

Abstract
Krokinobacter eikastus rhodopsin 2 (KR2) is the first light-driven Na+ pump discovered, and is viewed as a potential next-generation optogenetics tool. Since the positively charged Schiff base proton, located within the ion-conducting pathway of all light-driven ion pumps, was thought to prohibit the transport of a non-proton cation, the discovery of KR2 raised the question of how it achieves Na+ transport. Here we present crystal structures of KR2 under neutral and acidic conditions, which represent the resting and M-like intermediate states, respectively. Structural and spectroscopic analyses revealed the gating mechanism, whereby the flipping of Asp116 sequesters the Schiff base proton from the conducting pathway to facilitate Na+ transport. Together with the structure-based engineering of the first light-driven K+ pumps, electrophysiological assays in mammalian neurons and behavioural assays in a nematode, our studies reveal the molecular basis for light-driven non-proton cation pumps and thus provide a framework that may advance the development of next-generation optogenetics.
Similar content being viewed by others
Main
Many organisms capture light energy and information using the rhodopsin family of proteins, which comprise the heptahelical transmembrane (7-TM) proteins called opsins covalently linked to retinal. Based on their primary sequences, the opsin genes are classified into two groups: the microbial and animal opsins. The animal rhodopsins primarily work as G-protein-coupled receptors, whereas the microbial rhodopsins have divergent functions, such as ion pumps, ion channels, sensors and kinases1,2,3. Recently, the pump- and channel-type rhodopsins have attracted broad attention, since these microbial rhodopsins can be used as powerful tools in the neuroscience field to control neuronal activity in a wide range of living animals (optogenetics)4,5,6.
As compared to the light-gated ion channel channelrhodopsin (ChR), the light-driven ion pumps have a long research history. Since the discoveries of the light-driven proton pump bacteriorhodopsin (BR) and the light-driven chloride pump halorhodopsin (HR)7,8, several light-driven ion pumps, such as proteorhodopsins (PRs), xanthorhodopsin (XR), and archaerhodopins (ARs), have been cloned and studied in diverse research fields, including optogenetics9,10,11,12. However, these light-driven ion pumps were basically classified into only two groups: outward proton pumps and inward chloride pumps, and no non-proton cation pumps have been discovered. Almost all of the known microbial rhodopsins are covalently bound to all-trans retinal (ATR) via the protonated Schiff base in the resting state, and the positively charged Schiff base proton in the middle of the ion transport pathway prevents cation transport (Extended Data Fig. 1). In the case of proton pumps, the Schiff base proton itself works as the substrate, and retinal photoisomerization alters the pKa values of the Schiff base and the carboxylates located on the extracellular side (historically called the ‘Schiff base counterions’), consequently leading to the proton transfer from the Schiff base to the extracellular side (Extended Data Fig. 1a)3,13. In the case of chloride pumps, Cl− binding stabilizes the protonated Schiff base and retinal photoisomerization flips the N–H dipole, thus driving the movement of Cl− from the extracellular environment to the intracellular side (Extended Data Fig. 1b)3,14. Therefore, it was widely believed that the absence of light-driven non-proton cation pumps was reasonable, because a non-proton cation would experience electrostatic repulsion from the Schiff base proton (Extended Data Fig. 1c).
However, in 2013, a new microbial rhodopsin, containing the unique NDQ (Asn112, Asp116 and Gln123) motif, was cloned from the marine flavobacterium Krokinobacter eikastus, and was characterized as the first light-driven Na+ pump15. Although the new rhodopsin, Krokinobacter eikastus rhodopsin 2 (KR2), transports protons in the absence of Na+ and Li+, under physiological conditions it works solely as an outward Na+ pump. A previous study revealed its cyclic photochemical reaction (photocycle) involving four spectroscopically distinguishable intermediates (K, L, M and O) and identified some of the functionally important residues15,16, but the mechanism of Na+ transport remained elusive. Understanding the structural basis of non-proton cation transport by KR2 would be enormously valuable, not only to enhance our knowledge of rhodopsin family proteins, but also to facilitate the design of novel light-driven ion pumps with desired ion selectivity, such as K+ and Ca2+ pumps. Such ion pumps, as well as Na+ pumps, would be useful as next-generation optogenetics tools.
Overall structure and comparison with BR
To understand the structural basis for Na+ transport, we expressed, purified and crystallized KR2 lacking the five carboxy-terminal amino acid residues (residues 1–275). The crystals were obtained by the lipidic cubic phase (LCP) method17 under low pH conditions (pH 4.0). The structure was determined by the molecular replacement method, using the coordinates of XR (PDB ID: 3DDL)18, and refined to 2.3 Å resolution (Extended Data Fig. 2a). The crystals belonged to the I222 space group, and contained one KR2 protomer in the asymmetric unit (Extended Data Fig. 2b). Except for the four disordered amino-terminal residues (residues 1–4), KR2 (residues 5–275), ATR, three lipids, one polyethylene glycol (PEG), and several water molecules are clearly resolved in the electron density map (Extended Data Fig. 2c).
The structure of KR2 is composed of an extracellular N-terminal helix (N-helix; residues 10–17) and 7-TM domains (residues 21–268), connected by three intracellular loops (ICL1 to ICL3) and three extracellular loops (ECL1 to ECL3) (Fig. 1). To understand the structure–function relationship of KR2 in detail, we compared the structure of KR2 with that of the best-characterized light-driven proton pump, BR (PDB ID: 1C3W)19. Consistent with the relatively high sequence similarity (46%) (Extended Data Fig. 3), the overall structure of KR2 superimposed well on that of BR (root mean square deviation (r.m.s.d.) of 1.83 Å over all Cα atoms) (Extended Data Fig. 4). However, KR2 has two distinct features on its extracellular side: the unique conformation of ECL1 and the presence of a short N-helix.

Crystal structure of KR2, viewed parallel to the membrane (left), and from the extracellular side (right). KR2 consists of the N-helix and the seven transmembrane helices (TM1–TM7) connected by extracellular loops (ECL1–ECL3) and intracellular loops (ICL1–ICL3). ATR, depicted by stick models, is coloured blue, and ECL1 is coloured green.
ECL1 and N-helix
In KR2, ECL1 (residues 86–106), forming the antiparallel β-sheet, has an entirely different orientation as compared to that in BR (Figs 1, 2a–c and Extended Data Fig. 4a). It protrudes into the extracellular side, and its conformation is rather similar to that in XR18 (Extended Data Fig. 4b). Notably, the four carboxylates (Glu90, Glu91, Asp98 and Asp102) on ECL1 are positioned separately and oriented towards the bulk solvent, although they are considered to contribute to the extracellular Na+-binding site, and to compose part of the exit pathway for Na+ conduction15 (Fig. 2b). To analyse the functions of these carboxylates in more detail, we first measured the level of Na+ binding to the E90Q/E91Q, D98N, and D102N mutants, using attenuated total reflection Fourier-transform infrared (atrFTIR) spectroscopy. In this experiment, we monitored the infrared spectral changes upon Na+ binding, and the difference spectra clearly showed that among the four carboxylates, only Asp102 contributes to the extracellular Na+-binding site (Extended Data Fig. 5a). We next examined the functions of the D102N mutant, and found that it has reduced thermostability, although its activity and kinetics are similar to those of wild-type KR2 (Fig. 2d, e and Extended Data Fig. 5b–e). These unexpected results suggested that only Asp102, among the four carboxylates, forms the extracellular Na+-binding site, and this site does not function as part of the Na+ conducting pathway, but contributes to the thermostability of KR2.

a–c, Overall architecture of KR2 (a), with magnified views of the green (b) and purple (c) highlighted regions. The black dashed lines and the red sphere represent hydrogen bonds and a water molecule, respectively. d, Pump activities of wild-type (WT) KR2 and mutants in E. coli cells. The activities were evaluated by measuring the pH changes caused by the proton motive force generated upon Na+ transport by KR2, without (light blue) and with (dark blue) the protonophore carbonyl cyanide m-chlorophenylhydrazone (CCCP), as previously reported15. Each tick mark of the y axis represents 0.2 pH change. Grey shading represents the dark condition; white indicates illumination. e, Thermostability assays of detergent-solubilized wild-type KR2 and mutant proteins. a.u., arbitrary units.
The second novel feature is that KR2 has an additional short N-helix, which is located at a similar position to ECL1 in BR (Fig. 2c and Extended Data Fig. 4a). This helix interacts with ECL1 (Extended Data Fig. 5f) and tethers it to the protein core through direct and water-mediated hydrogen bonds among Glu11 on the N-helix, Glu160 on TM4, and Arg243 (Glu204 in BR) on TM7 (Fig. 2c). While this short helix has not been found in other microbial rhodopsins, it is well conserved among the putative Na+-pump rhodopsins containing the NDQ motif (Extended Data Fig. 3)20. Thus, we tried to analyse the E11A, E160A, and R243A mutant proteins, as well as the N-helix-truncated (Δ1–18) mutant. We successfully expressed and purified E11A, E160A, and R243A, but not Δ1–18 (Extended Data Fig. 5e), and the thermostability assay revealed that these three mutant proteins were all quite unstable, as compared to the wild-type protein (Fig. 2e and Extended Data Fig. 5g). However, in contrast to their instabilities in detergent, all three mutants showed slightly decreased, but significant, pump activities in Escherichia coli cell membranes (Fig. 2d). These results indicate that the N-helix and the interaction between the N-helix and the protein core have important roles in the structural stability of KR2, rather than its Na+-pump activity. This is in contrast to BR, in which Glu204, the residue corresponding to Arg243 in KR2, has an important role in proton release to the extracellular side3.
Intracellular cavity
As compared to the extracellular region, the intracellular regions of BR and KR2 are similar, except for minor structural differences in the ICLs and the C-termini (Extended Data Fig. 4a). Notably, KR2 has a narrow opening between the intracellular ends of transmembrane domains 1 and 7. This hydrophilic cavity, formed by Tyr45, Phe46, Thr49, Ser60, Asn61 and Gly263, is filled with water molecules and leads to Gln123 (Fig. 3a–c). Since Gln123 is highly conserved among the putative Na+-pump rhodopsins (Extended Data Fig. 3), and the corresponding residue in BR, Asp96, is known to work as the substrate (proton) donor for the deprotonated Schiff base in the late intermediate3,13, it was assumed that Gln123 is also involved in substrate (Na+) binding in KR2. The flash photolysis experiment with the Q123A mutant revealed that the lifetimes of the late intermediates in the photocycle of the Q123A mutant (τL/M = 5.5 ms, τO = 22 ms) were more than three times longer than the reported lifetimes of wild type (τL/M = 1.0 ms, τO = 7.9 ms)15 (Fig. 3d–f). This result suggested that Gln123 captures Na+ from the bulk solvent and contributes to the formation of the transient intracellular Na+-binding site, and is consistent with the previous study showing that the Q123A mutant has significantly decreased pump activity15.

a, Overall architecture of KR2, depicted by a surface representation. The region around the intracellular cavity is highlighted in the orange rectangle. b, c, Magnified views of the highlighted region in a, with (c) and without (b) the surface representation. The water molecules are shown as red spheres. d–f, Photocycle of the Q123A mutant. d, e, Transient absorption spectra of the Q123A mutant (d) and time traces of the absorption changes (e) at specific probe wavelengths. f, Photocycle scheme for the Na+ pump, determined from the analysis of the results shown in d and e.
Schiff base region in two states
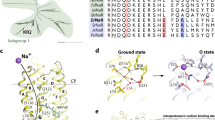
We next focused on the retinal binding pocket and the region around the Schiff base. In contrast to the relatively conserved retinal binding pocket (Supplementary Discussion and Extended Data Fig. 8), remarkable differences exist in the Schiff base regions between the structures of BR and KR2 (Fig. 4). In BR in the resting state, the protonated Schiff base points towards the extracellular side and hydrogen bonds with a key water molecule between the Schiff base counterions, Asp212 and Asp85. Asp212 is fixed by hydrogen bonds with Tyr57 and Tyr185, while Asp85, which works as the proton acceptor from the Schiff base in the M intermediate, interacts with Thr89 (Fig. 4a). In contrast, Asp85 and Thr89 are replaced by Asn112 and Asp116, respectively, in KR2, and the substitution of Tyr57 with Leu75 results in the loss of the hydrogen bond with Asp251 (Asp212 in BR). The key water molecule between Asn112 and Asp251 is located near Asp251 and Arg109 (Arg82 in BR), and is no longer hydrogen bonded with the Schiff base and Asn112 (Fig. 4b) (Supplementary Discussion and Extended Data Fig. 9). The orientation of the Schiff base is different from that in BR21, and in KR2 it points towards the area between Asp116 and Asn112, rather than towards the water (Fig. 4b and Extended Data Fig. 4c, d). We focused on Asp116, which is oriented away from the Schiff base and forms hydrogen bonds with Asn112 and Ser70 (Ile52 in BR). The estimated pKa value of Asp116, calculated using PROPKA22, was about 7.0, suggesting that Asp116 may be protonated in the crystal. To analyse the protonation state of Asp116 in more detail, we measured the absorption spectra of wild-type KR2 and D-to-N mutants of the titratable residues around the Schiff base, Asp116 and Asp251. We measured their spectra at pH 4.0 (the crystallization condition) and 7.0 (the physiological condition), and found that the peak wavelength (λmax) of the wild-type spectrum at pH 4.0 was red-shifted (536 nm), as compared to that at pH 7.0 (526 nm). This result suggested that Asp116 and/or Asp251 are deprotonated at pH 7.0, but protonated at pH 4.0. A similar red-shift was observed in the D251N mutant (526 nm at pH 7.0, and 540 nm at pH 4.0), but the λmax of the D116N mutant was already significantly red-shifted at pH 7.0 (558 nm) (Fig. 4d). These results strongly suggested that Asp116 is protonated in our structure under acidic conditions, and is deprotonated in the resting state under neutral conditions. Since the previous spectroscopic analysis revealed that Asp116 functioned as a proton acceptor in the M intermediate15, it was assumed that our structure determined under acidic conditions had a similar conformation to that of the M intermediate, in which Asp116 was protonated. This notion is in good agreement with the previous BR study, in which the authors demonstrated that the acid-induced structural change of BR resembled the change observed upon the formation of the M intermediate23.

a–c, Stereo views of the Schiff base regions of BR (a) and KR2 under acidic (b) and neutral (c) conditions. Red spheres and black dashed lines represent water molecules and hydrogen bonds, respectively. Grey dashed lines and numbers indicate the distances between Asn112 and the water in Å. d, The absorption spectra of wild-type (WT) KR2 and mutants. e, Photographs of the same KR2 crystal before (left) and after (right) the soaking experiment. f, Pump activities of wild-type KR2 and mutants in E. coli cells, without (light blue) and with (dark blue) CCCP. Each tick mark of the y axis represents 0.2 pH change.
To obtain structural information about KR2 under neutral conditions, we soaked the KR2 crystals at pH 7.5–8.5 for 48 h. The soaking led to the clear colour change of the crystal from purple to red (Fig. 4e), and thus we surmised that this soaking experiment changed the pH in the crystal and deprotonated Asp116. Using one of these red crystals, we determined the crystal structure of KR2 under neutral conditions, at 2.3 Å resolution. While the overall architectures of KR2 under both pH conditions were essentially identical (r.m.s.d. of 0.22 Å over all Cα atoms), there were some notable differences around the Schiff base (Fig. 4c). Under acidic conditions, the water molecule between Asn112 and Asp251 is located closer to Asp251 and Arg109, and these two residues interact by a water-mediated hydrogen bond. However, under the neutral conditions, the distance between the water and Asn112 was reduced from 5.1 to 3.1 Å, and thus Arg109 forms a direct hydrogen bond with Asp251. Since Asp251 and Arg109 are reportedly highly conserved and required for the Na+-pump activity, these structural differences suggest that the rearrangement of the water upon the M intermediate formation modulates the interaction between Asp251 and Arg10915,16.
Consistent with the pH-dependent peak shift of the absorption spectra observed in the purified proteins and crystals (Fig. 4d, e), the key difference was found in Asp116. In contrast to the observations made under acidic conditions, the electron density for the side chain of Asp116 indicated the coexistence of two alternative rotamers, one pointing towards Ser70 and Asn112 (rotamer 1), and the other pointing towards the Schiff base nitrogen (rotamer 2). Rotamers 1 and 2 represent the protonated and deprotonated forms of Asp116, respectively, and on the basis of the occupancy refinement, we estimated a 65:35 mixture (Extended Data Fig. 6). These results suggest that the deprotonated Asp116 is oriented towards the protonated Schiff base in the resting state (rotamer 2 in Fig. 4c), and Asp116 changes its conformation and hydrogen bonds with Asn112 and Ser70 after it accepts a proton from the Schiff base in the M intermediate (Fig. 4b and rotamer 1 in Fig. 4c). Since the proton transfer to Asp116 and its conformational change move the positively charged proton away from the Schiff base, these two events probably sequester the proton from the ion-conducting pathway and facilitate Na+ transport. A previous study reported that the D116N mutant completely abolished Na+ transport activity and implied that the proton transfer step was a prerequisite to Na+ transport activity15, thus we analysed whether the conformational change of the protonated Asp116 was functionally important. We measured the Na+ transport activities of the S70T, S70A, N112A, and N112D mutants and found that while the S70T and N112D mutants still retained Na+ transport activities, the S70A and N112A mutants lost Na+ transport activity (Fig. 4f). These results indicate that the hydrogen bonds among Asp116, Ser70 and Asn112, which fix the conformation of the protonated Asp116, are essential for Na+ transport.
Working model of Na+ transport
Based on these results, we propose a KR2 transport model (Fig. 5). In the resting state, a proton is bound to the Schiff base nitrogen, and this positive charge is located in the centre of the ion transport pathway (Fig. 5a), which prevents cation transport. After the retinal isomerization and the N–H flipping, the proton is transferred from the Schiff base to Asp116. This event would occur during the transition from the K to M intermediate (Fig. 5b). In the M intermediate, the protonated Asp116 flips away from the Schiff base and forms hydrogen bonds with Ser70 and Asn112. This flipping moves the positively charged proton away from the centre of the ion transport pathway, reduces the energy barrier to Na+ transport, and thereby facilitates Na+ conduction (Fig. 5c). The proton would re-associate with the Schiff base in the O intermediate (the next intermediate after the M intermediate)15, thus it would prevent the reverse flow of Na+ (Fig. 5d, Supplementary Discussion and Extended Data Fig. 10).

The water, proton and Na+ are depicted by the red, green and orange spheres, respectively. a, In the resting state, the positive charge of the Schiff base proton blocks Na+ transport. b, After retinal isomerization and N–H flipping, the Schiff base proton is transferred to Asp116. c, In the M intermediate, the protonated Asp116 flips away and forms hydrogen bonds with Ser70 and Asn112. The flipping of Asp116 reduces the energy barrier to Na+ transport, and thereby facilitates the Na+ conduction. d, The re-protonation of the Schiff base would inhibit the reverse flow of Na+.
KR2 application to optogenetics
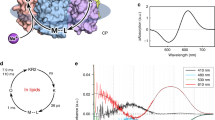
Like HR and AR3, KR2 is also expected to work as an inhibitory optogenetics tool. To test this possibility, we introduced a human-codon-optimized KR2 gene, fused with a membrane trafficking signal, enhanced yellow fluorescent protein (eYFP), and an endoplasmic reticulum export signal24, into cultured rat cortical neurons. The eYFP fluorescence was visible in neural cell bodies and membranous processes (Extended Data Fig. 7a). Voltage-clamp recordings showed that exposure to green light evoked a robust outward photocurrent, which was independent of the voltage (Fig. 6a, b). Next, we analysed whether the KR2-mediated photocurrent could inhibit neuronal spikes. Under current-clamp mode conditions, we injected a rectangular depolarizing current large enough to evoke repetitive spikes. As shown in Fig. 6c and Extended Data Fig. 7b, the generation of an action potential was completely blocked, while the membrane potential was hyperpolarized by KR2-activation with green light. The KR2-mediated inhibition of spiking was quite stable, and remained effective for more than one minute (Extended Data Fig. 7b).

a-c, Electrophysiology in cortical neurons. a, Voltage-clamp recording of the KR2 photocurrent. b, The current–voltage relationship. Values are means ± s.e.m. of 10 experiments. c, Current-clamp recording. Light illumination (green bar) inhibited the generation of the action potential. RMP, resting membrane potential. d, Effect of KR2 activation on C. elegans locomotion behaviour. The plot shows the change in the motion speed during and after light illumination (mean ± s.e.m., n = 9–10). e, Transverse section from the intracellular side. f, Pump activities of wild-type (WT) KR2 and mutants in E. coli cells suspended in 100 mM NaCl (top) or KCl (bottom), without (light blue) and with (dark blue) CCCP. Each tick mark of the y axis represents 0.5 pH change. g, Ion selectivities of wild-type KR2 and mutants. Values are means and standard deviation (s.d.) of three experiments. ND, not detected.
To examine whether KR2 could be applied to controlling neural activities in vivo, a KR2–Venus fusion protein was expressed in Caenorhabditis elegans neurons, using a pan-neuronal promoter. The Venus fluorescence was visible in neural cell bodies and membranous processes throughout the nervous system (Extended Data Fig. 7a). To test the effects of KR2 activation on neural activity, the velocity of forward locomotion was measured. Illuminating transgenic worms with green light in the presence of ATR specifically slowed down the worms’ locomotion (Fig. 6d), suggesting that the photoactivation of KR2 decreased the neural activity under the experimental conditions.
Based on these results, we propose that KR2 could indeed work as an inhibitory optogenetics tool in heterologous systems. To our knowledge, this is the first example of the successful application of a eubacterial rhodopsin to optogenetics. Unlike HR and AR3, KR2 is not expected to induce unintentional side effects, such as the activation of pH-sensitive channels25. The outward flow of Na+ would be more physiological than that of H+ or Cl−. Moreover, the voltage independent photocurrent of KR2 would allow efficient and reliable Na+ transport even under highly negative membrane potential conditions. Therefore, KR2 appears to have the potential to become an ideal inhibitory tool in some optogenetics experiments (Supplementary Discussion).
Engineering of K+-pumping KR2 variants
In the field of optogenetics, many variants of light-driven ion transporters have been engineered to expand the scope of their application. One of the most successful examples is the engineering of ChR variants with altered ion selectivity26,27,28. Thus, we next tried to engineer a KR2 variant with altered ion selectivity.
In our gating model of KR2, the flipping of Asp116 and the proton sequestration do not actively select Na+, but only facilitate the transfer of a non-proton cation. Thus, we assumed that it would be possible to engineer KR2 variants that preferentially transfer another cation, especially K+. Since the extracellular pathway of the ion-conduction was still ambiguous, we focused on two residues, Asn61 and Gly263, forming the intracellular vestibule (Figs 3b, c and 6e). We expressed several mutants of these two residues in E. coli, and found that some mutants showed outward K+ currents (Fig. 6e–g). Surprisingly, the double mutation N61P/G263W further increased the K+ conduction, and this mutant ( ) preferentially transferred K+ over Na+ (Fig. 6e–g). Although the K+/Na+ selectivity ratio is still not very high (1.17 ± 0.15), the intracellular K+ concentration is ten times higher than that of Na+, so
) preferentially transferred K+ over Na+ (Fig. 6e–g). Although the K+/Na+ selectivity ratio is still not very high (1.17 ± 0.15), the intracellular K+ concentration is ten times higher than that of Na+, so  is expected to work more efficiently as a K+ pump in vivo. Since no isolated or engineered light-driven K+ pump has been reported so far,
is expected to work more efficiently as a K+ pump in vivo. Since no isolated or engineered light-driven K+ pump has been reported so far,  provides a useful platform for the further engineering of KR2 variants to expand their applications.
provides a useful platform for the further engineering of KR2 variants to expand their applications.
Conclusion
The present crystal structures of KR2 and the accompanying functional analyses revealed the structural basis by which this microbial rhodopsin acquired Na+-pump activity, and enabled us to engineer the first light-driven K+ pump. Moreover, the electrophysiological and behavioural assays in mammalian neurons and nematodes showed the potential of KR2 as a new inhibitory optogenetics tool.
To fully understand the mechanism of Na+ transport by KR2, and to apply KR2 in several fields of neuroscience, additional studies are clearly needed. However, this study provides a valuable framework for the further development of optogenetics technology, as well as the basic mechanistic understanding of this new class of rhodopsin family proteins.
Methods
Cloning, protein expression and purification
The kr2 gene (GI: 478430804) was cloned from K. eikastus (NBRC 100814T) and subcloned into a pET-modified vector, with the resulting construct encoding a TEV cleavage site followed by a His8 tag at the C terminus15. For biochemical and spectroscopic experiments (pump activity assay, atrFTIR measurement, flash photolysis, quantification of KR2 expression in E. coli, thermostability assay), we mainly used a synthesized KR2 gene with codons optimized for E. coli expression16. The protein was expressed in C41 (DE3) E. coli cells, induced with 0.5 mM isopropyl β-d-thiogalactopyranoside (IPTG) and supplemented with 10 μM ATR (Sigma), for 3 h at 37 °C. The cells were disrupted and the membrane fraction was collected by ultracentrifugation (125,000g, 1 h). The protein was solubilized with 1.5% n-dodecyl-β-d-maltoside (DDM), in buffer containing 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 20 mM imidazole, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and 0.3% cholesteryl hemisuccinate (CHS). After Ni-NTA affinity chromatography, the collected fractions were treated with TEV protease and dialysed against buffer A (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.02% DDM, 0.004% CHS). The cleaved His8 tag and TEV protease were removed with Ni-NTA resin, and the protein was purified further by gel-filtration chromatography in the presence of 50 mM Tris-HCl (pH 8.0), 0.05% DDM, and 0.01% CHS. The purified protein was concentrated to 20 mg ml−1 using a centrifugal filter device (Millipore 100 kDa Mw cutoff), and frozen until crystallization.
Crystallization
The purified KR2 protein was mixed with monoolein (1-oleoyl-rac-glycerol) (Nu-Chek) in a 2:3 protein to lipid ratio (w/w). Aliquots (50–100 nl) of the protein–LCP mixture were spotted on a 96-well sandwich plate and overlaid by 800–1,000 nl of precipitant solution by the crystallization robot, mosquito LCP (TTP LabTech). Crystals of KR2 were initially obtained in the MemMeso crystallization screen (Molecular Dimensions), and optimized with 42% PEG 200, 0.1 M sodium acetate, pH 4.0, and 0.05 M MgCl2. The crystals were harvested directly from the LCP matrix using MiTeGen micromounts, and were flash-cooled in liquid nitrogen. The KR2 crystals under neutral conditions were prepared by adding soaking buffer (42% PEG 200, 0.1 M Tris-HCl, pH 8.0–9.0, and 0.05 M MgCl2) into the well of the plate containing KR2 crystals at pH 4.0. The final pH was 7.5–8.5.
Data collection and structure determination
X-ray diffraction data sets were collected on the BL32XU beamline at SPring-8, using a 10-μm-wide, 10-μm-high microbeam29. The diffraction data were processed with the XDS programs30, and the structure was solved by molecular replacement, using the structure of xanthorhodopsin (PDB ID: 3DDL) as a search model with Molrep31 in the CCP4 suite32. The resultant structure was iteratively refined in Refmac533 and Phenix34, and manually rebuilt in Coot35. In the Ramachandran plot, 97.0% of residues were in favoured regions and 2.6% of residues were in allowed regions. Final refinement statistics are summarized in Extended Data Fig. 2a. All figures were prepared with Cuemol (http://www.cuemol.org).
Measurement of Na+-pump activity in E. coli
E. coli cells expressing KR2 were cultured as described previously15. The cells were collected by centrifugation (4,800g, 3 min), washed three times with 100 mM NaCl, and re-suspended in the same solution for pump activity measurements. For the K+ transport assay, the cells were washed and suspended in 100 mM KCl in a similar manner. The cell suspension (7.5 ml) was placed in darkness and then illuminated using the output of a 1-kW tungsten–halogen projector lamp (Master HILUX-HR, Rikagaku, Japan) through a glass filter (Y-52, AGC Techno Glass, Japan) for 2.5 min at wavelengths >500 nm. The light-induced pH changes during the incubation were monitored with a pH meter (F-55, Horiba, Japan). Measurements were repeated under the same conditions after the addition of 10 μM CCCP.
atrFTIR measurement
Na+ binding to wild-type KR2 and mutants was studied by atrFTIR. Before the measurement, KR2 was reconstituted into 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC; molar ratio of KR2: DOPC = 1:20)15. The protein was placed on the surface of a diamond atrFTIR crystal (nine internal total reflections), and was dried in a gentle stream of N2. The sample was then rehydrated with the first solvent, containing 50 mM NaCl in 5 mM Tris–HCl (pH 8.0), at a flow rate of 0.5 ml min−1. The atrFTIR spectrum was recorded at 2 cm−1 resolution with an FTIR spectrometer (Agilent, California, USA) equipped with a liquid-nitrogen-cooled MCT detector. After exchange to the second solvent (50 mM KCl in 5 mM Tris–HCl, pH 8.0) and recording the FTIR spectrum, the difference FTIR spectrum was calculated by subtraction of the data obtained for the first and second solvents.
Flash photolysis
The transient absorption spectra of KR2 mutants were obtained by a flash photolysis system, using a multichannel detector (Hamamatsu Photonics, Japan). The purified sample was reconstituted into DOPC (molar ratio of KR2: DOPC = 1:20) and was suspended in 50 mM Tris–HCl buffer (pH 8.0), containing 100 mM NaCl15. The sample solution was placed in a quartz cuvette and excited with a beam of the second harmonics of a nanosecond-pulsed Nd3+-YAG laser (λ = 532 nm, INDI40, Spectra-Physics). The laser power was 3 mJ per pulse, and the repetition rate was 0.5 Hz, which is sufficiently slower than the photocycle rate of KR2, to avoid the excitation of transient photo-intermediates. The transient absorption spectra of KR2 were obtained by calculating the ratio of the transmitted probe light from a Xe arc lamp (L8004, Hamamatsu Photonics, Japan), with and without laser excitation. The time-evolution of the absorption change at a specific probe wavelength was obtained by combining the data points at different time-delays (tdelay = 80 μs–200 ms).
Quantification of KR2 expressed in E. coli
E. coli cells expressing KR2 were centrifuged and suspended in 50 mM Tris–HCl buffer (pH 8.0), containing 100 mM NaCl, to a final volume of 3 ml. Then, 200 μl of 1 mM lysozyme was added. The E. coli cells were disrupted by ultrasonication, and KR2 was solubilized in 3.0% DDM. The absorption change, representing the bleaching of KR2 by hydroxylamine, was measured with a UV-vis spectrometer (UV-2400PC, Shimadzu) with an integrating sphere (ISV-722, Jasco, Japan) for 0.5–1.5 h, after the addition of hydroxylamine to a final concentration of 500 mM15. Illumination was provided by the output from a 1-kW tungsten–halogen projector lamp (Master HILUX-HR, Rikagaku, Japan) through a glass filter (Y-52, AGC Techno Glass, Japan) at wavelengths >500 nm. The molecular extinction coefficient of rhodopsin (ε) was calculated by the ratio between the absorbances of rhodopsin and retinal oxime (ε = 33,900 M−1 cm−1), and the amount of KR2 expressed in E. coli cells was determined by the absorbance of the bleached rhodopsin and the ε value.
Measurement of thermostability
The thermostabilities of the KR2 mutants were estimated from the rates of colour bleaching of the pigment at 70 °C. A buffer containing 100 mM NaCl (or RbCl for wild-type KR2), 50 mM Tris-HCl (pH 8.0), and 0.1% DDM was used for the measurement. The concentration of the KR2 mutants was adjusted to OD532 = 0.5, and the solution (0.8 ml) was immersed in a heat bath maintained at 70 °C for 0.5–128 min. After heating, the solution was rapidly cooled on ice, and the protein aggregates were removed by centrifugation (10,000g, 3 min). The absorption spectrum of the supernatant was measured with a UV-vis spectrometer (UV-2400PC, Shimadzu), and the fraction of bleached protein was estimated from the decrease of the absorption at λmax.
Measurement of absorption spectra
Absorption spectra of KR2 mutants reconstituted into DOPC were recorded with a UV-vis spectrometer equipped with an integrating sphere. The samples were suspended in buffers containing 150 mM NaCl and 10 mM citric acid monohydrate, MES, MOPS, HEPES, CHES or CAPS. The pH was adjusted to pH 4 or pH 7 by the addition of NaOH or HCl.
Molecular cloning of mammalian expression construct
The gene encoding human-codon-optimized KR2 (hKR2) was commercially synthesized (GenScript, Japan) and subcloned in-frame into the EcoRI-BamHI sites of ptdTomato-N1. To prepare the 3.0 version of hKR2, the tdTomato was replaced with the eYFP carrying the membrane trafficking and ER export signals24 derived from the pAAV-EF1α-DIO-eNpHR3.0-eYFP vector (a gift from K. Deisseroth, Stanford University). The expression construct driven by the CMV promoter was named phKR2-3.0-eYFP. For optimization of hKR2 expression in neurons, the CMV enhancer/promoter of phKR2-3.0-eYFP was removed by digestion with AseI and NheI, and a PCR-amplified CaMKIIα promoter was inserted. A PCR-amplified woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) was additionally inserted in the NotI site of the plasmid, by In-Fusion cloning (Takara Bio). The pLenti-CaMKII-hChR2-eYFP-WPRE plasmid (a gift from K. Deisseroth) was used as a PCR template for the preparation of the CaMKIIα promoter and WPRE. The expression construct driven by the CaMKIIα promoter was named pCaMKIIα-hKR2-3.0-eYFP-WPRE. All constructs were verified by sequencing.
Mammalian cell culture
Cortical neurons were isolated from embryonic day 16 Wistar rats (Japan SLC Inc., Shizuoka, Japan), using Nerve-Cells Dispersion Solutions (Sumitomo Bakelite, Tokyo, Japan) according to the manufacturer’s instructions, and were grown in culture medium (Sumitomo Bakelite, Tokyo, Japan) under a 5% CO2 atmosphere at 37 °C. The expression plasmids were transiently transfected into cortical neurons by calcium phosphate transfection at days in vitro (DIV) 5 or 6. Electrophysiological recordings were then conducted at DIV 21–22 with neurons identified as expressing eYFP fluorescence by a conventional epifluorescence system.
Electrophysiology in rat cortical neurons
All experiments were performed at room temperature (23 ± 2 °C). Photocurrents were recorded as previously described36 using an EPC-8 amplifier (HEKA Electronic, Lambrecht, Germany) under a whole-cell patch-clamp configuration. The data were filtered at 1 kHz, sampled at 10 kHz (Digdata1440 A/D, Molecular Devices Co., Sunnyvale, California, USA), and stored in a computer (pClamp10.3, Molecular Devices).
The internal pipette solution for whole-cell voltage or current-clamp recordings from cortical neurons contained 125 mM K-gluconate, 10 mM NaCl, 0.2 mM EGTA, 10 mM HEPES, 1 mM MgCl2, 3 mM MgATP, 0.3 mM Na2GTP, 10 mM Na2-phosphocreatine, and 0.1 mM leupeptin, adjusted to pH 7.4 with KOH. The extracellular artificial cerebrospinal fluid solution contained 125 mM NaCl, 2.5 mM KCl, 25 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM CaCl2, 1 mM MgCl2, and 11 mM glucose, and was bubbled with a gas mixture containing 95% O2 and 5% CO2. In all cortical neuron experiments, artificial cerebrospinal fluid contained 20 μM 6,7-dinitroquinoxaline-2,3-dione (DNQX, Tocris Bioscience, Ellisville, Missouri, USA), 25 μM d-(-)-2-amino-5-phosphonovaleric acid (d-AP5, Tocris), and 100 μM picrotoxin (Nacalai Tesque, Kyoto, Japan) to block all synaptic inputs. The directly measured liquid junction potential was 11.4 mV and was compensated for.
Optics used in electrophysiological experiments
Irradiation was performed using a SpectraX light engine (peak, 530–600 nm, Lumencor Inc., Beaverton, Oregon, USA) controlled by computer software (pCLAMP10.3, Molecular Devices). The power of the light was directly measured under microscopy by a visible light-sensing thermopile (MIR-100Q, Mitsubishi Oil Chemicals, Tokyo, Japan) and was 5.4 mW per mm2.
Confocal images of cortical neurons
Cortical neurons prepared for electrophysiological recordings (DIV 22 or 24) were imaged three-dimensionally with a z-axis interval of 0.48 μm, using a conventional confocal laser-scanning microscope (LSM510META, Carl Zeiss) equipped with a 40× objective.
Generation of C. elegans transgenic lines
K. eikastus KR2 tagged with the fluorescent protein Venus (KR2–Venus) was placed under the pan-neuronal H20 promoter37 by the Gateway recombination system, in which the promoter on an entry vector and the open reading frame on a destination vector were combined by an LR reaction38. All C. elegans transgenic lines for optical stimulation experiments carried the lite-1(xu7) mutation, to reduce the intrinsic phototaxis response39. The expression construct pH20p::KR2-venus (20 ng μl−1) was introduced into the animals using standard transgenic techniques40 with the transformation marker punc-122p::venus (10 ng μl−1), generating lite-1(xu7); peEx1615[H20p::KR2::venus unc-122p::venus] (JN1615). Animals that harboured only the transgenic marker, lite-1(xu7); peEx1627[unc-122p::venus] (JN1627), were used as a control.
Behavioural assay in C. elegans
The forward locomotion speed of the animals was measured using a worm-tracking system41. C. elegans animals were grown to young adults on standard media42 and further cultivated overnight in the presence or absence of 5 μM ATR. For each trial, roughly 30 transgenic animals were washed from the culture plate and transferred to an ATR-free test plate (25 mM potassium phosphate (pH 6), 1 mM CaCl2, 1 mM MgSO4, 50 mM NaCl, 2% agar). After 3 min acclimatization on the test plate, a 30 s pulse of green light (peak wavelength = 525 nm; 0.15 mW per mm2) was delivered by a ring-shaped light-emitting diode (CCS Inc.). The average locomotion speed of all animals on a test plate was calculated for each second.
Statistical methods
No statistical methods were used to predetermine sample size.
References
Kouyama, T. & Murakami, M. Structural divergence and functional versatility of the rhodopsin superfamily. Photochem. Photobiol. Sci. 9, 1458–1465 (2010)
Spudich, J. L., Sineshchekov, O. A. & Govorunova, E. G. Mechanism divergence in microbial rhodopsins. Biochim. Biophys. Acta 1837, 546–552 (2014)
Ernst, O. P. et al. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 114, 126–163 (2014)
Zhang, F. et al. The microbial opsin family of optogenetic tools. Cell 147, 1446–1457 (2011)
Deisseroth, K. Optogenetics. Nature Methods 8, 26–29 (2011)
Aston-Jones, G. & Deisseroth, K. Recent advances in optogenetics and pharmacogenetics. Brain Res. 1511, 1–5 (2013)
Oesterhelt, D. & Stoeckenius, W. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nature New Biol. 233, 149–152 (1971)
Matsuno-Yagi, A. & Mukohata, Y. Two possible roles of bacteriorhodopsin; a comparative study of strains of Halobacterium halobium differing in pigmentation. Biochem. Biophys. Res. Commun. 78, 237–243 (1977)
Béjà, O. et al. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289, 1902–1906 (2000)
Balashov, S. P. et al. Xanthorhodopsin: a proton pump with a light-harvesting carotenoid antenna. Science 309, 2061–2064 (2005)
Chow, B. Y. et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463, 98–102 (2010)
Grote, M. & O’Malley, M. A. Enlightening the life sciences: the history of halobacterial and microbial rhodopsin research. FEMS Microbiol. Rev. 35, 1082–1099 (2011)
Lanyi, J. K. Proton transfers in the bacteriorhodopsin photocycle. Biochim. Biophys. Acta 1757, 1012–1018 (2006)
Essen, L. O. Halorhodopsin: light-driven ion pumping made simple? Curr. Opin. Struct. Biol. 12, 516–522 (2002)
Inoue, K. et al. A light-driven sodium ion pump in marine bacteria. Nature Commun. 4, 1678 (2013)
Ono, H., Inoue, K., Abe-Yoshizumi, R. & Kandori, H. FTIR spectroscopy of a light-driven compatible sodium ion-proton pumping rhodopsin at 77 K. J. Phys. Chem. B 118, 4784–4792 (2014)
Caffrey, M. & Cherezov, V. Crystallizing membrane proteins using lipidic mesophases. Nature Protocols 4, 706–731 (2009)
Luecke, H. et al. Crystallographic structure of xanthorhodopsin, the light-driven proton pump with a dual chromophore. Proc. Natl Acad. Sci. USA 105, 16561–16565 (2008)
Luecke, H., Schobert, B., Richter, H. T., Cartailler, J. P. & Lanyi, J. K. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291, 899–911 (1999)
Yoshizawa, S. et al. Functional characterization of flavobacteria rhodopsins reveals a unique class of light-driven chloride pump in bacteria. Proc. Natl Acad. Sci. USA 111, 6732–6737 (2014)
Kandori, H. Hydration switch model for the proton transfer in the Schiff base region of bacteriorhodopsin. Biochim. Biophys. Acta 1658, 72–79 (2004)
Rostkowski, M., Olsson, M. H., Sondergaard, C. R. & Jensen, J. H. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 11, 6 (2011)
Okumura, H., Murakami, M. & Kouyama, T. Crystal structures of acid blue and alkaline purple forms of bacteriorhodopsin. J. Mol. Biol. 351, 481–495 (2005)
Gradinaru, V. et al. Molecular and cellular approaches for diversifying and extending optogenetics. Cell 141, 154–165 (2010)
Li, T., Yang, Y. & Canessa, C. M. A method for activation of endogenous acid-sensing ion channel 1a (ASIC1a) in the nervous system with high spatial and temporal precision. J. Biol. Chem. 289, 15441–15448 (2014)
Kleinlogel, S. et al. Ultra light-sensitive and fast neuronal activation with the Ca2+-permeable channelrhodopsin CatCh. Nature Neurosci. 14, 513–518 (2011)
Berndt, A., Lee, S. Y., Ramakrishnan, C. & Deisseroth, K. Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science 344, 420–424 (2014)
Wietek, J. et al. Conversion of channelrhodopsin into a light-gated chloride channel. Science 344, 409–412 (2014)
Hirata, K. et al. Achievement of protein micro-crystallography at SPring-8 beamline BL32XU. J. Phys. Conf. Ser. 425, 012002 (2013)
Kabsch, W. Xds. Acta Crystallogr. D 66, 125–132 (2010)
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D 66, 22–25 (2010)
Collaborative Computational Project, 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 (1994)
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 (2011)
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010)
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010)
Ishizuka, T., Kakuda, M., Araki, R. & Yawo, H. Kinetic evaluation of photosensitivity in genetically engineered neurons expressing green algae light-gated channels. Neurosci. Res. 54, 85–94 (2006)
Shinkai, Y. et al. Behavioral choice between conflicting alternatives is regulated by a receptor guanylyl cyclase, GCY-28, and a receptor tyrosine kinase, SCD-2, in AIA interneurons of Caenorhabditis elegans. J. Neurosci. 31, 3007–3015 (2011)
Matsuki, M., Kunitomo, H. & Iino, Y. Goα regulates olfactory adaptation by antagonizing Gqα-DAG signaling in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 103, 1112–1117 (2006)
Liu, J. et al. elegans phototransduction requires a G protein-dependent cGMP pathway and a taste receptor homolog. Nature Neurosci. 13, 715–722 (2010)
Mello, C. C., Kramer, J. M., Stinchcomb, D. & Ambros, V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959–3970 (1991)
Yoshida, K. et al. Odour concentration-dependent olfactory preference change in C. elegans. Nature Commun. 3, 739 (2012)
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974)
Ran, T. et al. Cross-protomer interaction with the photoactive site in oligomeric proteorhodopsin complexes. Acta Crystallogr. D 69, 1965–1980 (2013)
Matsui, Y. et al. Specific damage induced by X-ray radiation and structural changes in the primary photoreaction of bacteriorhodopsin. J. Mol. Biol. 324, 469–481 (2002)
Kolbe, M., Besir, H., Essen, L. O. & Oesterhelt, D. Structure of the light-driven chloride pump halorhodopsin at 1.8 Å resolution. Science 288, 1390–1396 (2000)
Gushchin, I. et al. Active state of sensory rhodopsin II: structural determinants for signal transfer and proton pumping. J. Mol. Biol. 412, 591–600 (2011)
Kato, H. E. et al. Crystal structure of the channelrhodopsin light-gated cation channel. Nature 482, 369–374 (2012)
Di Tommaso, P. et al. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17 (2011)
Acknowledgements
We thank M. Hattori and H. Nishimasu for useful discussions and critical comments on the manuscript; A. Kurabayashi, N. Sahara, and J. Sasaki for technical assistance; and S. Doki, K. Hirata and the beam-line staff members at BL32XU of SPring-8 for assistance in data collection. The synchrotron radiation experiments were performed at BL32XU of SPring-8, with approval from RIKEN. This work was supported by the Platform for Drug Discovery, Informatics and Structural Life Science, of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), by JSPS KAKENHI (grant nos. 11J06643, 24115508, 24655009, 25104009, 24681003, 24227004, and 25291011), by the FIRST program, PRESTO, CREST, JST, and by a Grant-in-Aid for JSPS Fellows.
Author information
Authors and Affiliations
Contributions
H.E.K. expressed, purified, and crystallized KR2, and collected the diffraction data and solved the structures. K.I., H.O., Y.K., R.Y. and M.K. performed the functional analyses of KR2 in E. coli. H.K. and Y.I. performed the behavioural analysis in C. elegans. S.H., T.I., M.R.H., J.I., A.D.M. and H.Y. performed the electrophysiology in mammalian cells. M.T. and N.T. helped to make KR2 constructs for expression in mammalian cells. R.T. helped to measure the expression levels of KR2 in mammalian cells. K.Y. assisted with structure solution. S.Y. and K.K. provided the KR2 gene and helped to organize the project. H.E.K., K.I., R.I., H.K. and O.N. wrote the manuscript. H.K. and O.N. directed and supervised all of the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Ion transport mechanisms of light-driven proton pumps and chloride pumps.
a, The proton transport mechanism for light-driven proton pumping in BR3,4. The Schiff base proton works as the substrate, and avoids the electrostatic repulsion between itself and the substrate. b, The chloride transport mechanism for light-driven chloride pumping in HR3,4. The numbering corresponds to the residue numbers in HR from Halobacterium salinarum. c, The electrostatic repulsion problem has to be solved by the light-driven cation pumps, rather than the proton pumps. The substrate cation (A+) must avoid the electrostatic repulsion with the Schiff base proton.
Extended Data Figure 2 Crystallography.
a, Table describing data collection and refinement statistics of KR2 in acidic and neutral conditions. b, The crystal packing of KR2, viewed parallel to the membrane. c, Electron-density map for the retinal binding pocket of KR2. A stereo view of the 2Fo − Fc maps (blue mesh, contoured at 1.5σ) for the retinal binding pockets of KR2 under the acidic conditions is shown. Water molecules are depicted by red spheres.
Extended Data Figure 3 Structure-based sequence alignment of microbial rhodopsins.
The sequences are KR2, putative Na+-pump rhodopsin (NaR) from Dokdonia sp. PR095 (DsNaR, GenBank ID: AEX55013.1), putative NaR from Nonlabens dokdonensis DSW6 (NdNaR, GenBank ID: AGC76155.1), putative NaR from Truepera radiovictrix (TrNaR, NCBI reference sequence: YP_003706581.1), putative NaR from Gillisia limnaea (GlNaR, NCBI reference sequence: WP_006989277.1), putative NaR from Indibacter alkaliphilus (IaNaR, NCBI reference sequence: WP_009036080.1), xanthorhodopsin from Salinibacter ruber (Xanthorhodopsin, PDB ID: 3DDL)18, blue proteorhodopsin from Med12 (Blue_Proteorhodopsin, PDB ID: 4JQ6)43, bacteriorhodopsin from Halobacterium salinarium (Bacteriorhodopsin, PDB ID: 1IW6)44, halorhodopsin from Halobacterium salinarium (Halorhodopsin, PDB ID: 1E12)45, sensory rhodopsin II from Natronomonas pharaonis (Sensory_rhodopsinII, PDB ID: 3QAP)46, and the chimaeric channelrhodopsin between ChR1 from Chlamydomonas reinhardtii and ChR2 from Chlamydomonas reinhardtii (C1C2, PDB ID: 3UG9)47. The sequence alignment between KR2, xanthorhodopsin, blue proteorhodopsin, bacteriorhodopsin, halorhodopsin, sensory rhodopsin II and C1C2 was created using the T-Coffee server48. Secondary structure elements for KR2 are shown as coils and arrows. TT represents turns. Identical and conservatively substituted residues are highlighted in red. The N-helix residues and the residues tethering the N-helix to the protein core (Glu160 and Arg243 in KR2) are coloured green. Asp112 and Asp116 in the NDQ motif, and the Ser residue interacting with Asp116 (Ser70 in KR2) are coloured blue. Carboxylates on ECL1 are coloured yellow. The glutamine (Gln123 in KR2) that contributes to the putative intracellular Na+ binding site is coloured purple. The glycine and tyrosine residues that form the hydrophobic hole near the β-ionone ring of ATR are coloured orange. The asterisk under the alignment indicates the lysine that forms the Schiff base with ATR.
Extended Data Figure 4 Structural comparisons between KR2, BR and XR.
a, b, Side views (left) and extracellular views (right) of the superimposed structures of KR2 and BR (a) or KR2 and XR (b). The ATRs are shown as stick models. c, d, Structural comparison of the Schiff base orientations between KR2 and BR. c, Stereo view of the KR2 structure and its electron density in the Schiff base region. The 2Fo − Fc map (blue mesh, contoured at 2.0σ) is shown. d, Stereo view of the BR structure in the same region. The water molecule is depicted by a red sphere, and the hydrogen bond between the Schiff base and the water molecule is represented by the black dashed line.
Extended Data Figure 5 Functional characterization of wild-type KR2 and its mutants.
a, Na+ binding to wild-type (WT) KR2 and three ECL1 mutants. atrFTIR difference spectra of wild-type KR2 and three ECL1 mutants (E90Q/E91Q, D98N, and D102N) upon the exchange of 50 mM NaCl/KCl. Dotted lines are the duplicated difference spectra of wild type. b–d, Photocycle of the KR2 D102N mutant. b, c, Transient absorption spectra of the KR2 D102N mutant (b) and time traces of the absorption changes (c) at specific probe wavelengths. d, Photocycle scheme for the Na+ pump, determined from the analysis of the results shown in b and c. The lifetime of O-decay of wild-type KR2 is only shown for the major component (90.6% of total O-accumulation), and the previously reported minor component (9.4%) with τ = 112 ms is not discussed here15. Values are means and s.d. calculated from the traces shown in c. e, Expression levels of wild-type KR2 and its mutants. Blue bars indicate the expression levels of wild-type KR2 and its nine mutants (mean ± s.d., n = 3). The expression of the Δ1–18 mutant was too low to be determined. f, Left, magnified view of the interactions between the N-helix and TM4, TM7 and ECL1. Right, stereo view illustrating the interactions between the N-helix and TM4, TM7 and ECL1. Hydrogen bonds are shown by dashed lines and water molecules are represented by red spheres, the residue on the β-sheet is represented as a green stick model. g, Time constants of the decay of detergent-solubilized wild-type KR2 and mutant proteins in thermostability assays, shown as means ± s.d. estimated by the least squares fitting technique with an exponential function. All mutant proteins were assayed in buffer containing 100 mM NaCl (or RbCl for wild-type KR2), 50 mM Tris-HCl (pH 8.0), and 0.1% DDM.
Extended Data Figure 6 The electron densities around Asp116 under neutral conditions.
a–c, Stereo views of the structures and densities in the Schiff base region. The 2Fo − Fc maps (blue mesh, contoured at 1.0σ) and Fo − Fc maps (green and red meshes, contoured at 3.0 and −3.0σ, respectively) are shown. a, Structural refinement with Asp116 rotamer 1 resulted in a strong negative peak, suggesting its alternative conformation. b, The refinement with Asp116 omitted also suggested the presence of a mixture of Asp116 with different conformations. c, Asp116 with both rotamers 1 and 2 fits well in the electron density.
Extended Data Figure 7 Electrophysiological and behavioural analyses.
a, Membrane expression and localization of KR2 in mammalian cortical neurons (left, scale bar 20 μm) and C. elegans (right, scale bar 100 μm). b, KR2-dependent stable hyperpolarization. A current pulse was injected into a KR2-expressing neuron to evoke repetitive action potentials. The membrane potential was hyperpolarized for 10 s (top), 30 s (middle) and 60 s (bottom) when the neuron was illuminated by green light (5.4 mW per mm2, green bar). The resting potential was −74 mV throughout the experiment. c, The recovery of the locomotion speed in KR2-expressing C. elegans after cessation of illumination with green light. The green bar represents the light illumination.
Extended Data Figure 8 The retinal binding pockets and their surfaces in BR, XR and KR2.
a, Magnified views of the retinal binding pockets in BR (left panel), XR (middle panel) and KR2 (right panel). The residue that occludes the hydrophobic pore is indicated by the red dashed circle. b, Surface representations of the pocket, from the same viewpoint as in a. The blue and red dashed circles indicate the hydrophobic pores of XR and KR2, respectively. c, Surface representations of the pocket, from the same viewpoint as in a and b, showing the (putative) interaction between the second chromophore and the protein in the crystal structure of XR with salinixanthin (PDB ID: 3DDL) (left panel), and the putative binding site of the second chromophore, carotenoid (right panel).
Extended Data Figure 9 Putative water molecule between Asn112, Trp113 and Asp251.
Stereo view of the structures and densities in the Schiff base region, including the putative water molecule between Asn112, Trp113 and Asp251. The 2Fo − Fc maps (blue mesh, contoured at 1.5σ) are shown. The black dashed lines and the numbers are the possible hydrogen bonds and their distances (in Å) between the putative water molecule and the neighbouring residues.
Extended Data Figure 10 Calculated absorption spectra of wild-type KR2 in the O intermediate state.
The O absorption spectra of wild-type KR2 in the solution containing NaCl (red) or LiCl (yellow), calculated from the previously reported spectra15. The green dotted line represents the dark absorption spectrum of wild-type KR2 in the solution containing NaCl. The number above each spectrum is the peak wavelength (λmax).
Supplementary information
Supplementary Information
This file contains a Supplementary Discussion and Supplementary References. (PDF 205 kb)
Rights and permissions
About this article

Cite this article
Kato, H., Inoue, K., Abe-Yoshizumi, R. et al. Structural basis for Na+ transport mechanism by a light-driven Na+ pump. Nature 521, 48–53 (2015). https://doi.org/10.1038/nature14322
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14322
- Springer Nature Limited
This article is cited by
-
Current analytical approaches for characterizing nanoparticle sizes in pharmaceutical research
Journal of Nanoparticle Research (2024)
-
Light-Driven Sodium Pump as a Potential Tool for the Control of Seizures in Epilepsy
Molecular Neurobiology (2023)
-
Functional characterization of xanthorhodopsin in Salinivibrio socompensis, a novel halophile isolated from modern stromatolites
Photochemical & Photobiological Sciences (2023)
-
Acoustic levitation and rotation of thin films and their application for room temperature protein crystallography
Scientific Reports (2022)
-
Exploration of natural red-shifted rhodopsins using a machine learning-based Bayesian experimental design
Communications Biology (2021)