the top hits from the primary screen segregated into two specific classes containing either a 1,3-diazole with mono-substitution at the 1-position or a sterane base structure (Extended Data Fig. 3a–d). We selected miconazole and clobetasol, the top overall performing hits in each of the imidazole and sterane classes respectively, for further mechanistic and functional testing after confirming that both drugs readily crossed the blood–brain barrier in mice (Fig. 1f, Extended Data Fig. 2a and Supplementary Table 1). Miconazole is a topical antifungal agent functioning through cytochrome P450 inhibition, and clobetasol is a potent topical corticosteroid, but their functions in OPCs were unknown.
To test whether miconazole or clobetasol enhance remyelination in vivo, we used a toxin-induced model whereby focal demyelinated lesions are generated in dorsal white matter of the spinal cord of adult mice by localized injection of lysolecithin (lysophosphatidylcholine (LPC)). In lesioned animals, demyelination is complete within 4 days, after which OPCs are recruited into the lesion. Widespread remyelination does not normally start until 14–21 days post lesion (d.p.l.), which provides a defined window from days 4 to 14 to test the efficacy of drugs to enhance the extent and rate of remyelination16. Both miconazole (10 or 40 mg per kg (body weight)) and clobetasol (2 mg per kg) treatment induced a marked improvement within the lesions of treated mice compared with vehicle-treated controls. At 8 d.p.l. both drugs induced a significant increase in the number of newly generated CC1+ oligodendrocytes in the lesion core (Fig. 2a, b). This was coincident with extensive MBP staining in the lesions of miconazole- and clobetasol- but not vehicle-treated animals at both 8 and 12 d.p.l. (Fig. 2a). Electron micrographs and tissue sections stained with toluidine blue demonstrated that miconazole and clobetasol each induced a striking increase in the extent of remyelination (Fig. 2c, d and Extended Data Fig. 4a, b). At 12 d.p.l., lesions of vehicle-treated mice consisted mostly of unmyelinated axons (6% myelinated) while those of clobetasol- and miconazole-treated mice contained >70% remyelinated axons throughout the extent of the lesion (Fig. 2d). Analysis of myelin thickness relative to axon diameter (g ratio) at 12 d.p.l. revealed that miconazole- and clobetasol-induced myelin was thinner than intact myelin, a defining characteristic of remyelination (Fig. 2d).

a, Representative immunohistochemical images of treated mice showing newly generated oligodendrocytes (CC1, red) and MBP (green) within the lesion (approximated by white dashed outline) at eight and 12 d.p.l. Scale bar, 200 μm. b, Quantification of CC1+ oligodendrocytes per lesion area at 8 d.p.l. Values are mean ± s.e.m.; n = 3 mice per group. Two-tailed t-test, *P < 0.05. c, Representative electron micrographs showing remyelinated axons within lesions of drug-treated mice at 12 d.p.l. Scale bar, 2 μm. d, Scatter plot of g ratios of lesion axons at 12 d.p.l.; n = 100 calculated from two mice per group compared to wild-type intact axons. Percentage of lesion axons myelinated is indicated in the legend. Source data are provided for Fig. 2b, d.
We also evaluated whether miconazole or clobetasol could promote precocious myelination during development, in the absence of injury or disease. We treated mice at postnatal day 2—a time point that precedes widespread CNS myelination—daily for 4 days with drug or vehicle. In miconazole- and clobetasol-treated mice, we found a significant increase in the number of CC1+ oligodendrocytes in the lateral corpus callosum compared with vehicle-treated mice (Extended Data Fig. 5a). Additionally, we found a significantly larger portion of the corpus callosum was populated by MBP+ fibre tracts in miconazole- and clobetasol-treated mice (Extended Data Fig. 5b). This suggests that clobetasol and miconazole enhance myelination in the absence of damage or disease. Collectively, the LPC demyelination and developmental mouse models demonstrate that miconazole and clobetasol each function to induce the differentiation of endogenous OPCs in the CNS and promote enhanced myelination.
To determine whether the drugs were working at a particular stage of the OPC differentiation process, we seeded OPCs in differentiation conditions and treated them with either miconazole or clobetasol at different time points (0, 16, 24, or 48 h), and assayed MBP expression at 72 h. For both miconazole and clobetasol, the number of MBP+ oligodendrocytes present at 72 h was dependent on drug treatment within the first 24 h of differentiation (Fig. 3a). In agreement with these data, treatment of differentiating OPCs with either drug for different durations (24, 48, 56, and 72 h) induced a progressive, time-dependent increase in the number of MBP+ oligodendrocytes (Fig. 3b). These data suggest that both drugs function directly on OPCs early in the differentiation process. Additionally, neither drug showed a significant impact on astrocyte formation from OPCs in vitro, suggesting they probably function as direct inducers of oligodendrocyte differentiation (Fig. 3c).

a, Percentage MBP+ oligodendrocytes generated from OPCs at 72 h with treatments initiated at time points indicated; n = 6 wells per condition with >6,000 cells scored per well. b, Percentage MBP+ oligodendrocytes generated from OPCs treated simultaneously and analysed at time points indicated; n = 8 wells per condition with >1,700 cells scored per well. c, Percentage GFAP+ astrocytes generated from OPCs at 72 h of treatment; n = 4 wells per condition >2,900 cells scored per well. d, Heat map depicting biochemical inhibition of muscarinic receptors M1–M5 displayed as percentage inhibition with minimum (green) and maximum (red). e, Western blot of total glucocorticoid receptor and its phosphorylation at Ser220 (p-GR) in OPCs treated for 1 h. f, Percentage MBP+ oligodendrocytes generated from OPCs 72 h after treatment; n = 6 wells per condition with >1,400 cells scored per well. g, Western blot of total ERK1/2 and their phosphorylation at Thr202/Tyr204 or Thr185/Tyr187 (p-ERK1/2) in cells (OPCs or mouse embryonic fibroblasts) treated for 1 h. FGF served as a positive control for p-ERK1/2 induction. h, Western blot of total ERK1/2 and p-ERK1/2 in OPCs treated for 1 h in the presence of the indicated pathway inhibitors. All graphs depict mean ± s.e.m. Full western blots are available in Supplementary Fig. 2. Source data are provided for Fig. 3a–d, f.
Muscarinic receptor antagonists such as benztropine and clemastine have recently been identified as remyelinating agents8,9. Therefore we tested whether miconazole or clobetasol function through the muscarinic acetylcholine pathway using functional cellular reporter assays of all muscarinic receptor subtypes (M1–M5). Neither miconazole nor clobetasol inhibited any of the five muscarinic receptor subtypes (Fig. 3d). We then profiled whether clobetasol or miconazole biochemically inhibited the activity of 414 different kinase isoforms. Neither clobetasol nor miconazole inhibited any of the kinases tested, suggesting their activity is not based on direct inhibition of protein kinases (Supplementary Table 2).
To explore the signalling pathways in OPCs influenced by these drugs, we performed genome-wide RNA sequencing and phosphoproteomic analyses on mouse OPCs treated with drug or vehicle (Extended Data Fig. 6a–c and Supplementary Table 3). Miconazole or clobetasol treatment altered OPC transcript expression and phosphoproteins within hours, and influenced expression of genes in signalling pathways involved in oligodendrocyte maturation and myelination. Clobetasol potently modulated genes downstream of multiple nuclear hormone receptors, including glucocorticoid receptor, which are known to be important regulators of myelin gene expression13,17. Since glucocorticoid receptor signalling is also known to enhance Schwann-cell-mediated myelination in the peripheral nervous system18, we tested whether the activity of clobetasol on OPCs was mediated by glucocorticoid receptor signalling. Treatment of OPCs with clobetasol for 1 h increased the phosphorylation of glucocorticoid receptor at Ser220, an activating post-translational modification (Fig. 3e). RU486, a competitive glucocorticoid receptor antagonist, blocked clobetasol-induced glucocorticoid receptor phosphorylation and oligodendrocyte differentiation (Fig. 3e, f) suggesting that the activity of clobetasol in OPCs is mediated through the glucocorticoid receptor signalling axis.
For miconazole, pathway analyses showed that proteins in the mitogen-activated protein (MAP) kinase pathway were most strongly affected (Extended Data Fig. 7a, b and Supplementary Table 3). Most prominent was the strong and sustained phosphorylation of both extracellular signal-regulated kinases ERK1 and ERK2 (ERK1/2) at canonical activation sites, which we validated by western blotting (Fig. 3g). In mice, genetic loss of ERK1/2 in the oligodendrocyte lineage results in normal numbers of OPCs and oligodendrocytes but widespread hypomyelination, while constitutive activation of ERK1/2 results in a profound increase in the extent of remyelination after toxin-induced demyelinating injury19,20. In contrast to miconazole, treatment of OPCs with clobetasol or benztropine did not induce ERK1/2 phosphorylation (Fig. 3g). Miconazole treatment of a non-neural cell type, mouse fibroblasts, also showed no increase of ERK1/2 phosphorylation, indicating potential cell-type specificity (Fig. 3g). PD0325901, a small molecule inhibitor of ERK’s upstream MAP-kinase kinase (MEK), blocked the ability of miconazole to induce ERK1/2 phosphorylation, suggesting that miconazole functions through a MEK-dependent mechanism in OPCs (Fig. 3h). We also treated mouse OPCs with voriconazole, a triazole-containing antifungal cytochrome P450 inhibitor with 80% structural similarity to miconazole, which failed to induce changes in ERK1/2 phosphorylation (Fig. 3g). This was consistent with the observation that voriconazole did not promote the differentiation of OPCs into oligodendrocytes (Extended Data Fig. 7c). Taken together, these results suggest that the effect of miconazole on OPCs is independent of cytochrome P450 inhibition.
We then assessed whether clobetasol and miconazole treatment would enhance the differentiation of human OPCs into oligodendrocytes. We generated human OPCs from human embryonic stem cells (hESCs) and human-induced pluripotent stem cells (hiPSCs) (Extended Data Fig. 8a–c)21,22. We then treated human OPCs with DMSO, clobetasol, or miconazole for 21 days followed by staining for MBP, imaging, and HCA (Extended Data Fig. 8d–g). Both drugs enhanced human OPC differentiation, with miconazole exhibiting the most reproducible and potent effects.
To interpret the potential impact of clobetasol or miconazole as therapeutics in immune-mediated MS models, we tested effects on immune cell survival and function. We found that only clobetasol, as expected from its known corticosteroid properties, altered naive T-cell differentiation and both the proliferation and secretion of cytokines by proteolipid protein (PLP139–151)- or myelin oligodendrocyte glycoprotein (MOG35–55)-sensitized lymph node cells (Extended Data Fig. 9a–j). As such, only clobetasol, but not the solely remyelinating drugs miconazole or benztropine, showed efficacy in reducing disease severity in the immune-driven relapsing–remitting PLP139–151 experimental autoimmune encephalomyelitis (EAE) model (Fig. 4a). The positive effect of clobetasol in this model resulted from its immunosuppressive effects as evidenced by the severe reduction of T cells within the spleen (Fig. 4b).

a, Scoring of disease severity in relapsing remitting PLP139–151-induced EAE mice treated beginning on day 13 (black arrow) and ending on day 29; n = 10 mice per group. Graph depicts mean daily disease score ± s.e.m. b, Flow-cytometric-based quantification of spleen cell numbers at day 29 from the PLP139–151 EAE cohort in a. Values are mean ± s.e.m.; n = 4 or 5 mice per group. c, Scoring of disease severity in chronic progressive MOG35–55-induced EAE mice treated daily for 10 days beginning at the peak of disease on day 15 (black arrow); n = 12–16 mice per group. Graph depicts mean daily disease score ± s.e.m. d, Mean improvement in disease score per animal (peak score minus ending score) of MOG35–55 EAE cohort in c. Also shown are external validation results in MOG35–55 EAE from an independent contract laboratory. n = 12 mice per group. For all EAE experiments, drugs were dosed daily by intraperitoneal injection: clobetasol (2 mg/kg), miconazole (10 mg/kg), benztropine (10 mg/kg), or FTY720 (1 mg/kg). All EAE disease scoring was as follows: 0, no abnormality; 1, limp tail; 2, limp tail and hind limb weakness; 3, hind limb paralysis; 4, hind limb paralysis and forelimb weakness; and 5, moribund. Two-tailed t-test, *P < 0.05 and **P < 0.01 for drug-treated groups compared with their respective vehicle-treated group. Source data are provided for Fig. 4a–d.
We also used a second EAE mouse model, MOG35–55-induced, in which the immune response was relatively controlled and disease pathology recapitulated chronic progressive demyelination. We used a therapeutic, rather than prophylactic, treatment regimen to evaluate whether drugs could reverse, rather than prevent, disease. Miconazole- and clobetasol-treated animals all exhibited a marked improvement in function, with nearly all animals regaining use of one or both hind limbs (Fig. 4c, d). In contrast, vehicle-treated mice exhibited chronic hindlimb paralysis over the treatment period. Benztropine treatment also resulted in functional improvement, but to a lesser extent than miconazole and clobetasol (Fig. 4c, d). Overt functional recovery of miconazole- and clobetasol-treated mice correlated with histological improvements in the spinal cord. Specifically, drug-treated mice showed restoration of MBP expression and a reduction in the extent of demyelination in the spinal cord, whereas vehicle-treated mice showed sustained areas of white matter disruption (Extended Data Fig. 10a–e).
Although the immunosuppressive effect of clobetasol makes it challenging to evaluate its remyelinating potential in EAE directly, its consistent and robust induction of OPC differentiation in vitro, and enhancement of remyelination in non-immune-driven in vivo assays, suggests that it serves a role in both immunomodulation and promotion of myelination. In contrast, miconazole did not modulate immune cell function and our data indicate that it acts as a direct remyelinating agent. Given the potential of miconazole as a remyelinating therapeutic, we contracted a separate laboratory to provide independent validation of its efficacy in the MOG35–55-induced EAE preclinical model. The laboratory independently validated the preclinical efficacy of miconazole in MOG35–55-induced EAE to reduce disease severity in treated mice (Fig. 4d).
Since the approval in 1993 of interferon (IFN)-β-1b for the treatment of MS, therapeutic development has centred on the generation of additional immunomodulatory agents. Despite the effectiveness of many of these drugs to modulate CNS inflammation in patients with MS, none of them prevent chronic progressive disease and disability—largely because of their inability to stop or reverse the failure of remyelination in the CNS. We developed an advanced high throughput screening platform to discover effective remyelinating therapeutics. This pluripotent stem-cell-based system provides unprecedented scalability, purity, and genotypic flexibility to screen for compounds that enhance OPC differentiation and myelination. Using this platform we identified two drugs approved by the US Food and Drug Administration, miconazole and clobetasol, with newly discovered functions to modulate OPC differentiation directly, enhance remyelination, and significantly reduce disease severity in mouse models of MS. Since miconazole and clobetasol are currently only approved for topical administration in humans, significant optimization of dosing, delivery, and potentially chemical structure will be required to enhance the on-target pharmacology in OPCs while diminishing any potential off-target side effects. However, the ability of miconazole and clobetasol to cross the blood–brain barrier raises the exciting possibility that these drugs, or modified derivatives, could advance into clinical trials for the currently untreatable chronic progressive phase of MS.
Methods
No statistical methods were used to predetermine sample size.
Mouse OPC preparation
OPCs used in this study were generated from two separate EpiSC lines, EpiSC9 (female) and 129O1 (male), using in vitro differentiation protocols and culture conditions described previously3,23. Cultures were regularly tested and shown to be mycoplasma free. To ensure uniformity throughout all in vitro screening experiments, EpiSC-derived OPCs were sorted to purity by fluorescent activated cell sorting at passage five with conjugated CD140a-APC (eBioscience, 17-1401; 1:80) and NG2-AF488 (Millipore, AB5320A4; 1:100) antibodies. Sorted batches of OPCs were expanded and frozen down in aliquots. OPCs were thawed into growth conditions for one passage before use in screening assays.
In vitro phenotypic screening of OPCs
EpiSC-derived OPCs were seeded onto poly-d-lysine 96-well Viewplate or CellCarrier plates (PerkinElmer) coated with laminin (Sigma, L2020; 10 µg ml−1) using electronic multichannel pipetors. For the primary screen, 30,000 cells were seeded per well in screening medium (DMEM/F12 supplemented with N2 (R&D Systems), B-27 (Life Technologies), neurotrophin 3 (R&D Systems; 10 ng ml−1), cAMP (Sigma; 50 μM), IGF-1 (R&D Systems; 100 ng ml−1), noggin (R&D Systems; 100 ng ml−1)) and allowed to attach for 2 h before addition of drug. NIH Clinical Collection I and II (http://www.nihclinicalcollection.com) drugs were added to assay plates with 0.1 μl pin replicators (Molecular Devices, Genetix; X5051), resulting in a final primary screening concentration of 5 µM. Thyroid-hormone-positive controls and DMSO vehicle controls were included in each assay plate. Cells were incubated under standard conditions (37 °C, 5% CO2) for 3 days and fixed with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS). Fixed plates were permeabilized with 0.1% Triton X-100 and blocked with 10% donkey serum (v/v) in PBS for 2 h. Cells were labelled with MBP antibodies (Abcam, ab7349; 1:100) for 1 h at room temperature (∼22 °C) followed by detection with Alexa Fluor-conjugated secondary antibodies (1:500) for 45 min. Nuclei were visualized by DAPI staining (Sigma; 1 µg ml−1). All plates for the primary screen were processed and analysed simultaneously to eliminate variability. Donepezil was identified in the primary screen; however, the drug was not available at the time of dose–response testing and was excluded from further testing.
Dose–response testing of drug hits followed the same procedure with the following modifications to eliminate any artefacts in the primary screen: independently sourced drugs; a distinct batch of EpiSC-derived OPCs from a mouse of opposite sex; multi-dose testing; cytotoxicity analysis; an alternative marker of mature oligodendrocytes proteolipid protein 1 (PLP1, antibody clone AA3 provided by B. Trapp; 1:5,000); and an alternative high content assay endpoint parameter (percentage of oligodendrocytes differentiated instead of process intensity and length parameters). All drugs were tested in quadruplicate at seven different doses (ranging from 333 nM to 6.7 μM) and classified into tiers on the basis of their half-maximum effective concentration (EC50) to induce OPC maturation, and their toxicity (concentration at which 50% of the cells were lost). Tier A drugs (n = 3) consisted of nanomolar dose effectors with little to no detectable toxicity at doses tested. Tier B drugs (n = 4) showed nanomolar effects but demonstrated toxicity at high doses. Tier C and D drugs required high doses to see an effect, demonstrated toxicity at low doses, or failed to show a dose-dependent response.
HCA of in vitro screen
For the 5 μM in vitro screen, stained plates were imaged on the Opera confocal imaging system (PerkinElmer) and a set of 24 ×10 fields were collected from each well, resulting in an average of 10,000 cells being scored per well. For the dose–response (6.7 μM, 5 μM, 3.3 μM, 1.7 μM, 666 nM, 500 nM, and 333 nM) in vitro assays, plates were imaged on the Operetta High Content Imaging and Analysis system (PerkinElmer) and a set of 14 ×20 fields captured from each well resulting in an average of 3,300 cells being scored per well. Analysis (PerkinElmer Acapella, Harmony, and Columbus software) began by identifying intact nuclei stained by DAPI; that is, those traced nuclei that were larger than 50 μm2 in surface area and possessed intensity levels that were typical and less than the threshold brightness of pyknotic cells. Each traced nucleus region was then expanded by 50% and cross-referenced with the mature myelin protein (MBP or PLP1) stain to identify oligodendrocyte nuclei, and from this the percentage of oligodendrocytes was calculated. Processes emanating from oligodendrocyte nuclei were identified using the CSIRO2 analysis module within a custom Acapella script. Maximum mean process length (denoted ‘process length’) and mean process intensity (denoted ‘process intensity’) were generated on a per well basis. For the 5 μM in vitro screen, values were calculated and normalized to 100 for thyroid hormone (positive control)-treated wells and to 0 for DMSO (vehicle)-treated wells, on a per plate basis.
Phenotypic validation testing of OPCs
Briefly, OPCs were grown and expanded in laminin-coated flasks before harvesting for plating. Cells were dispensed in screening media (see above for details) using a Multidrop Combi dispenser (Thermo Fisher) into laminin/poly-l-ornithine-coated sterile, 1,536-well, black clear-bottom tissue culture plates (Brooks Automation), to a final density of 2,000 cells per well. Plates were sealed with gasketed stainless steel lids with holes for gas exchange (Wako USA). Following cell attachment, library compounds were transferred by pintool (Wako USA) using 10 nl slotted pins. Library compounds were serially diluted in DMSO, and were added to plates to yield final concentrations of 0 (DMSO only), 4, and 20 μM compound. After incubation for 72 h at 37 °C, cells were fixed, washed, and stained similar to the 96-well OPC assay protocol, although all aspiration steps were performed using a Biotek EL406 Microplate Washer Dispenser (Biotek) equipped with a 1,536-well aspiration manifold. Dispense steps were performed with both peristaltic pump cassettes (for gentle reagent additions) and syringe pump manifolds (for faster bulk dispenses). Cells were stained with DAPI (Sigma; 1 µg ml−1) and MBP antibody (Abcam, ab7349; 1:100). Plates were then imaged using an InCell 2000 Analyzer High Content Imager (GE Healthcare Bio-Sciences). Well images were analysed using InCell Analyzer Workstation software, and the MBP signal was quantified with a process detection algorithm, using total process skeleton length to qualify activity.
Ex vivo cerebellar slice cultures
Whole cerebellum was collected from C57BL/6 mice at postnatal day 7 and embedded in agarose. Sagittal slices were cut on a microtome (Leica) at 300 µm. Slices were cultured in a DMEM–Basal Medium Eagle’s base with 15% heat-inactivated horse serum, modified N2, and PDGF-AA. After 1 day in culture, slices were treated daily for 5 days with test drugs or vehicle (DMSO). Drugs tested were clobetasol (5 μM), hydroxyzine (5 μM), clotrimazole (2 μM), miconazole (1 μM), ketoconazole (1 μM), vesamicol (5 μM), propafenone (2 μM), dicyclomine (5 μM), benztropine (2 μM), haloperidol (5 μM), and medroxyprogesterone (5 μM). The identity of the drugs was blinded to the experimenter. Slices were then lysed for western blot or fixed in 4% PFA and processed for HCA as detailed below.
Immunohistochemistry
Immunohistochemistry was performed as previously described3. In short, tissue sections or whole slices were washed three times in PBS, blocked in PBS containing Triton X-100 (0.1%) and normal donkey serum (NDS, 2% for sections and 10% for cerebellar slices) and incubated with primary antibody overnight. For MBP immunohistochemistry, the primary antibody solution consisted of 2% NDS, 2% bovine serum albumin, and 0.1% saponin. For all other antibodies, the primary antibody solution consisted of 2% NDS and 0.1% Triton X-100. Primary antibodies used included rat anti-MBP (Abcam, ab7349; 1:100), mouse anti-APC CC1 clone (Millipore, MABC200; 1:500), and rabbit anti-IBA1 (Wako Chemicals, 019-19741; 1:1,000). The tissue was then washed in PBS and incubated in secondary antibodies for 2 h. Secondary detection was performed with Alexa Fluor-conjugated secondary antibodies (1:500) for 1 h. Luxol fast blue staining was performed as previously described7.
High content screen of cerebellar slices
MBP-stained cerebellar slices were analysed by confocal image on an Operetta system using PhenoLOGIC machine-learning technology within Harmony software. The software was trained to identify elongated fibres more characteristic of axonal ensheathment and to exclude regions of small fibres or diffuse background fluorescence on the basis of texture features. MBP-positive surface area was collected and normalized to the total surface area for the group of slices treated with each drug. A minimum of six slices were treated per drug, which included an equal distribution of medial and lateral slices.
Western blotting of cerebellar slices
Cerebellar slices (each biological replicate using 12 slices per condition; six each from two separate animals) were collected in PBS and centrifuged. The PBS was aspirated and the pellet resuspended in 100 µL lysis buffer (20 mMTris, 137 mM NaCl, 5.0 mM EDTA pH 8.0, 10% glycerol, 1% NP40, pH to 8.0 with HCl), incubated on ice for 20 min, centrifuged, and the supernatant collected. Protein concentration was determined by a Pierce BCA protein assay kit (Thermo Fisher). Equal amounts of protein were applied to NuPAGE 12% Bis-TRIS gels (Life Technologies), and electrophoretically transferred onto a PVDF membrane (Life Technologies). The membranes were incubated with rabbit anti-MBP (Millipore, AB980; 1:500) and consequently probed with horseradish peroxidase (HRP)-conjugated goat anti-rabbit (1:5,000) or incubated with HRP-conjugated mouse anti-β-actin (Sigma, A3854; 1:10,000) to ensure even loading of samples. Enhanced chemiluminescence was performed with a West Pico kit (Thermo Fisher) and relative optical density was measured using ImageJ (NIH).
Chemoinformatics
Structure–activity searches of azoles and steranes were performed with Canvas program (Schrodinger Software, release 2014-1: Canvas, version 1.9). Tanimoto similarity between voriconazole and miconazole was calculated by ROCS (OpenEye Scientific Software).
Pharmacokinetics
C57BL/6 adult female mice were dosed intraperitoneally with miconazole (10 mg/kg or 40 mg/kg) or clobetasol (10 mg/kg). After 1 or 6 h, 100 µl of plasma was collected then each animal was perfused with PBS. Brains were collected, weighed, and rinsed with PBS. Water (0.5 ml) was added to the brain samples, which were then homogenized. Plasma and brain samples were each diluted fivefold with blank rat plasma. Three hundred microlitres of internal standard solution was added to the samples, vortexed, and centrifuged. Five microlitres of each sample was injected into an API-4000Qtrap mass spectrometer and quantified (Climax Labs).
Focal demyelination and drug treatment
Focal demyelination in the spinal cord was induced by the injection of 1% LPC solution. Ten- to 12-week-old C57BL/6 female mice were anaesthetized using isoflurane and a T10 laminectomy was performed. One microlitre of 1% LPC was infused into the dorsal column at a rate of 15 ml h−1. The animals were euthanized either at day 8 or day 12 after the laminectomy (n = 6–9 per group). Animals that were euthanized at day 8 received vehicle or drug daily by intraperitoneal injection between days 3 and 7. Animals used in the day 12 experiments received vehicle or drug daily by intraperitoneal injection between days 4 and 11. Drugs were dissolved in DMSO and then diluted with sterile saline for injection. Mice were deeply anaesthetized using ketamine/xylazine rodent cocktail and then euthanized by transcardial perfusion with 4% PFA for histological analysis or 4% PFA, 2% gluteraldehyde, and 0.1 M sodium cacodylate for electron microscopy. PFA fixed tissue was equilibrated in 30% sucrose, embedded in OCT, and cryosectioned at 20 µm thickness and processed for CC1 and MBP immunohistochemistry. ImageJ was used to measure area of the lesion and CC1+ cells within the lesion were scored manually. For CC1 scoring, sections were taken from the centre of each lesion to control for lesion variability.
Electron microscopy
Samples were processed as previously described3,24. In short, samples were osmicated, stained en bloc with uranyl acetate, and embedded in EMbed 812, an Epon-812 substitute (EMS). Sections (1 µm) were cut and stained with toluidine blue and visualized on a light microscope (Leica DM5500B). Additional thin sections were cut, carbon-coated and imaged either on a JEOL JEM-1200-EX electron microscope or a T12 electron microscope (FEI).
Developmental myelination
Mouse pups of strain CD1 were administered 2 mg/kg clobetasol, 10 mg/kg miconazole, or vehicle (DMSO in saline) by daily intraperitoneal injections from postnatal day 2–6. Some clobetasol treated animals exhibited sickness on the basis of this treatment, with low body weight, and some animals of the cohort died before end of treatment. On postnatal day 6 the pups were anaesthetized using ketamine and xylazine and euthanized by transcardial perfusion with 4% PFA. Tissue was fixed overnight in 4% PFA, equilibrated in 30% sucrose, and embedded in OCT. Sections (20 μm) were cut and processed for CC1 and MBP immunohistochemistry. ImageJ was then used to count and measure area of the corpus callosum as well as measure the extent of the corpus callosum length covered by MBP+ processes. Eight coronal sections containing corpus callosum rostral to the hippocampus from at least three animals per group were used for these analyses. To quantitate extent of MBP in the corpus callosum, a line was drawn through the centre of the corpus callosum from the lateral tip to the dorsal most extent of MBP expression in the corpus callosum. The length of this line was measured and then the dorsal-most point of the line was extended to the dorsal tip of the corpus callosum and measured to yield the length of the lateral callosum. The two numbers were divided to get the MBP/corpus callosum proportion. A two-tailed t-test was used to compare drug- with vehicle-treated groups.
Muscarinic receptor antagonism
Miconazole, clobetasol, and benztropine (all at 1 µM in DMSO) were sent to Select Screen (Life Technologies) with identities coded. GeneBLAzer or Tango assays were performed to determine level of acetylcholine muscarinic receptor M1, M3, M5 (GeneBLAzer), M2 and M5 (Tango) antagonism.
Kinase profiling
LanthaScreen, Z′-LYTE, and Adapta kinase assays were performed by Select Screen (Life Technologies). LanthaScreen Eu kinase assays were performed in Greiner low-volume 384-well plates. Assay buffer consisted of 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, 1 mM EGTA. Each well consisted of this mixture: 4.0 µl of 4 µM test drug in assay buffer, 8 µl of 2× kinase/Eu antibody mixture, and 4 µl of 4× Alexa Fluor 647 tracer. Plates were incubated for 60 min at room temperature (∼22 °C), then Alexa Fluor 647 emission (665 nm) and Europium emission (615 nm) read on a fluorescent plate reader. Data were analysed by generating the emission ratio (665 nm/615 nm) for each test point and normalizing 0% to control wells with no known inhibitor and 100% to control wells with highest concentration of known inhibitor.
Z′-LYTE assays were performed in Corning, low volume 384-well plates. Assay buffer consisted of 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, 1 mM EGTA. Each well consisted of this mixture: 2.5 µl 4× of 4 µM drug in assay buffer, 5 µl 2× peptide/kinase mixture, 2.5 µl 4× ATP solution. Plates were then incubated at room temperature (∼22 °C) for 60 min. Then 5 µl of a development reagent that contained a protease that selectively digested the non-phosphorylated peptide was added and the plates incubated for 60 min. Coumarin emission (445 nm) and fluorescein emission (520 nm) were read on a fluorescent plate reader. Data were analysed by normalizing out background fluorescence then generating the emission ratio (445 nm/520 nm) for each test point. Data were further normalized to 0% in control wells with no ATP and 100% in control wells with synthetically phosphorylated peptide of the same sequence.
Adapta assays were performed in Corning, low volume 384-well plates. Assay buffer consisted of 30 mM HEPES. Each well consisted of this mixture: 2.5 µl 4× of 4 µM drug in assay buffer, 2.5 µl 4× ATP solution, 5 µl 2× substrate/kinase mixture. Plates were then incubated at room temperature (∼22 °C) for 60 min. Then 5 µl of a development reagent that contained europium-anti-ADP antibody and ADP tracer were added and the plate incubated for 60 min. Alexa Fluor 647 emission (665 nm) and europium emission (615 nm) were read on a fluorescent plate reader. Data were analysed by generating the emission ratio (665 nm/615 nm) for each test point and normalizing 0% to control wells with no ATP in the kinase reaction and 100% to control wells with ADP.
Raw data from all kinase assays can be found in Supplementary Table 2.
HCA of astrocyte induction
For the astrocyte experiments in Fig. 3c, the experimental setup was identical to the PLP1-based primary validation screen except plates were stained for GFAP (DAKO, Z0334; 1:5,000). BMP4 (R&D Systems; 50 ng ml−1) and LIF (Millipore; 103 U ml−1) were used as the positive control for astrocyte induction. Assay plates were imaged on the Operetta High Content Imaging and Analysis system and a set of 14 ×20 fields captured. Columbus Data Management and Analysis System software (PerkinElmer) was used to quantify the percentage of GFAP+ astrocytes in each well using a method similar to that developed for oligodendrocytes.
Global phosphoproteomics
Quantitative global phosphorylation studies were performed on OPCs across two different time points (1 and 5 h after treatment) with miconazole, clobetasol, or DMSO treatment using a label-free ultra-high-performance liquid chromatography–tandem mass spectrometry (LC–MS/MS) workflow without fractionation. Briefly, for each sample 30 million cells were lysed with 2% SDS solution with protease and phosphatase inhibitor (Thermo Fisher), and detergent was removed on 200 µl of the cell lysate using the FASP cleaning procedure25. Each sample was digested by a two-step Lys-C/trypsin proteolytic cleavage and subjected to phospho-enrichment using a commercially available TiO2 enrichment spin tips (Thermo Fisher). LC-MS/MS analysis used a UPLC system (NanoAcquity, Waters) that was interfaced to an Orbitrap ProVelos Elite MS system (Thermo Fisher). Fold-change calculations were determined from peptide intensities for each drug versus DMSO at each time point. Phosphopeptides with greater than twofold change were imported into Ingenuity Pathway Analysis to elucidate signalling pathways perturbed with drug treatment.
RNA sequencing and analysis
Cells were lysed directly in 1 ml TRIzol (Invitrogen) and stored at −80 °C. Once all samples were collected, samples were thawed on ice and separated with chloroform using Phase Lock Gel tubes (5 PRIME). RNA was isolated using the miRNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s protocol. One microgram of each sample was then poly-A selected, fragmented, and library prepared using the TruSeq RNA Sample Prep Kit (Illumina) according to the manufacturer’s protocol. Samples were indexed using TruSeq adapters. One hundred base-pair paired-end reads were generated for each sample on an Illumina HiSeq 2500 instrument at the Case Western Reserve University Genomics Core facility. Between 5 million and 13 million reads were generated per sample for drug time course experiments. EpiSC RNaseq data were previously published (GEO accession number GSE57403)26. EpiSCs, EpiSC OPCs, and in vivo OPCs were sequenced to depths of 51,271,458 reads, 61,072,460 reads, and 62,530,709 reads, respectively. For in vivo isolated OPCs, CD140a+ cells were immunopanned from the CNS of mouse pups at postnatal day 7 as described previously27. Cells were then cultured for 5 days in identical culture conditions to EpiSC-derived OPCs before analysis.
Reference genome files were retrieved from Illumina iGenomes (http://ccb.jhu.edu/software/tophat/igenomes.shtml). Reads were aligned to the mm9 genome using Tophat version 2.0.8 with default settings28. Expression values of known RefSeq genes were calculated in units of fragments per kilobase per million reads (FPKM) using Cufflinks version 2.0.2 (ref. 29). Expression values were tabled to eliminate background signal by converting all values below 0.25 to 0, and subsequently adding 0.25 to all values. FPKMs were quantile normalized to correct for inter-sample variation. To identify genes whose expression was perturbed by drug treatments, duplicate samples of OPCs were treated with drug or vehicle for 2, 6, or 12 h. RNA sequencing data were tested for differential expression by comparing treatments to vehicle at each time point using Cuffdiff version 2.0.2 (ref. 30). The collective list of changed genes for each drug was evaluated with Ingenuity Pathway Analysis (application build 261899, content version 18030641).
Western blotting of mouse OPCs
EpiSC-derived OPCs were seeded into poly-l-ornithine/laminin coated six-well plates and allowed to attach for 2 h in DMEM/F12 without additional factors. Cells were treated with indicated inhibitors or DMSO for 1 h—SCH772984 (ChemieTek, 1 µM), SB590885 (Tocris, 10 nM), LY294002 (Tocris, 10 µM), and PD0325901 (Stemgent, 1 µM). Cells were then stimulated with drug or FGF2 (R&D Systems; 20 ng ml−1) for 1 h and then lysed in 200 µL RIPA buffer (0.15 M NaCl, 0.05 M Tris, pH = 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 10% glycerol, HALT protease and phosphatase inhibitor (Thermo Fisher) added just before use) and incubated on ice for 20 min. Lysates were centrifuged at 4 °C and supernatant collected. Protein concentrations were determined by Pierce BCA protein assay kit (Thermo Fisher). Equal amounts of protein were resolved in a reduced manner on NuPAGE 4–12% Bis-Tris gels (Life Technologies) and transferred onto PVDF membranes (Life Technologies). Blots were blocked in either 5% BSA (phosphoprotein) or 5% milk (non-phosphoprotein). Primary antibodies were all from the same vendor (Cell Signaling) and included phospho-Erk1/2 (4370S, clone D13.14.4E; 1:2,000), ERK1/2 (9107S, clone 3A7; 1:2,000), phospho-glucocorticoid receptor (4161S; 1:1,000), and glucocorticoid receptor (12041, clone D6H2L; 1:1,000) followed by incubation with HRP-conjugated secondary antibodies and chemiluminescent enhancement by West Pico substrate (Thermo Fisher).
Generation and screening of human OPCs
Human OPCs were generated from skin fibroblast-derived human iPSC line (CWRU43, Tesar laboratory) and hESC lines H7 (NIH Human Embryonic Stem Cell Registry WA07; NIH approval number NIHhESC-10-0061) and H9 (NIH Human Embryonic Stem Cell Registry WA09; NIH approval number NIHhESC-10-0062) as previously described21,22. iPSC- and hESC-derived OPCs were characterized by Sox10 (R&D Systems, AF2864; 1:100) staining, and then seeded in 96-well plates at 40,000 cells per well for drug testing. Cells were cultured with 1 μM miconazole, 5 μM clobetasol, or vehicle (DMSO) for 21 days, with fresh media changes with drug or vehicle every 2 days. Plates were fixed and stained with MBP (Abcam, ab7349; 1:100) then imaged on the Operetta system. We analysed results with slight modification to HCA Acapella scripts used for mouse oligodendrocytes.
Naive CD4+ T-cell assays
Naive CD4+ T cells (CD4+ L-selectinhi cells) were purified using AutoMacs Magnetic Bead cell separation technology (Miltenyi Biotech) from total lymph node cells isolated from unprimed mice with purity ranging from 98 to 99.9%. For in vitro activation, 5 × 105 naive CD4+ T cells were activated in the presence of plate-bound anti-CD3 (1 µg ml−1) plus Th1- (200 U ml−1 interleukin-2 (IL-2), 40 U ml−1 IL-12, 10 µg ml−1 anti-IL-4) or Th17- (10 ng ml−1 TGF-β1, 50 ng ml−1 IL-6, 1 μg ml−1 anti-IFN-γ, 1 μg ml−1 anti-IL-4, 1 μg ml−1 anti-IL-2) promoting conditions. On day 4, the cultured T cells were collected and the percentage of viable cytokine positive cells assessed by flow cytometry. The cells were stained with a LIVE/DEAD Fixable Violet Dead Cell Stain Kit, for 405 nm excitation (Life Technologies), anti-CD4-APC/Cy7 (clone RM4-5), anti-IFN-γ-PerCP/Cy5.5 (clone XMG1.2), and anti-IL-17-APC (clone eBio17B7) (eBioscience). Viable cells (5 × 105) were analysed per individual sample using a BD Canto II cytometer (BD Biosciences), and the data were analysed using FloJo version 9.5.2 software (Tree Star).
Ex vivo lymphocyte recall assays
Female SJL/J (Harlan Laboratories) or C57BL/6 mice were housed under SPF conditions. Six- to seven-week-old female mice were immunized subcutaneously with 100 μl of an emulsion containing 200 μg of Mycobacterium tuberculosis H37Ra (BD Biosciences) and 50 µg of PLP139–151 (SJL/J) or MOG35–55 (C57BL/6) distributed over three sites on the flank. For ex vivo culture draining, lymph nodes on day 8 were collected and cells were activated in the presence of anti-CD3 (1 µg ml−1) in the absence or presence of clobetasol, miconazole, or benztropine (10−9–10−5 M). To assess total cellular proliferation, cultures were pulsed with tritiated thymidine (1 µCi) at 24 h and cultures were harvested at 72 h. In replicate wells, culture supernatants were harvested at 72 h after culture, and the level of IFN-γ and IL-17 were assessed via Luminex assay (Millipore).
PLP139–151-induced relapsing remitting EAE
Six- to seven-week-old female SJL/J mice were induced with PLP139–151 as for ex vivo recall assays. Mice were allowed to progress to disease onset at day 13 before being randomized into treatment groups (n = 10 mice per group). Mice were then monitored for paralysis and treated daily by intraperitoneal injection with vehicle (DMSO in sterile saline), benztropine (10 mg/kg), clobetasol (2 mg/kg), or miconazole (10 mg/kg) beginning on day 13 and ending on day 29. This period fully encompassed the acute phase of disease onset followed by remission and the primary disease relapse. Treatments were blinded to the experimenters performing the assays. Mice were followed for disease severity in a blinded fashion with disease scoring as follows: 0, no abnormality; 1, limp tail; 2, limp tail and hind limb weakness; 3, hind limb paralysis; 4, hind limb paralysis and forelimb weakness; and 5, moribund.
MOG35–55 chronic progressive EAE model
EAE was induced by immunizing 10-week-old C57BL/6 female mice with 100 μl injection of MOG35–55/complete Freund’s adjuvant emulsion (Hooke Laboratories). One hour after immunization, mice were given 100 ng of pertussis toxin by 100 μl intraperitoneal injection. A second dose of pertussis toxin was administered the next day. EAE onset was monitored daily and scored using a clinical scale where 0 represented no abnormality; 1, limp tail; 2, limp tail and hind limb weakness; 3, hind limb paralysis; 4, hind limb paralysis and forelimb weakness; and 5, moribund. Mice that appeared moribund or exhibited forelimb paralysis were immediately euthanized and not used for the study. Once mice reached peak of disease (∼day 15; clinical score = 3) they were randomized into treatment groups, and drug or vehicle (DMSO in saline) was administered intraperitoneally daily for 10 days (n = 12–16 mice per group). Doses for each drug were miconazole (10 mg/kg), clobetasol (2 mg/kg), and benztropine (10 mg/kg). At these doses, no drugs showed any overt evidence of toxicity to any of the animals. Experimenters were blinded to the identity of the treatments and animals were scored daily. Cumulative disease scores for each animal were calculated during the treatment period, and a two-tailed t-test compared drug- with vehicle-treated groups. The extent of recovery for each animal was calculated as the difference between the peak disease score and the score at the end of each experiment, and a two-tailed t-test was used to compare each treatment with vehicle. External validation of MOG35–55 EAE experiments (n = 12 mice per group) was performed at Hooke Laboratories with experimenters blinded to the identity of the substances. FTY720 (fingolimod, 1 mg/kg), a known immunomodulatory drug, was used as a positive control during external validation of miconazole (10 mg/kg).
Animal welfare
All animal experiments were performed in accordance with protocols approved by the Case Western Reserve University and Northwestern University Institutional Animal Care and Use Committees.
Accession codes
Primary accessions
Gene Expression Omnibus
Data deposits
RNAseq datasets have been deposited in GEO under accession number GSE63804. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper. Correspondence and requests for materials should be addressed to P.J.T. (paul.tesar@case.edu) or R.H.M. (rhm3@gwu.edu).
References
Goldman, S. A., Nedergaard, M. & Windrem, M. S. Glial progenitor cell-based treatment and modeling of neurological disease. Science 338, 491–495 (2012)
Chang, A., Tourtellotte, W. W., Rudick, R. & Trapp, B. D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 346, 165–173 (2002)
Najm, F. J. et al. Rapid and robust generation of functional oligodendrocyte progenitor cells from epiblast stem cells. Nature Methods 8, 957–962 (2011)
Tesar, P. J. et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199 (2007)
Brons, I. G. et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195 (2007)
Mi, S. et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann. Neurol. 65, 304–315 (2009)
Bai, L. et al. Hepatocyte growth factor mediates mesenchymal stem cell-induced recovery in multiple sclerosis models. Nature Neurosci. 15, 862–870 (2012)
Deshmukh, V. A. et al. A regenerative approach to the treatment of multiple sclerosis. Nature 502, 327–332 (2013)
Mei, F. et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nature Med. 20, 954–960 (2014)
Fancy, S. P. et al. Overcoming remyelination failure in multiple sclerosis and other myelin disorders. Exp. Neurol. 225, 18–23 (2010)
Franklin, R. J. & Ffrench-Constant, C. Remyelination in the CNS: from biology to therapy. Nature Rev. Neurosci. 9, 839–855 (2008)
Dubois-Dalcq, M., Ffrench-Constant, C. & Franklin, R. J. Enhancing central nervous system remyelination in multiple sclerosis. Neuron 48, 9–12 (2005)
Barres, B. A., Lazar, M. A. & Raff, M. C. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development 120, 1097–1108 (1994)
Woodruff, R. H. & Franklin, R. J. The expression of myelin basic protein exon 1 and exon 2 containing transcripts during myelination of the neonatal rat spinal cord–an in situ hybridization study. J. Neurocytol. 27, 683–693 (1998)
Woodruff, R. H. & Franklin, R. J. The expression of myelin protein mRNAs during remyelination of lysolecithin-induced demyelination. Neuropathol. Appl. Neurobiol. 25, 226–235 (1999)
Jeffery, N. D. & Blakemore, W. F. Remyelination of mouse spinal cord axons demyelinated by local injection of lysolecithin. J. Neurocytol. 24, 775–781 (1995)
Kumar, S., Cole, R., Chiappelli, F. & de Vellis, J. Differential regulation of oligodendrocyte markers by glucocorticoids: post-transcriptional regulation of both proteolipid protein and myelin basic protein and transcriptional regulation of glycerol phosphate dehydrogenase. Proc. Natl Acad. Sci. USA 86, 6807–6811 (1989)
Morisaki, S. et al. Endogenous glucocorticoids improve myelination via Schwann cells after peripheral nerve injury: an in vivo study using a crush injury model. Glia 58, 954–963 (2010)
Ishii, A., Fyffe-Maricich, S. L., Furusho, M., Miller, R. H. & Bansal, R. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J. Neurosci. 32, 8855–8864 (2012)
Fyffe-Maricich, S. L., Schott, A., Karl, M., Krasno, J. & Miller, R. H. Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J. Neurosci. 33, 18402–18408 (2013)
Hu, B. Y., Du, Z. W. & Zhang, S. C. Differentiation of human oligodendrocytes from pluripotent stem cells. Nature Protocols 4, 1614–1622 (2009)
Wang, S. et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 12, 252–264 (2013)
Najm, F. J. et al. Isolation of epiblast stem cells from preimplantation mouse embryos. Cell Stem Cell 8, 318–325 (2011)
Najm, F. J. et al. Transcription factor-mediated reprogramming of fibroblasts to expandable, myelinogenic oligodendrocyte progenitor cells. Nature Biotechnol. 31, 426–433 (2013)
Wisniewski, J. R., Nagaraj, N., Zougman, A., Gnad, F. & Mann, M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J. Proteome Res. 9, 3280–3289 (2010)
Factor, D. C. et al. Epigenomic comparison reveals activation of “seed” enhancers during transition from naive to primed pluripotency. Cell Stem Cell 14, 854–863 (2014)
Cahoy, J. D. et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278 (2008)
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009)
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnol. 28, 511–515 (2010)
Trapnell, C. et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature Biotechnol. 31, 46–53 (2013)
Acknowledgements
This research was supported by grants from the US NIH NS085246 (P.J.T. and R.H.M.), NS030800 (R.H.M.), and NS026543 (S.D.M.); New York Stem Cell Foundation (P.J.T.); Myelin Repair Foundation (P.J.T., R.H.M., and S.D.M.); Mt. Sinai Health Care Foundation (P.J.T.); and NIH predoctoral training grants T32GM008056 (R.T.K.) and F30CA183510 (T.E.M). Additional support was provided by the Cytometry & Imaging Microscopy, Proteomics, and Genomics core facilities of the Case Comprehensive Cancer Center (P30CA043703), the CWRU Council to Advance Human Health, and philanthropic support from the Goodman, Long, and Geller families. P.J.T. is a New York Stem Cell Foundation–Robertson Investigator. We are grateful to M. Hitomi, W. Harte, D. Adams, W. Seibel, M. Haag, P. Scacheri, J. Wanta, C. Fang, H. Olsen, T. LaFramboise, J. Song, F. Van den Akker, and M. Shoham for technical assistance and discussion, A. Lager and M. Elitt for comments on the manuscript, B. Trapp for the PLP1 antibody, and B. Barres and B. Zuchero for RNA from in vivo isolated OPCs.
Author information
Authors and Affiliations
Contributions
F.J.N., R.H.M., and P.J.T. designed the overall screening strategy; F.J.N. and P.J.T. generated mouse OPCs and performed the primary screen; F.J.N., K.Q., H.T., and R.P. designed analysis scripts for the primary screen; K.R.B., M.S., M.B.B., and A.J. performed 1,536-well external validation of primary screen; M.M., F.J.N., A.Z., E.S., C.K., and A.S. performed organotypic slice culture and in vivo assays; F.J.N. and K.Q. designed analysis scripts for the slice cultures; A.P.R., J.R.P., and S.D.M. designed and performed PLP EAE and immune cell experiments; R.T.K., Z.S.N., and F.J.N. generated and tested human OPCs; D.C.F., F.J.N., T.E.M., and P.J.T. generated and analysed RNAseq data; D.M.S., F.J.N., and P.J.T. generated and analysed the proteomics data; F.J.N., M.M., R.H.M., and P.J.T. analysed all of the data and wrote the paper. All authors edited and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Performance of the primary screen.
a, Representative flow cytometry plots showing co-expression of NG2 and CD140a in both batches of EpiSC-derived OPCs used for this study. The batches of EpiSC-derived OPCs were sorted to purity (circled areas of plots) before use in this study. b, RNaseq expression heat map showing downregulation of pluripotent stem cell transcripts and upregulation of OPC transcripts when EpiSCs were differentiated into OPCs. Fragments per kilobase exon per million reads (FPKM) for each transcript are shown compared with in vivo isolated mouse OPCs. c, Quantification of DMSO (v/v) tolerance of EpiSC-derived OPCs in 96-well plates shown as mean ± s.e.m. For reference, 0.05% (v/v) DMSO was used as vehicle for all in vitro experiments in this study; n = 16 wells per group with >690 cells scored per well. d, Quantification of cell viability of thyroid hormone (positive control) and DMSO vehicle treatments per well across all ten assay plates shown as mean ± s.e.m.; n = 80 wells per group with >6,800 cells scored per well. e, Signal to background (S/B) mean values with standard deviation (s.d.) of controls from the entire screen; n = 80 wells per group. f, Raw data of MBP process length from the primary screen for thyroid hormone treatment and DMSO vehicle across all plates shown as mean ± s.d.; n = 8 wells per group with >6,800 cells scored per well. g, Raw data of MBP process intensity from the primary screen for thyroid hormone treatment and DMSO vehicle shown as mean ± s.d.; n = 8 wells per group with >6,800 cells scored per well.
Extended Data Figure 2 Drug hit ranking and validation.
a, Chart ranking the 22 primary drug hits (single dose rank) into four tiers on the basis of calculation of EC50 to induce PLP1+ oligodendrocytes from OPCs and the concentration at which 50% of the cells were lost (50% Tox) calculated from a seven-point dose treatment; n = 4 wells per dose per drug using independently sourced drug and separate OPC batch from the primary screen. Tiers ranged from the most potent and least toxic effectors to the least potent and most toxic: tier a (green), tier b (grey), tier c (orange), and tier d (red). The 1,536-well format external validation of 14 out of 16 tested hits is also shown. Drugs were further ranked into groups of high (green), medium (grey), and low (orange) on the basis of their ability to increase MBP+ axonal ensheathment in mouse cerebellar brain slices relative to vehicle (DMSO)-treated controls as measured by HCA. NT, not tested. b, External validation whole 1,536-well images of MBP+ (green) oligodendrocytes generated from OPCs after 72 h of treatment. GE InCell HCA is shown with processes traced in yellow and nuclei in blue.
Extended Data Figure 3 Primary screen structure–activity analysis.
Chemoinformatic identification of two substructures consistently enriched in high-performing drugs in the OPC assay. Numerical activity rank in the primary screen is indicated with the top 22 shown in green, 23–50 shown in grey, and >51 in red. a, 1,3-Diazoles, mono-substituted at the 1-position showed consistent activity on OPCs. b, c, 1,3-Diazoles, poly-substituted at two or more of the R groups (b), or 1,2,4-triazoles, mono-substituted at the 1-position (c) showed no activity on OPCs. d, The sterane base structure showed enrichment in the top performing hits.
Extended Data Figure 4 Histological assessment of remyelination in the LPC-induced model of demyelination.
a, Representative electron micrographs showing remyelinated axons within lesions of miconazole treated mice at 8 d.p.l. Scale bar, 2 μm. b, Histological sections stained with toluidine blue showing the extent of remyelination in the lesions of treated animals at 12 d.p.l. Normal uninjured myelin appears to the left of the black dashed line demarcating the definitive lesion edge. Scale bar, 20 μm.
Extended Data Figure 5 Miconazole and clobetasol enhance myelination in vivo.
a, b, Representative immunohistochemical images of the lateral corpus callosum (CC) of postnatal day 6 mouse pups that had been injected intraperitoneally daily for 4 days previously starting on postnatal day 2 with vehicle, clobetasol (2 mg/kg), or miconazole (10 mg/kg). CC1 (red) marks newly generated oligodendrocytes (a) and MBP (green) shows the extent of developmental myelination (b). Clobetasol and miconazole treatment each induce a marked increase in the number of CC1-positive oligodendrocytes in the lateral corpus callosum (a) and a significant increase in the length of the corpus callosum covered with aligned MBP+ fibres (b). Scale bar, 200 μm. Two-tailed t-test, *P ≤ 0.05 and **P ≤ 0.01. Str, striatum. All graphs are presented as mean ± s.e.m.
Extended Data Figure 6 RNaseq time course of drug-treated OPCs.
a, Volcano plots of all genes from OPCs treated with clobetasol or miconazole relative to vehicle control, with differentially expressed genes highlighted (red). Significance (measured as −log10[q value]) is plotted in relation to expression change (log2[treatment/vehicle]). Time course was after 2, 6, and 12 h of drug treatment. b, Venn diagrams depicting the overlap of genes differentially expressed at any time point and increased in treatments versus vehicle (left), as well as those decreased in treatments versus vehicle (right). c, Significant canonical pathways perturbed by each drug treatment according to Ingenuity Pathway Analysis.
Extended Data Figure 7 Global phosphoproteomic analysis of miconazole-treated OPCs.
a, b, OPCs treated with miconazole for (a) 1 h or (b) 5 h followed by global phosphoproteomic analysis. Proteins highlighted in green were observed to have a twofold or greater increase in phosphorylation whereas those highlighted in red were observed to have a twofold or greater decrease in phosphorylation compared with time-point-matched vehicle-treated controls. Proteins highlighted in grey were detected in the analysis but were not changed compared with vehicle control. See Supplementary Table 3 for the full phosphoproteomic data set. c, Quantification of the percentage of MBP+ oligodendrocytes differentiated from mouse OPCs after 72 h of treatment with DMSO, miconazole (1 μM), or voriconazole (seven doses, 0.01–6.7 μM); n = 6 wells per condition with >6,000 cells scored per well. Graph presented as mean ± s.e.m. The chemical structure of voriconazole is shown.
Extended Data Figure 8 Miconazole and clobetasol enhance human OPC differentiation.
a, Representative phase contrast image of a hESC colony cultured on matrigel. b, Representative phase contrast image of hESC-derived OPCs. c, hESC-derived OPCs stain positive for Sox10. d, e, Representative images of hESC-derived OPCs (d) and hiPSC-derived OPCs (e) treated with vehicle (DMSO), miconazole (1 μM), or clobetasol (5 μM) for 21 days stained for MBP (red). f, g, HCA of hESC-derived (f) and hiPSC-derived (g) OPCs differentiated in the presence of drugs or vehicle over 21 days; n = 3–5 wells per condition with >120 cells scored per well. Graphs presented as mean ± s.e.m. Scale bars, 100 μm.
Extended Data Figure 9 Effects of miconazole and clobetasol on immune cell survival and function.
a–d, Quantification of cell proliferation (a, c) and differentiation (b, d) of naive CD4+ T cells from unprimed SJL/J mice after activation with plate-bound anti-CD3 under Th1 (a, b) or Th17 (c, d) cell driving conditions. e–j, Ex vivo recall assays quantifying cell proliferation (ΔCPM) (e, h), with IFN-γ (f, i) and IL-17 (g, j) cytokine production from lymphocytes of mice primed with PLP139–151 (e–g) or MOG35–55 (h–j). Cultures were treated with vehicle (DMSO), benztropine, clobetasol, or miconazole (10−9–10−5 M) and analysed after 4 days. Four independent replicates are shown for each assay.
Extended Data Figure 10 Histological improvements in MOG35–55EAE spinal cords after treatment with miconazole or clobetasol.
a, b, Representative images of luxol fast blue (LFB) staining (a) demonstrated a clear decrease in areas of white matter disruption in the spinal cords of drug-treated animals which coincides with increased MBP staining (b). c, IBA1 staining showed a small reduction of immune cell infiltration into the lesion areas, especially in clobetasol-treated animals, but not an abrogation. d, e, Representative images stained with toluidine blue (d) and electron micrographs (e) revealed a reduction in the areas of demyelination in drug-treated animals. Lesioned areas are outlined with black dotted lines. Insets in toluidine blue staining show higher magnification of myelination in the corresponding spinal cords. Scale bars, 100 μm (a–c, d) and 2 μm (e).
Supplementary information
Supplementary Information
This file contains Supplementary Table 1 and Supplementary Figures 1-2 (full scans of western blots presented in the Letter). (PDF 695 kb)
Supplementary Table 2
This table shows global kinase profiling. Results of the 414 kinase assays tested are listed in the first three tabs for each of the Z’LYTE, Adapta, and Lantha assays, respectively. (XLSX 83 kb)
Supplementary Table 3
This table shows global phosphoproteomics. Tab 1 is the raw data of all detected phosphorylated peptides after one and five hour treatments of DMSO, clobetasol, and miconazole of OPCs. Tab 2 contains peptides that met the two-fold change cutoff for each drug relative to the DMSO value at the same time point. Tab 3 shows IPA analysis of miconazole treatment after one and five hour treatments. (XLSX 563 kb)
Rights and permissions
About this article

Cite this article
Najm, F., Madhavan, M., Zaremba, A. et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 522, 216–220 (2015). https://doi.org/10.1038/nature14335
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14335
- Springer Nature Limited
This article is cited by
-
Extracellular vesicles produced by HIV-1 Nef-expressing cells induce myelin impairment and oligodendrocyte damage in the mouse central nervous system
Journal of Neuroinflammation (2024)
-
Artificial axons as a biomimetic 3D myelination platform for the discovery and validation of promyelinating compounds
Scientific Reports (2023)
-
Inflammation in multiple sclerosis: consequences for remyelination and disease progression
Nature Reviews Neurology (2023)
-
An in vivo accelerated developmental myelination model for testing promyelinating therapeutics
BMC Neuroscience (2022)
-
Higher levels of myelin are associated with higher resistance against tau pathology in Alzheimer’s disease
Alzheimer's Research & Therapy (2022)